Journal of Biophysical Chemistry
Vol.2 No.2(2011), Article ID:5149,11 pages DOI:10.4236/jbpc.2011.22015
Unfolded annealing molecular dynamics conformers for wild-type and disease-associated variants of alpha-synuclein show no propensity for beta-sheet formation
1Biological Sciences Department, California State Polytechnic University Pomona, Pomona, Canada;
2Department of Chemistry at Lakehead University and Thunder Bay Regional Research Institute, Thunder Bay, Canada;
*Corresponding Author: wely.floriano@lakeheadu.ca
Received 21 December 2010; revised 20 January 2011; accepted 28 January 2011.
Keywords: Alpha-Synuclein; Parkinson’s Disease; Lewy Body Formation; Beta Sheet Formation;Unfolding; Molecular Simulations
ABSTRACT
Aggregation of alpha-synuclein leads to the formation of Lewy bodies in the brains of patients affected by Parkinson’s disease (PD). Native human alpha-synuclein is unfolded in solution but assumes a partial alpha-helical conformation upon transient binding to lipid membranes. Annealing Molecular Dynamics (AMD) was used to generate a diverse set of unfolded conformers of free monomeric wild-type alpha-synuclein and PD-associated mutants A30P and A53T. The AMD conformers were compared in terms of secondary structure, hydrogen bond network, solvent-accessible surface per residue, and molecular volume. The objective of these simulations was to identify structural properties near mutation sites and the non-amyloid component (NAC) region that differ between wildtype and disease-associated variants and may be associated to aggregation of alpha-synuclein. Based on experimental evidence, a hypothesis exists that aggregation involves the formation of intermolecular beta sheets. According to our results, disease-associated mutants of alphasynuclein are no more propense to contain extended beta regions than wild-type alpha-synu- clein. Moreover, extended beta structures (necessary for beta sheet formation) were not found at or around positions 30 and 53, or the NAC region in any unfolded conformer of wild-type, A30P or A53T alpha-synuclein, under the conditions of the simulations. These results do not support the hypothesis that the mutant's higher propensity to aggregation results solely from changes in amino acid sequence leading to changes in secondary structure folding propensity.
1. INTRODUCTION
Alpha-synuclein gene (SNCA) encodes a protein (alpha-synuclein) that is believed to integrate presynaptic signaling and membrane trafficking [1]. Defects in the SNCA gene have been implicated in the pathogenesis of Parkinson’s Disease (PD) [2-7] . Alpha-synuclein is expressed in the brain, in the substantia nigra, and it is specifically found in the dopamine nigrostriatal presynaptic neurons. Research suggests that alpha-synuclein aids brain function, possibly by helping cells communicate with one another. Aggregation of the misfolded protein in the brain causes the formation of Lewy Bodies, dark colored masses in the brain, which are landmarks of PD [8]. The accumulation of alpha-synuclein in the brain results in loss of dopaminergic neurons, with consequent reduction of striatal dopamine levels [9]. These protein aggregates are toxic enough to cause damage to the part of the brain responsible for motor movements, which is physically translated as slower movement and tremors. Alpha-synuclein peptides are also a major component of amyloid plaques in the brains of patients with Alzheimer’s Disease (AD) [10] and it accumulates abnormally in several other neurodegenerative illnesses [11] . Mutations in SNCA are associated with rare familial cases of early-onset Parkinson’s disease. In particular, the mutations A30P and A53T have been linked to familial PD [12].
Drug treatments that are prescribed to PD patients includes dopamine receptor agonists [9,13-16] , which alleviate symptoms but do not prevent progression of the disease. Currently, there is no drug treatment that specifically targets the aggregation of alpha-synuclein, or prohibits its accumulation. A need exists for identifying novel approaches for prevention and treatment of PD. A better understanding of the mechanisms of misfolding and aggregation of alpha-synuclein may facilitate the development of new therapeutic strategies. In this work, we use molecular dynamics simulations to gain insights into the first steps of alpha-synuclein aggregation.
Native human alpha-synuclein is unfolded in solution but assumes a partial alpha-helical conformation upon transient binding to lipid membranes [17,18] . Experimental data has shown that native alpha-synuclein is more compact than other natively unfolded proteins [18-19] . It has been proposed that this partial condensation is driven by interactions between the highly charged C-terminus and a large hydrophobic central region of the protein sequence, and that this structure could inhibit the formation of alpha-synuclein aggregates [19]. NMR spectroscopy has been used to characterize the conformational properties of alpha-synuclein in solution as a free monomer and when bound to lipid vesicles and lipid-mimetic detergent micelles [18]. The N-terminal region of alpha-synuclein binds to synthetic lipid vesicles and detergent micelles in vitro and adopts a highly helical conformation, consistent with predictions based on sequence analysis. The C-terminal part of the protein does not associate with either vesicles or micelles, remaining free and unfolded. The experimentally determined micelle-bound alpha-synuclein structure (PDB code 1XQ8) consists of two alpha helices connected by an 8-residue long loop, in a candy-cane shape. The disease-associated mutations A30P and A53T are located within the alpha helical regions of the NMR-determined micelle bound structure. A30P mutation is located on the first helix of the protein, while A53T is on the second helix. Both mutations may change propensity of these segments to fold into helices when in close proximity of the membrane and when in solution. In particular, the A30P replacement introduces a kink on the second helix of the helical alpha-synuclein, which may impact its folding and/or normal function.
Although specific factors that lead to protein misfolding and aggregation are not well understood, the first step in the aggregation process is thought to be the formation of alpha-synuclein dimers, which further aggregate into oligomers (Yu and Lyubchenko, Early stages for Parkinson’s development: alpha-synuclein misfolding and aggregation). It has been observed experimentally that neurotoxic protofibrils have beta-sheet-rich oligomers consisting of 20-25 alpha-synuclein molecules [10,11,17] , and that the mutants A30P and A53T are more likely to form aggregates than wild-type alpha-synuclein ADDIN EN.CITE ADDIN EN.CITE.DATA [11] . It has also been suggested that a central hydrophobic region comprised of residues 61-95, referred as NAC (non-amyloid component), promotes fibril formation [11] . NMR spectroscopy showed that native alpha-synuclein assumes several conformations that shield the NAC region of the protein through long-range interactions between the Nand C-termini [20] . The mutations A30P and A53T seem to cause structural fluctuations that lead to loss of the native conformations [20] .
