Journal of Biophysical Chemistry
Vol.2 No.2(2011), Article ID:5138,12 pages DOI:10.4236/jbpc.2011.22014
MetalloPred: a tool for hierarchical prediction of metal ion binding proteins using cluster of neural networks and sequence derived features
Department of Biotechnology and Bioinformatics, Jaypee University of Information Technology, Waknaghat, India; *Corresponding Author: pknaik73@rediffmail.com; pradeep.naik@juit.ac.in
Received 19 January 2011; revised 13 February 2011; accepted 21 February 2011.
Keywords: Metalloprotein; Classification;Sequence Derived Parameters; Neural Networks
ABSTRACT
Given a protein sequence, how can we identify whether it is a metalloprotein or not? If it is, which main functional class and subclasses it belongs to? This is an important biological question because they are closely related to the biological function of an uncharacterized protein. Particularly, with the avalanche of protein sequences generated in the post genomic era and since conventional techniques are time consuming and expensive, it is highly desirable to develop an automated method by which one can get a fast and accurate answer to these questions. Here, a top-down predictor, called MetalloPred, is developed which consists of 3 level of hierarchical classification using cascade of neural networks from sequence derived features. The 1st layer of the prediction engine is for identifying a query protein as metalloprotein or not; the 2nd layer for the main functional class; and the 3rd layer for the sub-functional class. The overall success rates for all the three layers are higher than 60% that were obtained through rigorous cross-validation tests on the very stringent benchmark datasets in which none of the proteins has 30% sequence identity with any other in the same class or subclass. MetalloPred achieved good prediction accuracies and could nicely complement experimental approaches for identification of metal binding proteins. MetalloPred is freely available to be used in-house as a standalone and is accessible at http://www.juit.ac.in/assets/Metallopred/.
1. INTRODUCTION
Metalloprotein is a generic term for a protein that contains a metal ion cofactor. Metalloproteins have captivated chemists and biochemists, particularly since 1950s, when the first X-ray crystal structure of a protein, sperm whale myoglobin indicated the presence of an iron atom [1]. The metal ion is usually coordinated by nitrogen, oxygen or sulfur atoms belonging to amino acids in the polypeptide chain and/or a macro-cyclic ligand incorporated into the protein [2,3]. The presence of the metal ion allows metalloenzymes to perform functions such as redox reactions that cannot be performed by the limited set of functional groups found in amino acids [1]. Metalloproteins play important roles in structural stability and complex formation [4-8], gene expression regulation and alteration [9-12], DNA processing [13], signaling processes and cellular event [14], transport [11,15,16], metabolism control [15,17], antibody recognition [18] and other biological processes such as cellular respiration, photosynthesis, nitrogen fixation and antioxidant defense [19]. Approximately, 1/3 of structurally-determined proteins are metalloproteins [20]. Much effort has been devoted to understanding the structure and function of these proteins.
Traditionally the metalloproteins have been identified, based on experimental techniques such as absorbance spectroscopy [21], gel electrophoresis [22], metal-affinity columns and shift assay [23], chromatography [24], mass spectroscopy [22], NMR [9] and combined spectroscopic studies [25]. These techniques which require purified or semi-purified proteins of interest, do not facilitate identification of unknown proteins from a complex mixture, or require multi-step processes and very specialized equipment which limit their application ranges. Therefore, there is need to explore alternative methods for facilitating the identification of metalloproteins to complement these experimental methods. With the exponential growth of sequence data, an insurmountable task of characterizing these sequences with experimental methods is very cumbersome. It is thus desirable to explore automated computational methods for the annotation of novel protein sequences. Several sequenced-based computational methods have been explored based on similarity search, metal-binding sites sequence motifs [26,27] and multiple sequence alignments against known metalloproteins [28]. Because of the sequence, structural and functional diversity of metalloproteins [4-8,14-17], it is desirable to explore additional methods that predict metalloproteins directly from sequence or sequence-derived properties. For a newly-found protein sequence the most interesting thing people wish to know is about its biological function and hence the following questions are often asked: Is the query protein a metalloprotein or non-metalloprotein? If it is, which main functional class does it belong to? Or going further deeper, what about its sub-functional class? The present study was initiated in an attempt to develop a top–down approach to solve all these problems and make it accessible to the vast majority of experimental scientists by providing a user-friendly web-server In this study, we have developed cascade of artificial neural network (NN) prediction systems for metalloproteins. The generalized classification obtained by the method suggests that MetalloPred could be useful as a starting point in initial screening and ab initio prediction of metalloproteins, and, in combination with comparative studies on completed genomic sequences, it could give further insight into the evolution of protein structure and function.