A hypothesis can be deduced from the experimental evidence discussed above: free unfolded alpha-synuclein forms beta-sheets with other existing free alpha-synuclein molecules, leading to aggregation. To form intermolecular beta-sheets, one or more regions of the monomeric protein are likely to adopt extended beta conformation, which is required for beta-sheet formation. The regions more likely to be involved in the initial steps of aggregation are the regions immediately surrounding positions 30 and 53 (because mutants are more likely to aggregate), and/or the region comprising amino acids 61-95 (because shielding of this region is associated with reduced aggregation). To test this hypothesis, we used molecular dynamic simulations to generate an ensemble of unfolded single monomeric alpha-synuclein conformers. Since the formation of oligomers is thought to involve native unfolded alpha-synuclein, we first induced unfolding of the experimentally determined micellebound structure (PDB code 1XQ8) using high temperature MD. Unfolding was confirmed by analysis of secondary structure. Annealing MD (AMD) was subsequently used to generate a diverse pool of unfolded conformers, which were analyzed. Structural diversity of unfolded conformers was assessed by the degree of secondary structure remaining in the AMD conformers and the root mean square deviation in coordinates between conformers. Radius of gyration and 1H-15N chemical shifts were calculated for the AMD ensemble and compared to experimental values to validate the simulations. The same procedure adopted for the wild-type alpha-synuclein was used to generate unfolded conformers of the disease-associate mutants A30P and A53T. Secondary structure assessment of the MD conformers was used to determine the content of beta strands in various unfolded conformations of each variant of the protein. The native protein conformers for wild-type alpha-synuclein were compared to the disease-associated mutants to determine if the mutants show higher tendency to assume extended beta conformation and, hence to form beta sheets, and to identify the regions with higher propensity to form beta-strands. Solvent-accessible surface area, molecular volume, and hydrogen bond network analysis were used to examine whether unfolded conformers of mutant alpha-synuclein are more likely to expose the NAC region than the wild-type. A total of 290 structurally diverse unfolded conformers of each variant were analyzed.
2. METHODOLOGY
Overall Strategy. Our first step was to generate unfolded conformers of free monomeric native alpha-synuclein. For this step, the experimentally determined NMR structure of micelle-bound alpha-synuclein (PDB code 1XQ8) was used as starting point. This is due to the fact that no experimentally determined structure of unfolded (native) alpha synuclein is available. MD at constant temperature and pressure was used to unfold and equilibrate the native alpha-synuclein structure in solution. This step was repeated to generate unfolded structures for free monomeric disease-associated mutants A30P and A53T. Structures for the mutants were created by computationally mutating the corresponding residue in the NMR structure 1XQ8. The new side chain was placed using rotamer optimization, and the resulting structure was energy-minimized prior to MD. The last conformer from these equilibrium MDs were used as starting point for Annealing MD, which was performed with the objective of sampling the conformational space of unfolded alpha-synuclein and its unfolded mutants. Unfolded conformers for each alpha-synuclein variant were collected from MD and analyzed for secondary structure content, solvent-accessible surface, molecular volume, and hydrogen bond content. For all MDs, the protein was immersed in explicit water. The initial density of the water was set to 0.997g/mL (water at 298K). The pH of the simulations was set at 7 to simulate physiological conditions. The NaCl concentration in mass percent was set to 0.9% (physiological solution). The protein was placed in a cell under periodic boundary conditions. All MD simulations were performed using the program Yasara (www.yasara.org) with the Amber99 force field. Lennard-Jones forces and the direct space portion of the electrostatic forces were calculated using a cutoff of 7.86 Å. Electrostatic interactions were calculated using the Particle Mesh Ewald method as implemented in Yasara (www.yasara.org).
MD at Constant Number of Particles, Pressure, and Temperature (NPT). The MD simulations to unfold the initial experimental structure was performed with a 20 ps heating step from 0 K to 400 K, followed by 1800 ps at 400 K, and a cooling step from 400 K to 0 K in 20 ps. A previous MD study on the formation of alpha-synuclein pore-like structure [21] found that the experimentally determined micelle-bound structure is highly stable and retain its overall structure through MD at 310 K and 1atm, for 5 ns, in explicit water. For this reason, we used a higher temperature to induce unfolding (400 K). Intra/inter molecular forces were updated every 2 fs. Snapshots were saved every 10 ps. The pressure was set to the constant value of 1 atm. The choice of temperature was based on trial MD runs from 300 K in 50 K increments until significant loss of alpha-helix content was observed. For the last MD conformer of wild-type alpha-synuclein at 400 K, the secondary structure content dramatically decreased from the initial largely helical structure (61% alpha-helix content) to a disordered structure (6% alpha-helix, 40% coil, 54% turn). Similarly, the conformer at the end of the MD simulations for each alpha-synuclein variant was found to correspond to an unfolded state in solution. These last conformers were submitted to a steepest descent energy minimization followed by a simulated annealing minimization until the energy converged, and then were used as starting conformer for Annealing MD. The minimization protocol is described in detail elsewhere [22]. A total of 180 NPT MD conformers were generated and analyzed for each variant (wild-type, A30P and A53T).
Annealing MD. A total of 10 cycles of heating and cooling were performed for each case (wild-type, and A30P and A53T mutants). In each cycle, the system was heated from 0 K to 400 K in 20 ps, remained at 400K for 100 ps, and then cooled down to 0 K in 20 ps. Snapshots were saved every 5000 steps (10 ps). The pressure was kept constant at 1 atm and a multiple time step of 1 fs for intramolecular and 2 fs for intermolecular forces was used. A total of 110 unfolded AMD conformers were generated and analyzed for each wild-type alpha-synuclein, A30P and A53T variants (100 conformers from the 100 ps equilibration phases and 10 from the energyminimized (0 K) end points of each cycle).
Secondary Structure Determination. The secondary structure content for each conformer was determined using the program DSSP [23], and further processed by a custom-build script to generate secondary structure plots in html format. DSSP uses pattern recognition of hydrogen bonded and geometrical features to assign secondary structures to a protein structure. The resulting plots show the secondary structure assignment for each of the 140 amino acid in alpha synuclein. The plots are color-coded according to the type of secondary structure assigned to each residue (salmon: alpha helix; yellow: G-helix; cyan: P-helix; green: H-bonded turn; blue: bend; pink: beta bridge; orange:extended strand; gray: undetermined (random coil)).
Hydrogen Bond, Solvent-accessible Surface Area and Molecular Volume Calculations. Hydrogen-bond network (HB), solvent-accessible surface area (SASA) and molecular volume (MV) were determined using a custom-made Yasara script. This script calculated all the non-covalent interactions between hydrogen acceptors and donors, the residues involved in these interactions, the distance between them (in angstrom), the energy (kcal/mol) associated to the interaction, the overall number of donors, acceptors, and the total energy for each snapshot. SASA was calculated using effective solvation radii instead of normal van der Waals (VDW) radii to account for hydrogen bonds between water probe and solute. The molecular (solvent excluded) volume was calculated using a Gaussian approximation, as implemented in Yasara (www.yasara.org).
3. RESULTS AND DISCUSSION
Equilibrium NPT MD Simulations. A total of 180 snapshots taken during the MD simulation at constant temperature and pressure were used to determine the protein’s structural stability under the simulation conditions, and whether the MD reached equilibrium. The analysis of Energy vs. Time plots showed that all three cases reached equilibrium after 300 ps (these and similar plots are not presented, but can be obtained from the authors upon request). Temperature vs. time plots displayed very small fluctuations in temperature over the entire trajectories. The initial and final MD conformers for each case (wild-type, A30P and A53T) are shown in Figure 1. At the end of the unfolding MD, wild-type alpha-synuclein still resembles somewhat the initial “candy-cane” structure, while the mutants have assumed different shapes. The alpha-helix content, shown in Table 1, significantly decreased for all three variants from 61% for the original structure (PDB code 1XQ8) to 6% for wild-type, 9% for A30P, and 3% for A53T alpha-synuclein. Although the wild-type alpha-synuclein preserved its candy-cane shape somewhat, it did so without retaining much of its original secondary structure content.