2. MATERIALS AND METHODS
2.1. Preparation of Dataset
All metalloproteins used in this study are collected from a comprehensive search of protein data bank (www.rcsb.org). A total of 14625 metalloprotein sequences were obtained and have been classified into calcium-binding (3466), magnesium-binding (2886), potassium-binding (173), sodium-binding (157), cobalt-binding (200), copper-binding (887), manganese-binding (968), molybdenum-binding (134), nickel-binding (147), vanadium-binding (11), zinc-binding (4861) and ironbinding (328). This data set was further refined by discarding protein sequences having length less than 20 amino acids, as they are very unlikely form a proper pocket to coordinate with metal ion. Some proteins were found to bind with more than one metal ion and have been discarded. With the aim of avoiding prejudiced learning in the networks, we scaled the sequences such that the inequality in the number of protein sequences in each class may be compromised. We reduced the proteins in each class with a similarity cutoff of 30% using BLASTClust [29]. A negative dataset consisting of 5738 protein sequences, representing non-class members is also selected from PDB database. These datasets are divided into training, testing and independent evaluation sets (Table 1).
2.2. Feature Extraction
Following three types of discrete feature vectors were constructed for each protein sequence.
1) Amino acid composition: given the sequence of a protein, its amino acid composition was computed and then used to generate a set of 20 features representing composition of 20 standard amino acids in the protein sequences that include A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, and Y. These features have been widely used in predicting different structural classes
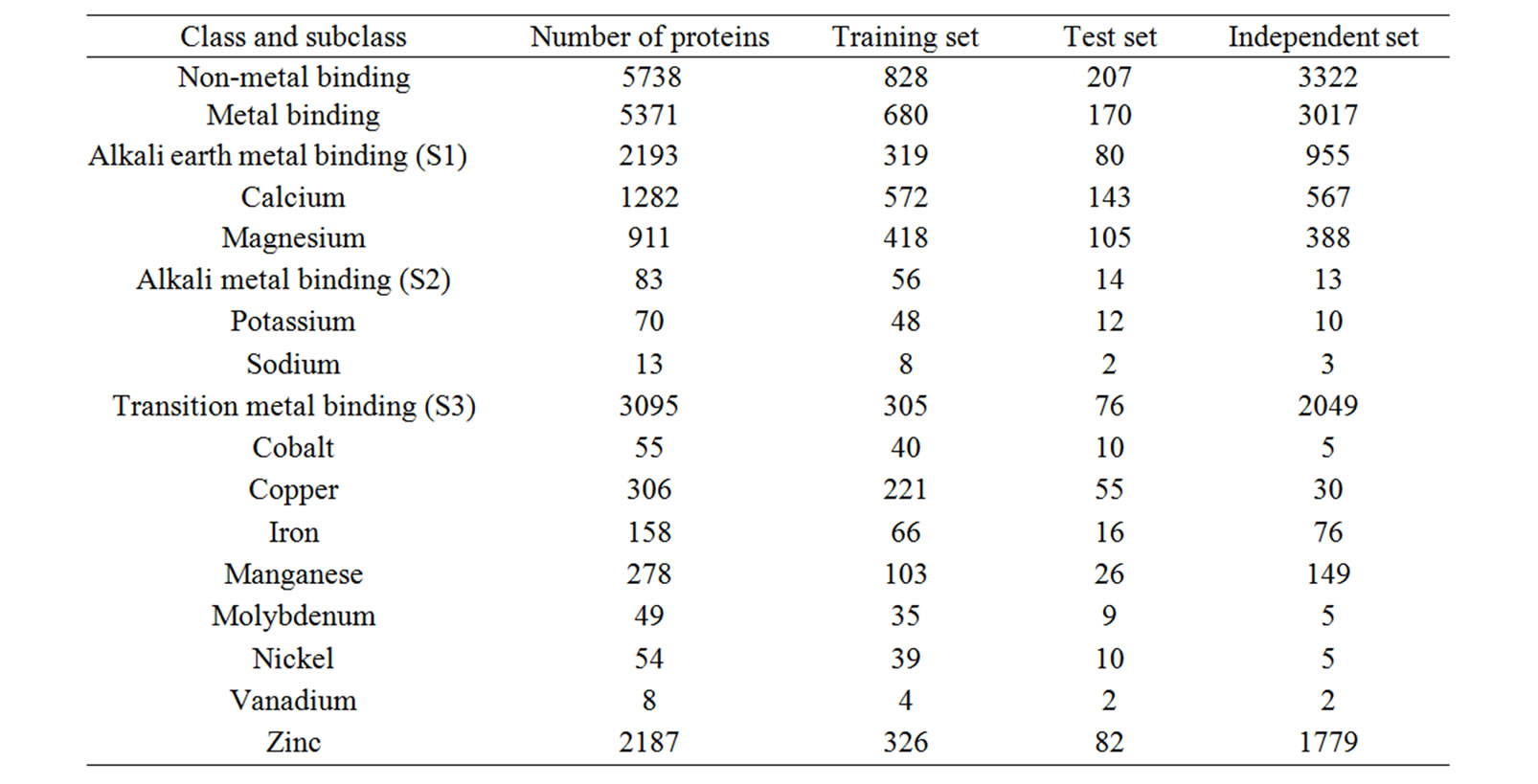
Table 1. Number of proteins used for training and validation of MetalloPred.