The average, minimum, and maximum values for molecular volumes and solvent-accessible surface area for all NPT MD conformers are shown in Table 1. All three cases (wild-type, A30P and A53T) showed an increase in molecular volume, relative to the initial experimentally determined micelle-bound conformation (column “1XQ8” in Table 1), consistent with the transition from a membrane-bound helical conformation to an unfolded and more globular structure. The mutant A30P has the highest increase in molecular volume over the duration of the trajectory (4.7% compared to 4.1% for wild-type and 4.0% for A53T). On average, the unfolded conformers have lower SASA than the helical micelle-bound one used as starting point of the simulations. All three cases lost 15% - 20% of its initial HB content by the end of the
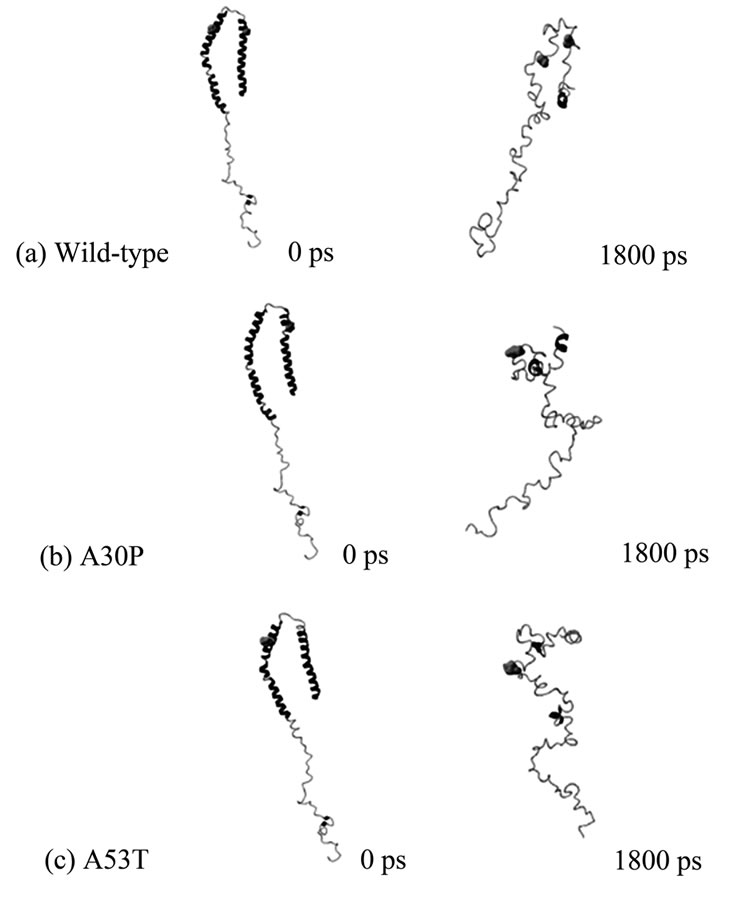
Figure 1. Initial (0ps) and final (1800ps) NTP MD conformers of (a) Wild-type; (b) A30P; and (c) A53T alpha-synuclein. Positions 30 (right) and 53 (left) are shown as van der Waals spheres, colored in green. Wild-type alpha-synuclein is unfolding during the simulation but still retains its “candy-cane” structure, which is not observed in the unfolded mutants. The final MD conformers correspond to solution (native) structures of wild-type and disease-associated mutants of alpha-synuclein.
simulation. Wild-type alpha-synuclein had the most change in the number of intramolecular hydrogen bonds, while A30P has the least change in the number of hydrogen bonds.
Temperature-induced MD unfolding from a micelle-bound to a solution conformation thus involved transitioning to higher molecular volumes and less solvent-exposed surface area for all three alpha-synuclein variants studied here. This is consistent with hydrophobic residues originally interacting with a membrane environment (hence more exposed) being gradually shielded from the solvent as the protein becomes more globular.
The number of conformers displaying a particular type of secondary structure at positions 30 and 53 for disease-associated mutants compared to wild-type alpha-synuclein is shown in Figure 2. These bar graphs were generated to give an overall idea of the content of secondary structures the mutated residues assume throughout the entire simulation. From these graphs, the overall picture is that there is no propensity to assume extended beta conformation at these positions in alpha
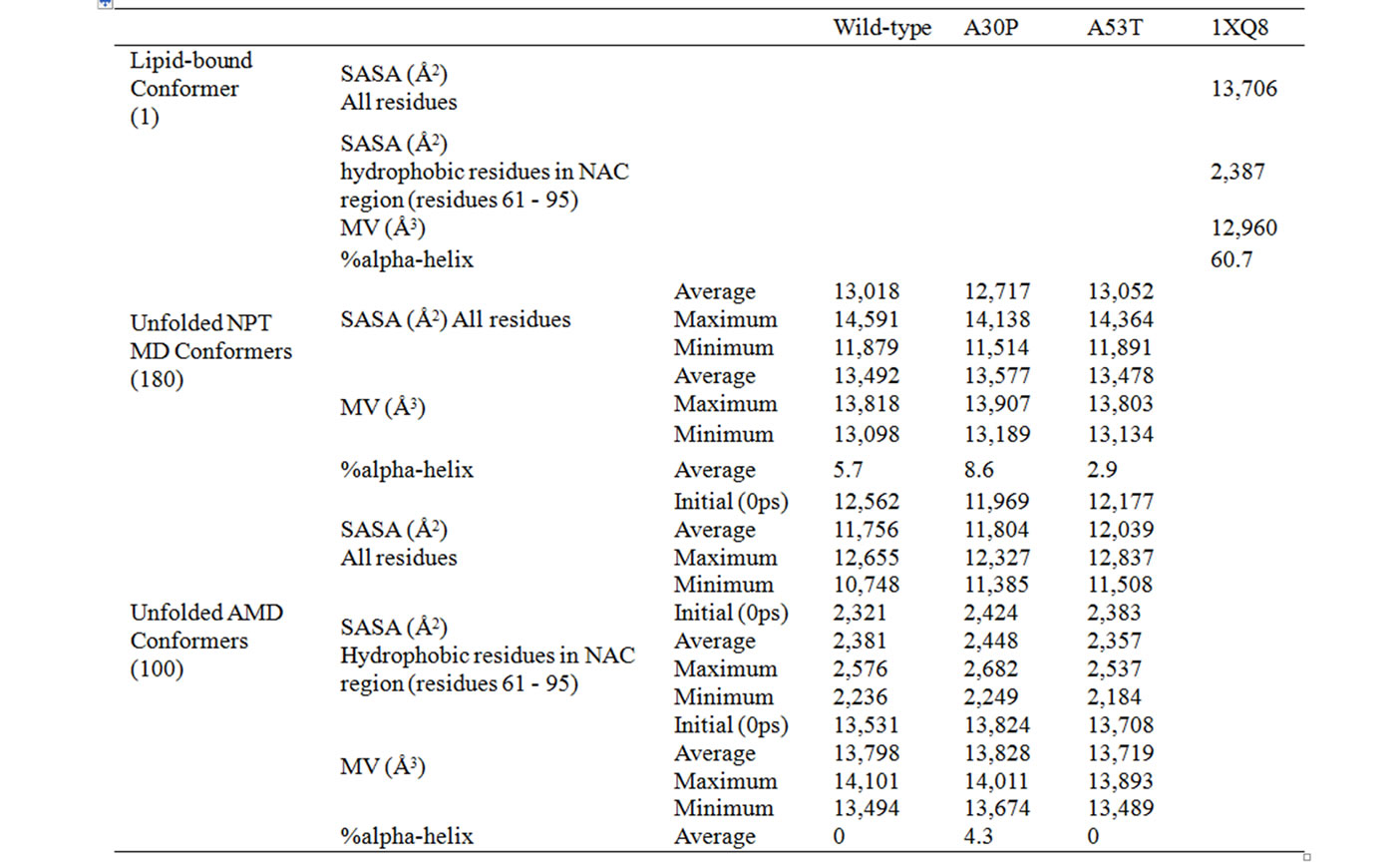
Table 1. Average values for molecular volumes (MV), solvent-accessible surface areas (SASA), and secondary structure content for wild-type, A30P and A53T alpha-synuclein conformers obtained by NPT MD and AMD. SASA is reported for all residues or for only the hydrophobic residues (A, G, I, L, M, P, Y, V) in the NAC region (residues 61 to 95). Percent of alpha-helical secondary structures were determined using Yasara [24].