[30-32] and subcellular localization [33-36] of proteins. The formula used to calculate amino acid composition is:
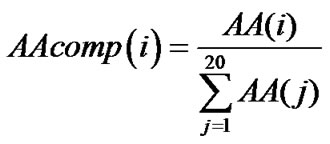
where AA(i) = Frequency of ith amino acid.
2) Physicochemical properties: twelve sequence derived properties for each protein sequence was calculated using EMBOSS (EBI) package [37]. The parameters include molecular weight, total charge, isoelectric point, mole percentages of tiny (A, C, G, S, T); small (A, C, D, G, N, P, S, T, V); aliphatic (I, L, V); aromatic (F,H,W,Y); non-polar (A, C, F, G, I, L, M, P, V, W, Y); polar (D, E, H, K, N, Q, R, S, T), charged (D, E, H, K, R); acidic (D, E) and basic (H, K, R) amino acids.
3) Pseudo amino acid composition (PseAA)
This class of descriptor consists of a set of 37 features, 20 of which are weighted amino acid compositions and rest 17 are correlation factors calculated among amino acids for each protein sequence [38].
A protein sequence P with L amino acid resides can be represented as:
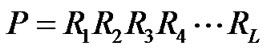 (1)
(1)
where R1 represents the 1st residue of the protein P, R2 the 2nd residue, and so forth. According to the simplest discrete model, the amino acid composition of the protein P based on the equation (1) can be expressed as:
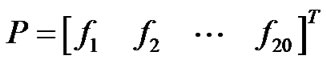 (2)
(2)
where 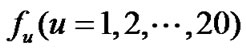 are the normalized occurrence of frequencies for the 20 native amino acids in P, and T the transposing operator. The additional 17 features are a series of rank-different correlation factors along a protein chain and were calculated as follows.
are the normalized occurrence of frequencies for the 20 native amino acids in P, and T the transposing operator. The additional 17 features are a series of rank-different correlation factors along a protein chain and were calculated as follows.
A protein sequence P consisting of L amino acid resides can be represented as:

(3)
where 20 + λ components are given by
 (4)
(4)
where w is the weight factor and τk is the k-th tier correlation factor that reflects the sequence order correlation between all the k-th most contiguous residues as formulated by
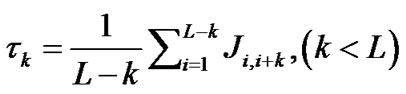 (5)
(5)
with
 (6)
(6)
where Φξ(Ri) is the ξ-th function of the amino acid Ri, and Г the total number of the functions considered. Φ1(Ri), Φ2(Ri) and Φ3(Ri) represented respectively the hydrophobicity value [39], hydrophilicity value [40], and side chain mass of amino acid Ri (Table 2); while Φ1(Ri+k), Φ2(Ri+k) and Φ3(Ri+k) are the corresponding values for the amino acid Ri+k. Therefore, the total number of functions considered is Γ = 3.
It can be seen from equation (3) that the first 20 components, i.e., p1, p2,  , p20 are associated with the conventional AA composition of protein, while the remaining components p20+1,
, p20 are associated with the conventional AA composition of protein, while the remaining components p20+1,  , p20+λ are the correlation factors that reflect the 1st tier, 2nd tier,
, p20+λ are the correlation factors that reflect the 1st tier, 2nd tier,  , and the λth tier sequence order correlation patterns. It is through these additional λ factors that the important sequence-order information is incorporated.
, and the λth tier sequence order correlation patterns. It is through these additional λ factors that the important sequence-order information is incorporated.
2.3. System Architecture and Component of NN Topology
The overall classification system consists of three layers of successive multilayer feed forward (acyclic) artificial NNs (Figure 1), each one with a single hidden layer at which the computation takes place. Some com
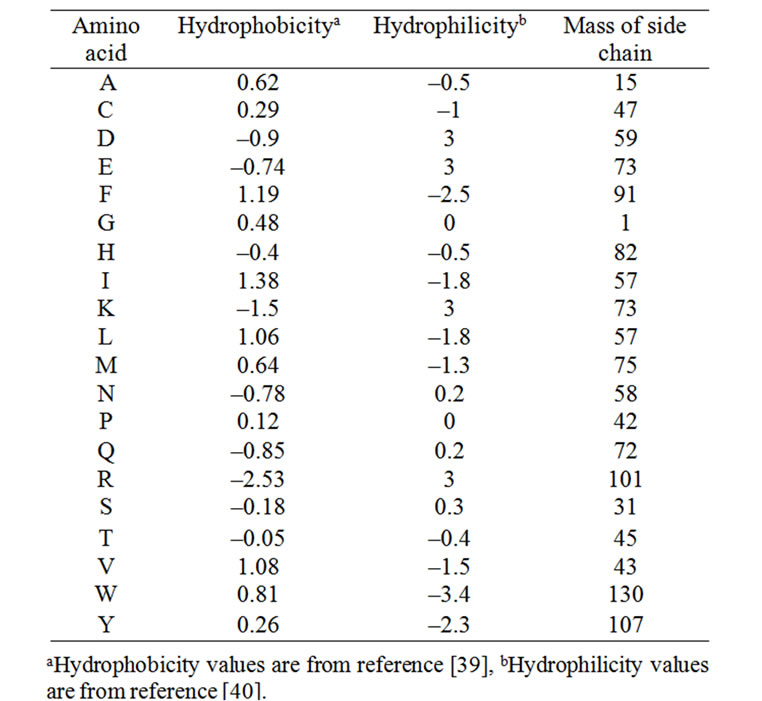
Table 2. Hydrophobicity, hydrophilicity and mass of side chain scales for 20 amino acids used in calculating pseudo amino acid composition (PseAA).
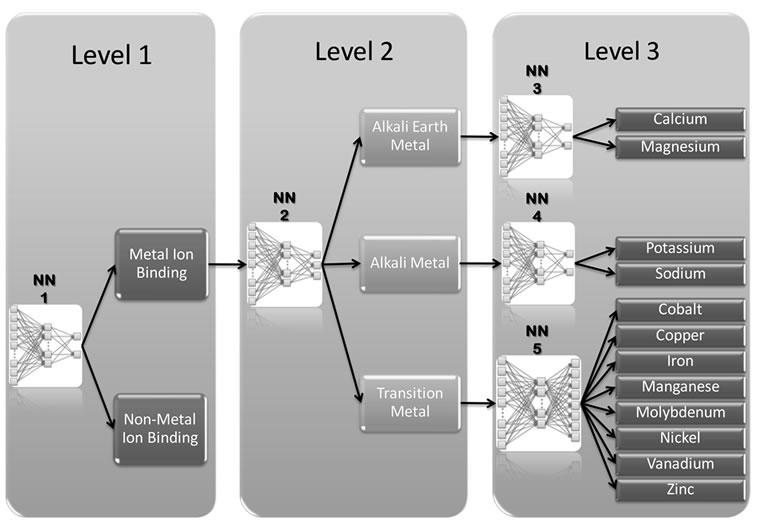
Figure 1. A schematic drawing to classify metalloproteins and non-metalloproteins as well as the three main functional classes of metalloproteins and their subclasses.
mon features shared by all NNs are the following:
1) There is full connectivity as every node in each network layer is connected to every other node in the adjacent forward layer.
2) There are a small number of nodes in the hidden layer responsible for the actual learning process carried out by each component network.
3) The activation function on each node is a nonlinear, sigmoid logistic function of the weighted sum of all synaptic weights (plus a constant bias).
NN1 is binary classifiers which classify an input protein sequence as a metalloprotein or non-metalloprotein. If the input protein sequence is classified as a metalloprotein then it is processed by NN2 which gets classified into one of the three main classes of metal binding proteins (alkali earth metal, alkali metal or transition metal). Each class consists of an independent NN [(alkali earth metal (NN3), alkali metal (NN4) and transition metal (NN5)] for classification of input protein sequence specifically into metal ion it binds. We have used three categories of sequence derived features such as physicochemical properties, amino acid composition and pseudo amino acid composition for training of NNs. Using these parameters independently and with combination we have developed seven neural network clusters: NNprop, NNAAcomp, NNpseAA, NNpseAA + prop, NNAAcomp + prop, NNpseAA + AAcomp and NNpseAA + AAcomp + prop. Before the learning process, all network synaptic weights are initialized to small random values which have been optimized to final weights during the learning process based on backpropagation algorithm [41].
An important issue in the design of a NN classification system is the network’s generalization, that is, its ability to give correct predictions when it is presented with unseen examples. With a small number of training samples and a relatively large number of synaptic weights, there is always the possibility that the network’s free parameters will adapt to the special features of the training data (over-fitting). A straightforward way to overcome this problem is to use a sufficient number of training examples (usually more than 30 times the number of adjustable network parameters). However, the protein classes are unbiased and it is not possible to have these many numbers. Therefore to control the over fitting in our application, we have employed non-convergent criteria (early stopping method); the training process is stopped before the finishing of optimization procedure. We follow the common method which is to withhold and use part of the training data (20%) as an internal validation set. Training is stopped at the point at which the classification error on the holdout subset begins to rise.
In the prediction phase, just like the forward pass in learning, network weights are globally fixed (those obtained after the convergence of the training process) and the NN is presented with an unknown example for classification. In the same hierarchical manner, the input signal propagates once in the forward direction and the output value constitutes the network’s decision based on the already studied training examples. The prediction accuracy of the models has been validated using self test, jackknife test and independent data set. For jackknife test we randomized the test set for 100 times and recorded average performance accuracy.