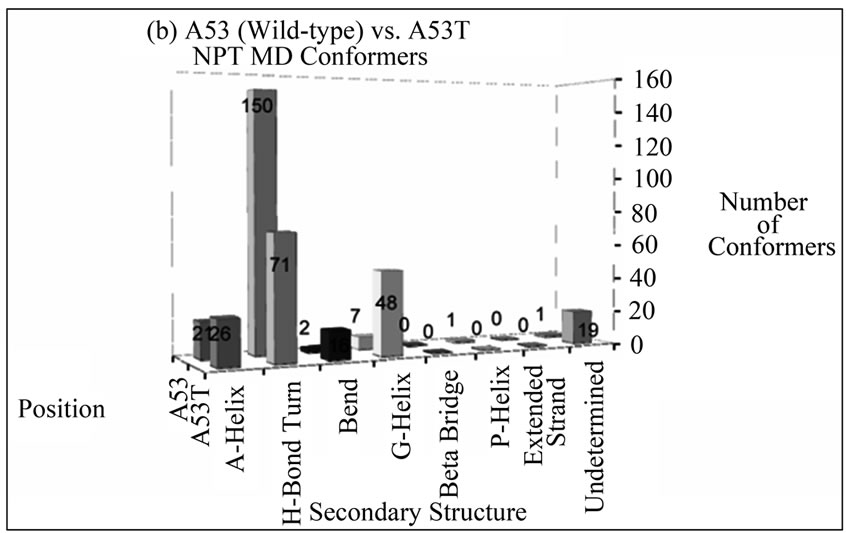
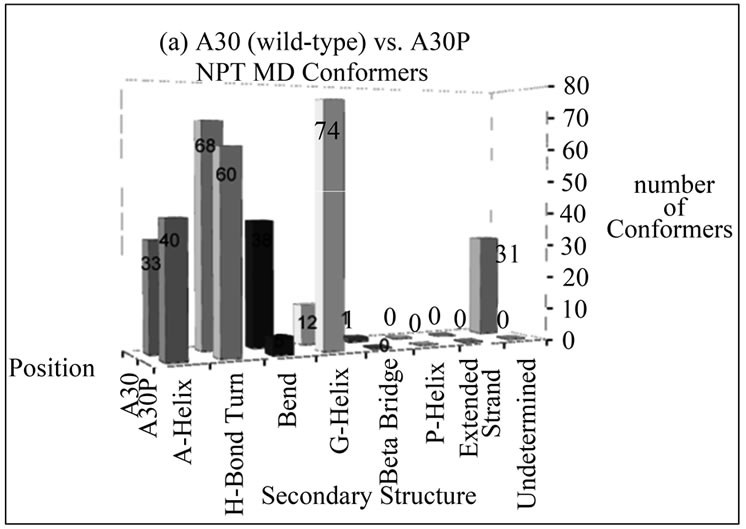
Figure 2. Number of NTP MD conformers folded into a particular type of secondary structure at positions (a) 30 and (b) 53 in wild-type (A30 and A53) or disease-associated mutants (A30P and A53T) of alpha-synuclein. The mutant MD conformers display larger helical content at these positions compared to wild-type alpha-synuclein.
synuclein variants, and the mutants are more likely to retain helical conformation (alpha or G) in both positions than wild-type alpha-synuclein.
Annealing MD. The Potential Energy trend for wild-type alpha-synuclein during annealing MD (AMD) was to decrease over time. Each cycle started with a higher energy conformer than it ended. A30P had a few cycles (2, 3, 4, and 8) producing conformers higher in energy than the ones from the other cycles. The values for Potential Energy for A53T followed the same trend observed for wild-type alpha-synuclein, with the late conformers being lower energy than the early-cycle ones. Smaller energy fluctuations are observed within a cycle than across cycles. The root mean square deviation
(RMSD) in Carbon alpha coordinates was calculate to assess how much each AMD conformer differ from the initial unfolded conformation. All three cases showed an increase in RMSD relative to the initial conformation throughout the AMD. The larger RMSD value among AMD conformers was 10Å for wild-type, 12Å for A30P, and 13Å for A53T. The overall behavior of the energy the AMD conformers being lower than the initial NPT MD conformer, along with the RMSD distribution indicates that AMD was successful at creating a diverse set of stable unfolded conformers.
Alpha-synuclein changed conformation without much loss of intermolecular hydrogen bonds. There was a shift in partners but not a great variation in number. Overall, the mutants had the most change in hydrogen bonds, with bonds broken and re-made in different parts of the protein while folding to specific conformers. The number of HBs ranged from 80 to 142 for wild-type, 75 - 140 for A30P, and 90 - 146 for A53T alpha-synuclein unfolded conformers.
The secondary structure assignment per residue is presented in Figure 3 for the energy-minimized average conformer of the AMD ensembles of each alphasynuclein variant. Secondary structure assignments for the original micelle-bound structure and for the last NPT MD conformers are also presented in Figure 3. Percents of alpha-helical secondary structures determined using Yasara are shown in Table 1. The number of unfolded AMD conformers assuming a particular secondary structure at positions 30 or 53 for wild-type, A30P, and A53T alpha-synuclein is shown in bar-graph representations in Figure 4.