3. RESULTS AND DISCUSSION
To assess the performance of the MetalloPred, we applied several tests. We created a new independent test set with well-characterized protein sequences from all level of classes and sub-classes (Table 1) to evaluate the performance of the new integrated system. In addition we have also performed sub-sampling test (self test) and jackknife test for examining the accuracy of MetalloPred. All these validation tests are commonly used for performance evaluation of a predictor. Jackknife test is deemed the most rigorous and objective [30] and hence has been increasingly adopted by investigators in examining the quality of various prediction methods [42-44]. A direct comparison with results from previous metal binding protein prediction studies may not be most appropriate because of the differences in the protein classes predicted, datasets, protein descriptors, prediction methods and parameters.
3.1. Performance of 1st Layer of Neural Network
The performance and validation results of NN1 are given in Table 3" target="_self"> Table 3. An overall accuracy of 99.74% and
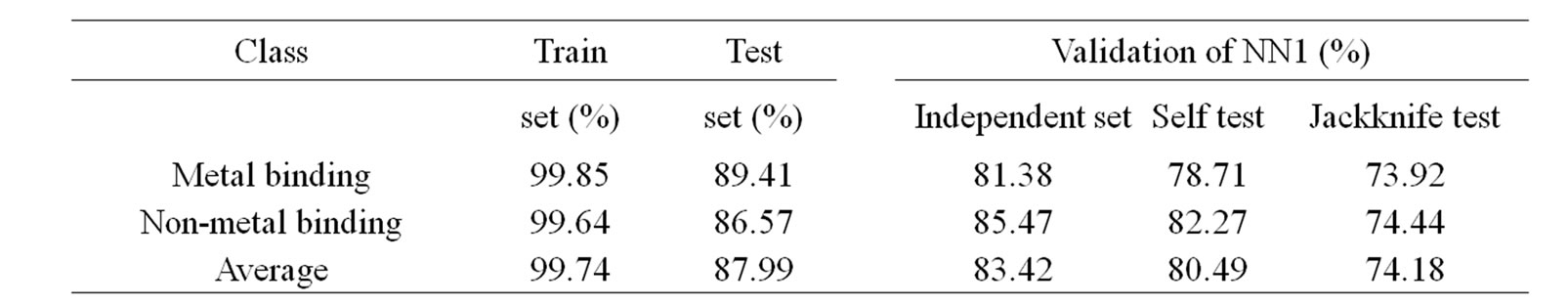
Table 3. Performance accuracy and validation results of 1st layer of MetalloPred based on combination of pseudo amino acid composition, amino acid composition and physicochemical properties.
87.99% for the training and test set data using combination of sequence derived features such as pseudoamino acid composition, amino acid composition and physicochemical properties. While considering the validation techniques by using an independent data set, self test and jackknife test, the overall accuracy of the 1st layer of MetalloPred is 83.42%, 80.49% and 74.18% respectively. The details of the performance accuracy and validation results based on different types of sequence derived feature have been represented in supplementary Table 1.
3.2. Performance of 2nd Layer of Neural Network
The overall success rate in identifying the metalloproteins among their three main functional classes is 99.25% (using training set) and 81.91% (using test set) (Table 4). Similarly the overall performance accuracy based on three types of validation tests has been found to be 75.16% (using independent data set), 73.24% (using self test) and 64.23% (using jackknife test). The corresponding results by Metallopred on the data set for three major classes of metalloproteins using different types of sequence derived features are given in supplementary Table 2.
3.3. Performance of 3rd Layer of Neural Network
The performance accuracy and validation results of NNs in identifying subclasses of alkali earth metal (NN3), alkali metal (NN4) and transition metal (NN5) binding proteins using the combination of all sequence derived features has been given in Table 5. The corresponding results by MetalloPred on the detection of calcium and magnesium metal binding proteins are 91.72% (training set), 91.07% (test set), 77.46% (independent data set), 81.1% (self test) and 75.51% (jackknife test) on the data set ‘S1’. Similarly for the data set ‘S2’ the performance accuracy for the detection of potassium and sodium binding proteins are 97.95% (training set), 96.4% (test set), 79.2% (independent data set), 91.57% (self test) and 83.33% (jackknife test). The overall accuracy of detection of cobalt-binding, copper-binding, iron-binding, manganese-binding, molybdenum-binding, nickel-binding, vanadium-binding and zinc-binding is 98.88% (training set), 95.39% (test set), 84.06% (independent data set), 71.39% (self test) and 67.98% (jackknife test) using the data set ‘S3’. The details of the performance accuracy have been represented in supplementary Table 3.
For the current data sets in which none of the protein sequence has ≥30% sequence identity to any other in a same class or subclass, the overall success rates by the MetalloPred in identifying the main functional classes of metalloproteins and their subclasses is very high. In an earlier study, contribution of individual feature property to protein classification is investigated by separately conducting classification by the use of each feature property [45-47].
The same method was employed here. An analysis on the classification of the group of all metal binding proteins seems to suggest that, in order of prominence, the hydrophobicity and hydrophilicity play a more prominent role than other feature properties. Hydrophobicity has been shown to be important for metal-protein interactions such that metal binding sites usually appear in clusters with hydrophobic environment. High-affinity metal binding sites in some proteins are located at sequence segments with specific amino acid composition, and specific sequence motifs have been used for predicting metal-binding proteins [48-50]. It was also found that polarity and solvent accessibility of the binding site influences the functional properties of metal-binding proteins. Therefore, our prediction results are consistent with these experimental findings. Overall MetalloPred is a very powerful predictor in identifying metalloproteins, their main classes, and their subclasses.