The alpha-helix content for the AMD ensembles is significantly lower than the content found for micelle-bound alpha-synuclein. The average AMD conformer for wild-type and A53T mutant have 0% alpha-helix, while the A30P mutant retains 4% alpha-helical structures. Some content of 3 - 10 helix is also observed (6% for wild-type, 4% for A30P, and 3% for A53T alpha-synuclein). Extended beta conformation (necessary for beta sheet formation) was not observed around positions 30 or 53, or in the NAC region (residues 61 - 95) in any unfolded conformer of wild-type, A30P or A53T alpha-synuclein. In comparison to wild-type, the mutant A30P has higher propensity to form alpha helices at and

Figure 3. Secondary structure assignment per residue for the micelle-bound alpha-sybuclein (PDB code 1XQ8), last conformer from NPT MD, and energy-minimized time average AMD conformers of (a) wild-type; (b) A30P; and (c) A53T alpha-synuclein. Secondary structures are color-coded as follows: salmon: alpha helix; yellow: G-helix; cyan: P-helix; green: H-bonded turn; blue: bend; pink: beta bridge; orange: extended strand; gray: undetermined (random coil). Positions 30, 53 and the NAC region (residues 61 to 95) are enclosed by thick lines. Assignment was based on torsion angles and hydrogen bond patterns as determined by the program DSSP [23].
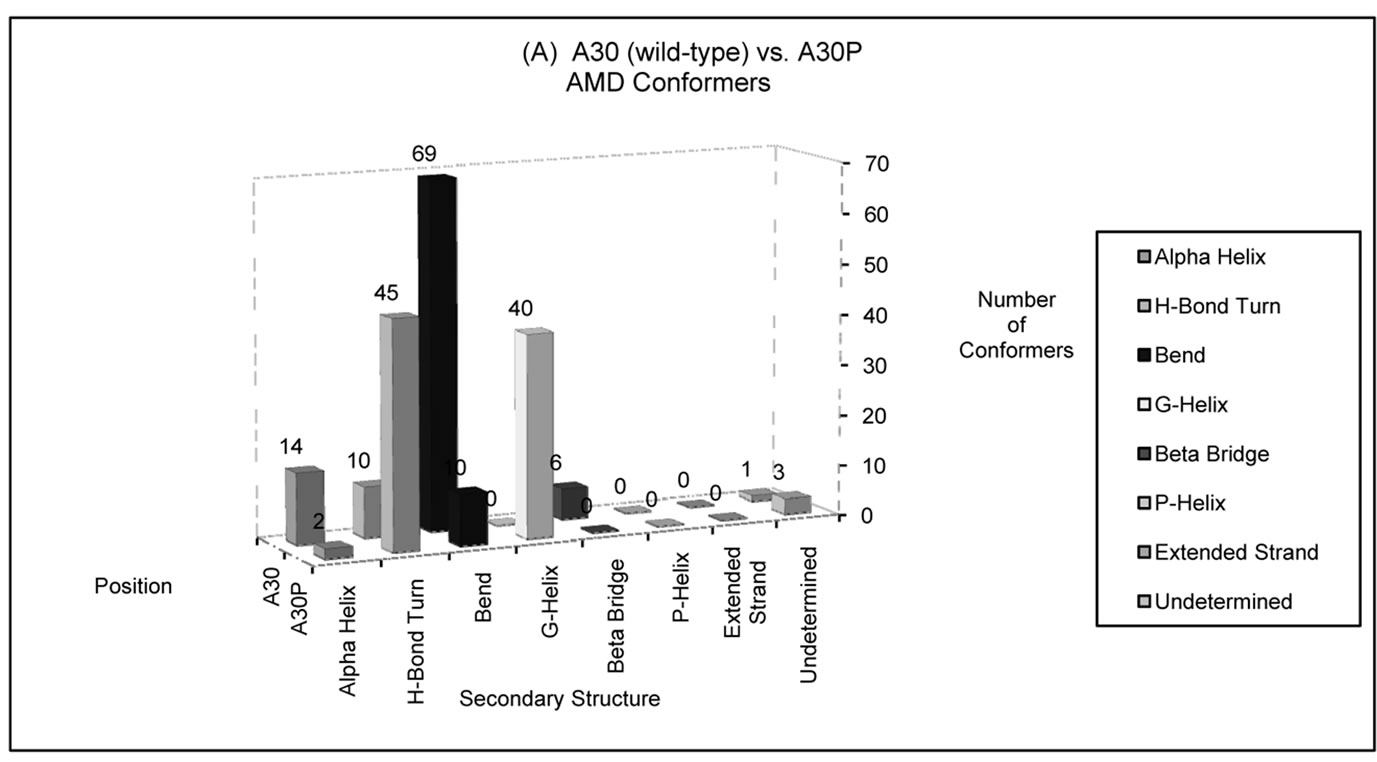

Figure 4. Number of AMD conformers folded into a particular type of secondary structure at positions (a) 30 and (b) 53 in wild-type (A30 and A53) or disease-associated mutants (A30P and A53T) of alpha-synuclein. The disease-associated AMD conformers display larger helical content at these positions compared to wild-type alpha-synuclein. Position 30 has a higher propensity to fold into a helical structure among A30P conformers, compared to the wild-type conformers. In contrast, wild-type conformers are more likely to assume helical conformations at position 53 than unfolded conformers of A53T alpha-synuclein.
around position 30. In contrast, wild-type conformers were more likely to assume helical conformations at position 53 than unfolded conformers of PD-associated mutant A53T.
The average values for molecular volumes and solvent-accessible surface area for all AMD conformers are shown in Table 1. The molecular volume of the average AMD conformer was 6% - 7% higher than the folded (micelle-bound) conformer for all three cases. A53T is the most compact variant overall. Solvent-Accessible Surface Areas for all residues and for hydrophobic residues in the NAC region (residues 61 - 95) of wild-type,
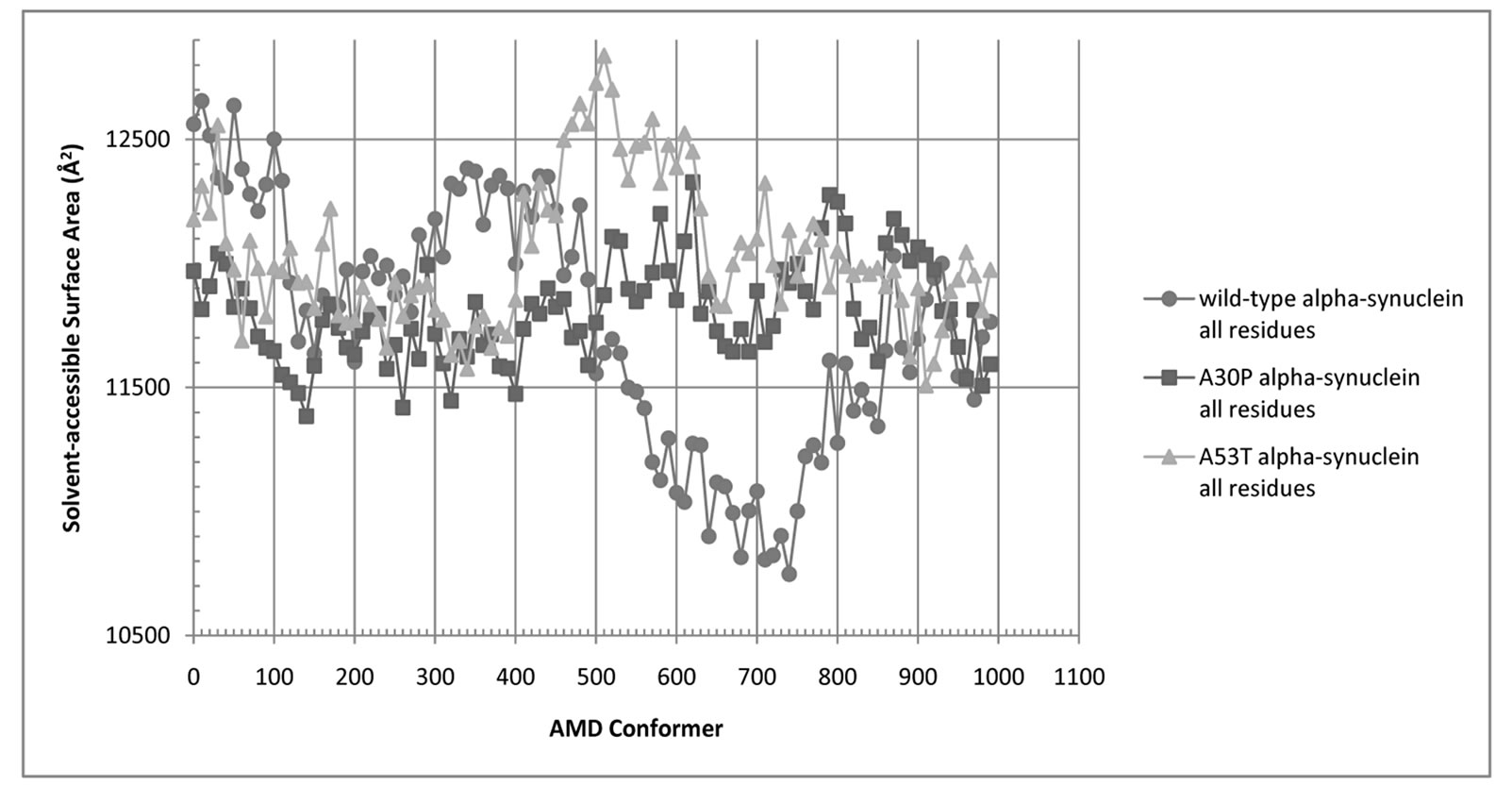
Figure 5. Solvent-Accessible Surface Area (SASA) for all residues of wild-type (blue circles), A30P (red squares), and A53T alpha-synuclein (green triangles).
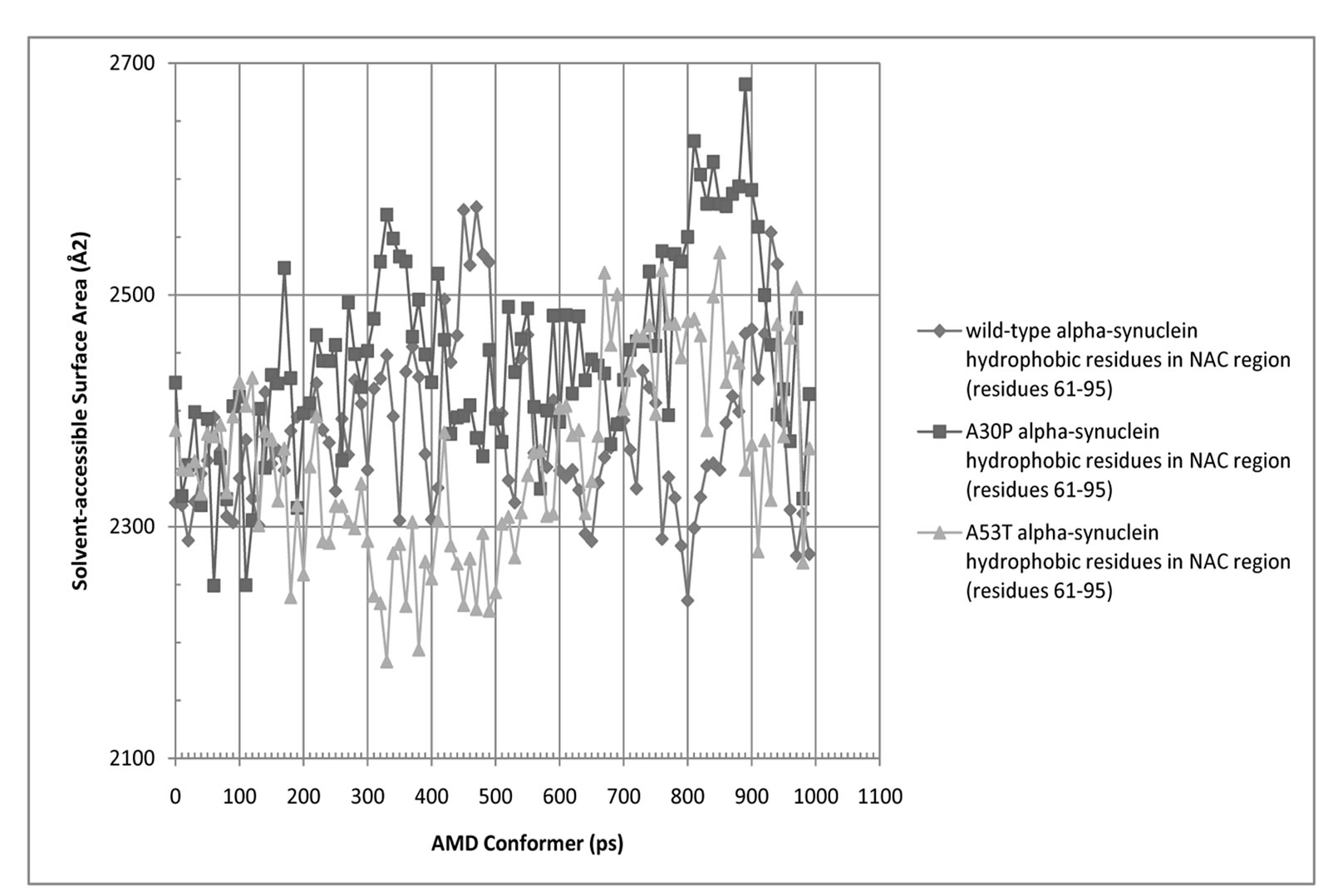
Figure 6. Solvent-Accessible Surface Area (SASA) for hydrophobic residues in the NAC region (residues 61 - 95) of wild-type, A30P, and A53T alpha-synuclein.
and A30P and A53T alpha-synuclein are shown in Figures 5 and 6. The average SASA values in Å2 for wild-type, A30P and A53T are, respectively, 11756, 11804, and 12039. The micelle-bound folded alpha-synuclein is 8% to 13% more exposed to solvent than the average unfolded variants, which is consistent with the globular nature of the unfolded conformers versus the helical candy-cane shaped folded protein. On average, the hydrophobic residues in the NAC region ranged from 2% (A30P) more to 2% less (A53T) exposed to solvent than in the micelle-bound structure. The NAC region tends to be more exposed in A30P than in the other variants. The average NAC region SASA value for unfolded wild-type is comparable to micelle-bound alpha-synuclein, which indicates most AMD conformers did not display significant shielding of that region. Some conformers, however, had up to 8.5% lower exposure of the hydrophobic residues in the NAC region than micelle-bound alpha-synuclein, which is consistent with experimental observations ADDIN EN.CITE ADDIN EN.CITE.DATA [20] .