4. CONCLUSIONS
From a practical point of view, the most important aspect of a prediction model is its ability to make correct predictions. Till date most of the available methods use the 3-D structure of the protein to predict and classify metal ion binding proteins. This is a very tedious job and requires much costlier endeavors. The sequence of a protein is an important determinant for the detailed molecular function of proteins and would consequently also be useful for prediction of metal ion binding protein and
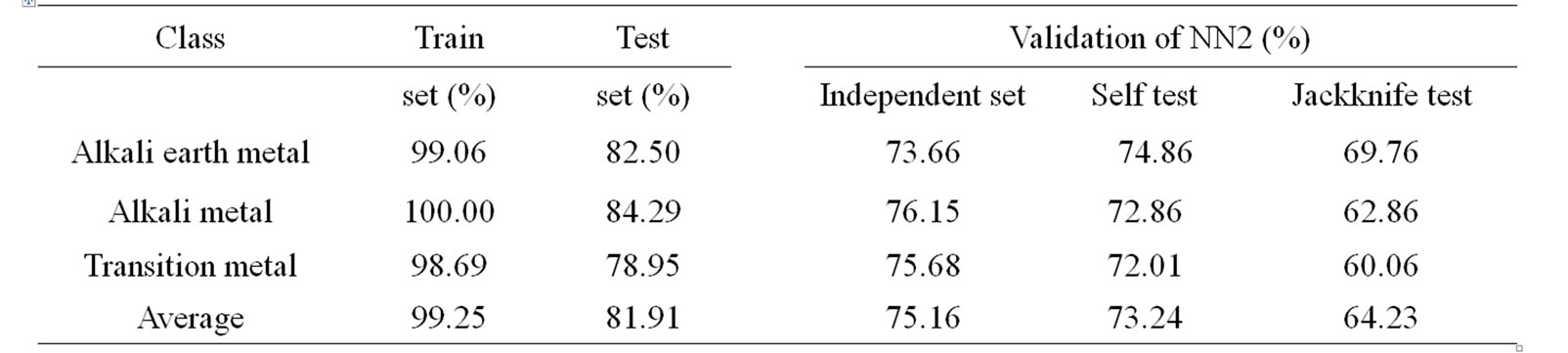
Table 4. Performance accuracy and validation results of 2nd layer of MetalloPred based on combination of pseudo amino acid composition, amino acid composition and physicochemical properties.

Table 5. Performance accuracy and validation results of 3rd layer of MetalloPred based on combination of pseudo amino acid composition, amino acid composition and physicochemical properties.
their classes. Additionally much encouraging results have been predicted using the sequence derived parameters technique. Therefore, a much accurate and reliable method is to predict the metal ion binding proteins and metal ion binding protein classes based on both strategies. Cascade of neural networks used in this study appears to be a potentially useful tool for the prediction of metalbinding proteins of different classes. The prediction accuracy may be further enhanced with the further expansion of our knowledge about metal-binding proteins, particularly for those small metal-binding classes, more refined representation of the structural and physicochemical properties of proteins and the improvement of prediction algorithms such as the better treatment of imbalanced dataset in the next version of our prediction tool.
REFERENCES
- Finkelstein, J. (2009) Metalloproteins. Nature, 460, 813-813. doi:10.1038/460813a
- Rosette, M. and Malone, R. (2002) Bioinorganic chemistry a short course. John Wiley and Sons, New York.
- Gregolinski, J., Starynowicz, P., Hua, K.T., Lunkley, J.L., Muller, G. and Lisowski, J. (2008) Helical lanthanide (III) complexes with chiral nonaaza macrocycle. Journal of American Chemical Society, 130, 17761-17773. doi:10.1021/ja805033j
- Wintz, H., Fox, T., Wu, Y.Y., Feng, V., Chen, W., Chang, H.S., Zhu, T. and Vulpe, C. (2003) Expression profiles of Arabidopsis thaliana in mineral deficiencies reveal novel transporters involved in metal homeostasis. Journal of Biological Chemistry, 278, 47644-47653. doi:10.1074/jbc.M309338200
- Cox, E.H. and McLendon, G.L. (2000) Zinc-dependent protein folding. Current Opinion in Chemical Biology, 4, 162-165. doi:10.1016/S1367-5931(99)00070-8
- Michel, S.L. and Berg, J.M. (2002) Building a metal binding domain, one half at a time. Chemical Biology, 9, 667-668. doi:10.1016/S1074-5521(02)00160-6
- Guntinas, M.B., Bordin, G. and Rodriguez, A.R. (2002) Identification, characterization and determination of metal-binding proteins by liquid chromatography. Analytical and Bioanalytical Chemistry, 374, 369-378. doi:10.1007/s00216-002-1508-3
- Yang, W., Lee, H.W., Hellinga, H. and Yang, J.J. (2002) Structural analysis, identification, and design of calcium-binding sites in proteins. Proteins, 47, 344-356. doi:10.1002/prot.10093
- Jensen, M.R., Petersen, G., Lauritzen, C., Pedersen J. and Led, J.J. (2005) Metal binding sites in proteins: Identification and characterization by paramagnetic NMR relaxation. Biochemistry, 44, 11014-11023. doi:10.1021/bi0508136
- Wu, H., Yang, Y., Jiang, S.J., Chen, L.L., Gao, H.X., Fu, Q.S., Li, F., Ma, B.G. and Zhang, H.Y. (2005) DCCP and DICP: Construction and analyses of databases for copperand iron-chelating proteins. Genomics, Proteomics and Bioinformatics, 3, 52-57.