Validation of the MD ensembles using experimental parameters. To increase confidence in our results, we compared experimentally determined biophysical parameters (radius of gyration and 15N resonances) to predicted values derived from the AMD ensembles.
Radius of gyration was calculated from the center of mass for each AMD conformer of wild-type alpha-synuclein using Yasara, and compared to experimental values determined from small angle x-ray scattering at 23˚C (296 K) and various pHs [25]. The experimental radius of gyration at pH 7.5 and 23˚C for wild-type alpha-synuclein is 40 ± 1 Å, whereas at pH 3 the radius of gyration was found to be 30 ± 1 Å [25]. The average radius of gyration for the AMD ensemble of wild-type alpha-synuclein was 34.86 Å, which is within the experimental range. In contrast, the calculated radius of gyration for the mostly helical micelle-bound structure (PDB code 1XQ8) is 48.46 Å.
Chemical shifts were predicted using ShiftX [26] for the average AMD conformers and the experimentally determined micelle-bound structure 1XQ8. The micelle-bound structure was used as reference to assess the accuracy of the predictions. Experimentally determined 15N and 13C chemical shifts were downloaded from the Biological Magnetic Resonance Bank (BMRB) (http://www.bmrb.wisc.edu) for micelle-bound (folded), wild-type, A30P and A53T alpha-synuclein (BMRB accession numbers 5744, 16543, 16546, and 16548). The original data was obtained by solution NMR at 293K and pH 6.0 [27] for the disordered structures, and at 298K and pH 7.4 in sodium dodecyl sulfate micelles for folded alpha-synuclein [28]. Experimental values were directly compared to those predicted by ShiftX. Prolines were explicitly excluded from the linear regressions to avoid the possibility of missing data being interpreted as zeros, which can skew the correlations. The correlation coefficients, R2, for the 15N comparisons were: 0.54 for the experimentally determined structure 1XQ8, 0.45 for AMD wild-type, 0.46 for AMD A30P, and 0.39 for AMD A53T alpha-synuclein. All correlations involved at least 133 data points and were all deemed statistically significant by two-tailed P value tests using Prism (GraphPad Inc.). The relatively low R2 value for the experimentally determined structure 1XQ8 reflects the accuracy of ShiftX, whereas the R2 associated to the AMD conformers reflect both ShiftX’s accuracy and the ability of the AMD conformers to adequately represent the experimental ensemble. The fact that all R2 are statistically significant indicates that the structures used to calculate the chemical shifts are reasonable representations of the ensembles that generated the experimental data. Other factors that may account for differences between predicted and experimentally determined values are temperature and the absence of multiple alpha-synuclein molecules in the simulations. The predicted chemical shifts were also in general agreement with other published NMR studies [29-32] .
4. CONCLUSIONS
Parkinson’s disease is a degenerative neurological disorder characterized by rigidity, tremors, and bradykinesia. This is caused by loss of dopaminergic neurons. A landmark of PD is the formation of Lewy bodies, which are protein aggregates thought to play a role in neuronal loss. A major component of these aggregates is the protein alpha-synuclein, which is natively unfolded in solution. There are two mutations (A30P and A53T) that have been linked to the disease and seem to give alpha-synuclein a higher propensity to form aggregates in the substantia nigra ADDIN EN.CITE ADDIN EN.CITE.DATA [11] . A proposed mechanism for aggregation involves the initial formation of dimers and subsequent oligomerization through intermolecular beta-sheet formation. In order for aggregation to occur via this mechanism, the individual alpha-synuclein monomers need to fold one or more region of its peptide chain into extended beta conformation. Since disease-associated alpha-synuclein variants are more likely to aggregate, they should also be more likely to display extended beta structures than the wild-type protein.
The scope of this study was to investigate through molecular dynamics whether disease-associated mutants of alpha-synuclein are intrinsically more disposed to form intermolecular beta sheets compared to wild-type, thus explaining their observed propensity to form aggregates. Our results suggest that disease-associated mutants of free monomeric alpha-synuclein have no higher propensity to assume extended beta conformation than wild-type alpha-synuclein, at the conditions simulated. It is possible that other factors not considered in our simulations, such as the presence of metal ions or excess of free monomeric protein, alter the conformational landscape of alpha-synuclein, perhaps favoring the formation of extended beta structures under these conditions. Experimental evidence [9,33-35] exists for metal ions binding to alpha-synuclein that seems to support this hypothesis, and further simulations are necessary to investigate this possibility. Evidence also exists for modulation of alpha-synuclein conformation by other molecules such as dopamine derivatives [36] and pesticides [37-40] , and a molecular docking-MD study is underway to investigate these effects. Simulations involving multiple alpha-synuclein molecules are also needed to investigate whether the conformational changes necessary to promote aggregation are induced by the presence of other alpha-synuclein molecules, and how many are necessary to promote aggregation. Previous MD simulations of intermolecular beta sheets between peptide segments comprised of alpha-synuclein residues 71 - 82 [41] indicate that anti-parallel beta-sheet conformers are stable at 320 K and higher temperature, which seems to support this idea.
Studies such as this one provide valuable insights into the molecular basis of the disease and may aid the design of new strategies to prevent or control alpha-synuclein aggregation. Therapeutic strategies targeting causes, not only symptoms, are the best hope for PD patients.
5. ACKNOWLEDGEMENTS
The authors would like to thank Dr. Cristian Follmer (Federal University of Rio de Janeiro) for providing us with valuable insights into the alpha-synuclein and Parkinson’s disease world. This work was supported in part by the College of Science and the Biological Sciences Department at the California State Polytechnic University Pomona (WBF's previous affiliation).
REFERENCES
- Clayton, D.F. and George, J.M. (1999) Synucleins in synaptic plasticity and neurodegenerative disorders. Journal of Neuroscience Research, 58, 120-129.doi:10.1002/(SICI)1097-4547(19991001)58:1<120::AID-JNR12>3.0.CO;2-E
- Bueler, H. (2009) Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Experimental Neurology, 218, 235-246.
- Zhou, H.Y. and Chen, S.D. (2005) Parkin, Parkin substrates and Parkinson’s disease. Progress in Biochemistry and Biophysics, 32, 912-916.
- Dekker, M.C.J. et al. (2003) Parkinson’s disease: Piecing together a genetic jigsaw. Brain, 126, 1722-1733.doi:10.1093/brain/awg172
- Hattori, N. et al. (2000) Autosomal recessive juvenile parkinsonism: A key to understanding nigral degeneration in sporadic Parkinson’s disease. Neuropathology, 20, S85-S90. doi:10.1046/j.1440-1789.2000.00312.x
- Wszolek, Z.K. and Markopoulou, K. (1999) Molecular genetics of familial parkinsonism. Parkinsonism & Related Disorders, 5, 145-155.doi:10.1016/S1353-8020(99)00030-9
- Schapira, A.H.V. (1997) Pathogenesis of Parkinson’s disease. Baillieres Clinical Neurology, 6, 15-36.
- Spillantini, M.G. et al. (1997) Alpha-synuclein in Lewy bodies. Nature, 388, 839-840. doi:10.1038/42166
- Sit, S.Y. (2000) Dopamine agonists in the treatment of Parkinson’s disease—Past, present and future. Current Pharmaceutical Design, 6, 1211-1248. doi:10.2174/1381612003399581
- Khan, A. et al. (2005) Metals accelerate the formation and direct the structure of amyloid fibrils of NAC. Journal of Inorganic Biochemistry, 99, 1920-1927.doi:10.1016/j.jinorgbio.2005.06.018
- Lashuel, H.A. et al. (2000) Protofilaments, filaments, ribbons, and fibrils from peptidomimetic self-assembly: Implications for amyloid fibril formation and materials science. Journal of the American Chemical Society, 122, 5262-5277. doi:10.1021/ja9937831
- Jellinger, K.A. (2007) Morphological substrates of parkinsonism with and without dementia: A retrospective clinico-pathological study. Journal of Neural Transmission-Supplement, 72, 91-104.
- Cabedo, N. et al. (2009) An overview on benzylisoquinoline derivatives with dopaminergic and serotonergic activities. Current Medicinal Chemistry, 16, 2441-2467. doi:10.2174/092986709788682100
- Montastruc, J.L. et al. (1999) Treatment of Parkinson’s disease should begin with a dopamine agonist. Movement Disorders, 14, 725-730.doi:10.1002/1531-8257(199909)14:5<725::AID-MDS1003>3.0.CO;2-L
- Gottwald, M.D. et al. (1997) New pharmacotherapy for Parkinson’s disease. Annals of Pharmacotherapy, 31, 1205-1217.
- Uitti, R.J. and Ahlskog, J.E. (1996) Comparative review of dopamine receptor agonists in Parkinson’s disease. Cns Drugs, 5, 369-388.doi:10.2165/00023210-199605050-00006
- Tofaris, G.K. and Spillantini, M.G. (2007) Physiological and pathological properties of alpha-synuclein. Cellular and Molecular Life Sciences, 64, 2194-2201.doi:10.1007/s00018-007-7217-5
- Ulmer, T.S. et al. (2005) Structure and dynamics of micelle-bound human alpha-synuclein. Journal of Biological Chemistry, 280, 9595-9603. doi:10.1074/jbc.M411805200
- Dedmon, M.M. et al. (2005) Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. Journal of the American Chemical Society, 127, 476-477. doi:10.1021/ja044834j
- Bertoncini, C.W. et al. (2005) Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. Journal of Biological Chemistry, 280, 30649-30652.doi:10.1074/jbc.C500288200
- I.F. Tsigelny, et al. (2007) Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS Journal, 274, 1862-1877. doi:10.1111/j.1742-4658.2007.05733.x
- Krieger, E. et al. (2004) Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins, 57, 678-683. doi:10.1002/prot.20251
- Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogenbonded and geometrical features. Biopolymers, 22, 2577- 2637. doi:10.1002/bip.360221211
- Krieger, E. et al. (2002) Increasing the precision of comparative models with YASARA NOVA—A selfparameterizing force field. Proteins, 47, 393-402. doi:10.1002/prot.10104
- Uversky, V.N. et al. (2001) Evidence for a partially folded intermediate in alpha-synuclein fibril formation. Journal of Biological Chemistry, 276, 10737-10744. doi:10.1074/jbc.M010907200
- Neal, S. et al. (2003) Rapid and accurate calculation of protein H-1, C-13 and N-15 chemical shifts. Journal of Biomolecular Nmr, 26, 215-240.doi:10.1023/A:1023812930288
- Bodner, C.R. et al. (2010) Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry, 49, 862-871.doi:10.1021/bi901723p
- Chandra, S. et al. (2003) A broken alpha-helix in folded alpha-synuclein. Journal of Biological Chemistry, 278, 15313-15318. doi:10.1074/jbc.M213128200
- Fernandez, C.O. et al. (2004) NMR of alphasynuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. Embo Journal, 23, 2039-2046.
- Georgieva, E.R. et al. (2010) The Lipid-binding Domain of Wild Type and Mutant alpha-Synuclein compactness and interconversion between the broken and extended helix forms. Journal of Biological Chemistry, 285, 28261-28274. doi:10.1038/sj.emboj.7600211
- Sasakawa, H. et al. (2007) Ultra-high field NMR studies of antibody binding and site-specific phosphorylation of alpha-synuclein. Biochemical and Biophysical Research Communications, 363, 795-799.doi:10.1016/j.bbrc.2007.09.048
- Wu, K.P. et al. (2008) Characterization of conformational and dynamic properties of natively unfolded human and mouse alpha-synuclein ensembles by NMR: Implication for aggregation. Journal of Molecular Biology, 378, 1104-1115. doi:10.1016/j.jmb.2008.03.017
- Wolozin, B. and Golts, N. (2002) Iron and Parkinson’s disease. Neuroscientist, 8, 22-32.doi:10.1177/107385840200800107
- Golts, N. et al. (2002) Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. Journal of Biological Chemistry, 277, 16116-16123. doi:10.1074/jbc.M107866200
- Rasia, R.M. et al. (2005) Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proceedings of National Academy Sciences of USA, 102, 4294-4299. doi:10.1073/pnas.0407881102
- Latawiec, D. et al. (2010) Modulation of alpha-synuclein aggregation by dopamine analogs. Plos One, 5, e9234.doi:10.1371/journal.pone.0009234
- Gatto, N. et al. (2009) Alpha-Synuclein repeat polymorphisms and pesticide exposure in Parkinson’s disease. Epidemiology, 20, S124-S124.doi:10.1097/01.ede.0000362429.51721.26
- Betarbet, R. et al. (2006) Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiology of Disease, 22, 404-420. doi:10.1016/j.nbd.2005.12.003
- Cicchetti, F. et al. (2009) Environmental toxins and Parkinson's disease: What have we learned from pesticide-induced animal models? Trends in Pharmacological Sciences, 30, 475-483.doi:10.1016/j.tips.2009.06.005
- Richardson, J.R. et al. (2009) Elevated Serum Pesticide Levels and Risk of Parkinson Disease. Archives of Neurology, 66, 870-875. doi:10.1001/archneurol.2009.89
- Yoon, J. et al. (2009) Simulation studies on the stabilities of aggregates formed by fibril-forming segments of alpha-Synuclein. Journal Biomolecular Structure and Dynamics, 27, 259-270.