- Hantke, K. (2001) Iron and metal regulation in bacteria. Current Opinion in Microbiology, 4, 172-177. doi:10.1016/S1369-5274(00)00184-3
- Bouton, C.M. and Pevsner, J. (2000) Effects of lead on gene expression. Neurotoxicology, 21, 1045-1055.
- Feng, M., Patel, D., Dervan, J.J., Ceska, T., Suck, D., Haq, I. and Sayers, J.R. (2004) Roles of divalent metal ions in flap endonuclease-substrate interactions. Nature Structural and Molecular Biology, 11, 450-456. doi:10.1038/nsmb754
- Carafoli, E. (2002) Calcium signaling: A tale for all seasons. Proceeding National Academic of Science USA, 99, 1115-1122. doi:10.1073/pnas.032427999
- Harris, E.D. (2000) Cellular copper transport and metabolism. Annual Review of Nutrition. 20, 291-310. doi:10.1146/annurev.nutr.20.1.291
- O’Halloran, T.V. and Culotta, V.C. (2000) Metallochaperones: An intracellular shuttle service for metal ions. Journal of Biological Chemistry, 275, 25057-25060. doi:10.1074/jbc.R000006200
- Vallee, B.L. and Auld, D.S. (1990) Active-site zinc ligands and activated H2O of zinc enzymes. Proceeding National Academic of Science USA, 87, 220-224. doi:10.1073/pnas.87.1.220
- Zhou, T., Hamer, D.H., Hendrickson, W.A., Sattentau, Q.J. and Kwong, P.D. (2005) Interfacial metal and antibody recognition. Proceeding National Academic of Science U.S.A., 102, 14575-14580.doi:10.1073/pnas.0507267102
- Lieu, P.T., Heiskala, M., Peterson, P.A. and Yang, Y. (2001) The roles of iron in health and disease. Molecular Aspects of Medicine, 22, 1-87.doi:10.1016/S0098-2997(00)00006-6
- Barondeau, D.P. and Getzoff, E.D. (2004) Structural insights into protein metal ion partnerships. Current Opinion of Structural Biology, 14, 765-774. doi:10.1016/j.sbi.2004.10.012
- Reed, G.H. and Poyner, R.R. (2000) Mn2+ as a probe of divalent metal ion binding and function in enzymes and other proteins. Metal Ions Biological Systems, 37, 183-207.
- Binet, M.R., Ma, R., McLeod, C.W. and Poole, R.K. (2003) Detection and characterization of zincand cadmium-binding proteins in Escherichia coli by gel electrophoresis and laser ablation inductively coupled plasma-mass spectrometry. Analytical Biochemistry, 318, 30-38. doi:10.1016/S0003-2697(03)00190-8
- Herald, V.L., Heazlewood, J.L., Day, D.A. and Millar, A.H. (2003) Proteomic identification of divalent metal cation binding proteins in plant mitochondria. FEBS Letter, 537, 96-100. doi:10.1016/S0014-5793(03)00101-7
- Papoyan, A. and Kochian, L.V. (2004) Identification of Thlaspi caerulescens genes that may be involved in heavy metal hyperaccumulation and tolerance: Characterization of a novel heavy metal transporting ATPase. Plant Physiology, 136, 3814-3823. doi:10.1104/pp.104.044503
- Schnepf, R., Haehnel, W., Weighardt, K. and Hildebrandt, P. (2004) Spectroscopic identification of different types of copper centers generated in synthetic four-helix bundle proteins. Journal of American Chemical Society, 126, 14389-14399. doi:10.1021/ja0484294
- Etterna, T.J., Huynen, M.A., De Vos, W.M. and Van der Oost, J. (2003) TRASH: A novel metal-binding domain predicted to be involved in heavy-metal sensing, trafficking and resistance. Trends in Biochemical Science, 28, 170-173. doi:10.1016/S0968-0004(03)00037-9
- Rigden, D.J. and Galperin, M.Y. (2004) The DxDxDG motif for calcium binding: Multiple structural contexts and implications for evolution. Journal of Molecular Biology, 343, 971-984. doi:10.1016/j.jmb.2004.08.077
- Andreini, C., Banci, L., Bertini, I. and Rosato, A. (2006) Counting the zinc-proteins encoded in the human genome. Journal of Proteome Research, 5, 196-201. doi:10.1021/pr050361j
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403-410.
- Chou, K.C. and Zhang, C.T. (1995) Prediction of protein structural classes. Critical Review in Biochemistry and Molecular Biology, 30, 275-349. doi:10.3109/10409239509083488
- Klein, P. (1986) Prediction of protein structural class by discriminant analysis. Biochimica Biophysica Acta, 874, 205-215. doi:10.1016/0167-4838(86)90119-6
- Anfisen, C.B. (1973) Principles that govern the folding of protein chains. Science, 181, 223-230. doi:10.1126/science.181.4096.223
- Chou, K.C. (2000) Review: Prediction of protein structural classes and subcellular locations. Current Protein and Peptide Science, 1, 171-208. doi:10.2174/1389203003381379
- Hua, S. and Sun, Z. (2001) Support vector machine approach for protein subcellular localization prediction. Bioinformatics, 17, 721-728. doi:10.1093/bioinformatics/17.8.721
- Nakai, K. (2000) Protein sorting signals and prediction of subcellular localization. Advances in Protein Chemistry, 54, 277-344. doi:10.1016/S0065-3233(00)54009-1
- Zhou, G.P. and Doctor, K. (2003) Subcellular location prediction of apoptosis proteins. Proteins, 50, 44-48. doi:10.1002/prot.10251
- Rice, P., Longden, I. and Bleasby, A. (2000) EMBOSS: The European molecular biology open software suite. Trends in Genetics, 16, 276-277. doi:10.1016/S0168-9525(00)02024-2
- Chou, K.C. (2001) Prediction of protein cellular attributes using pseudo amino acid composition. Proteins, Structure, Function and Genetics, 43, 246-255. doi:10.1002/prot.1035
- Tanford, C. (1962) Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. Journal of American Chemical Society, 84, 4240-4247. doi:10.1021/ja00881a009
- Hopp, T.P. and Woods, K.R. (1981) Prediction of protein antigenic determinants from amino acid sequences. Proceeding of National Academic of Science USA, 78, 3824-3828. doi:10.1073/pnas.78.6.3824
- Rumelhart, D.E., Hinton, G.E. and Williams, R.J. (1986) Learning internal representations by error propagation. In: D.E. Rumelhart, J.L. McClelland, PDP Research Group Editors, Parallel distributed processing: Explorations in the microstructure of cognition. Foundations, MIT Press, Cambridge, 318-362.
- Zhou, G.P. (1998) An intriguing controversy over protein structural class prediction. Journal of Protein Chemistry, 17, 729-738. doi:10.1023/A:1020713915365
- Chou, K.C. and Cai, Y.D. (2002) Using functional domain composition and support vector machines for prediction of protein subcellular location. Journal of Biological Chemistry, 277, 45765-45769. doi:10.1074/jbc.M204161200
- Huang, Y. and Li, Y. (2004) Prediction of protein subcellular locations using fuzzy k-NN method. Bioinformatics, 20, 21-28. doi:10.1093/bioinformatics/btg366
- Lin, H.H., Han, L.Y., Zhang, H.L., Zheng, C.J., Xie, B. and Chen, Y.Z. (2006) Prediction of the functional class of lipid-binding proteins from sequence derived properties irrespective of sequence similarity. Journal of Lipid Research, 47, 824-831. doi:10.1194/jlr.M500530-JLR200
- Fierro-Monti, I. and Mathews, M.B. (2000) Proteins binding to duplexed RNA: One motif, multiple functions. Trends in Biochemical Science, 25, 241-246. doi:10.1016/S0968-0004(00)01580-2
- Perez-Canadillas, J.M. and Varani, G. (2001) Recent advances in RNA-protein recognition. Current Opinion in Structural Biology, 11, 53-58.doi:10.1016/S0959-440X(00)00164-0
- Hunt, J.A., Ahmed, M. and Fierke, C.A. (1999) Metal binding specificity in carbonic anhydrase is influenced by conserved hydrophobic core residues. Biochemistry, 38, 9054-9062. doi:10.1021/bi9900166
- Rapisarda, V.A., Chehin, R.N., De Las Rivas, J., Rodriguez-Montelongo, L., Farias, R.N. and Massa, E.M. (2002) Evidence for Cu(I)-thiolate ligation and prediction of a putative copper-binding site in the Escherichia coli NADH dehydrogenase-2. Archives Biochemistry and Biophysics, 405, 87-94.doi:10.1016/S0003-9861(02)00277-1
- Abbott, J.J., Pei, J., Ford, J.L., Qi, Y., Grishin, V.N., Pitcher, L.A., Phillips, M.A. and Grishin, N.V. (2001) Structure prediction and active site analysis of the metal binding determinants in gamma-glutamylcysteine synthetase. Journal of Biological Chemistry, 276, 42099-42107. doi:10.1074/jbc.M104672200
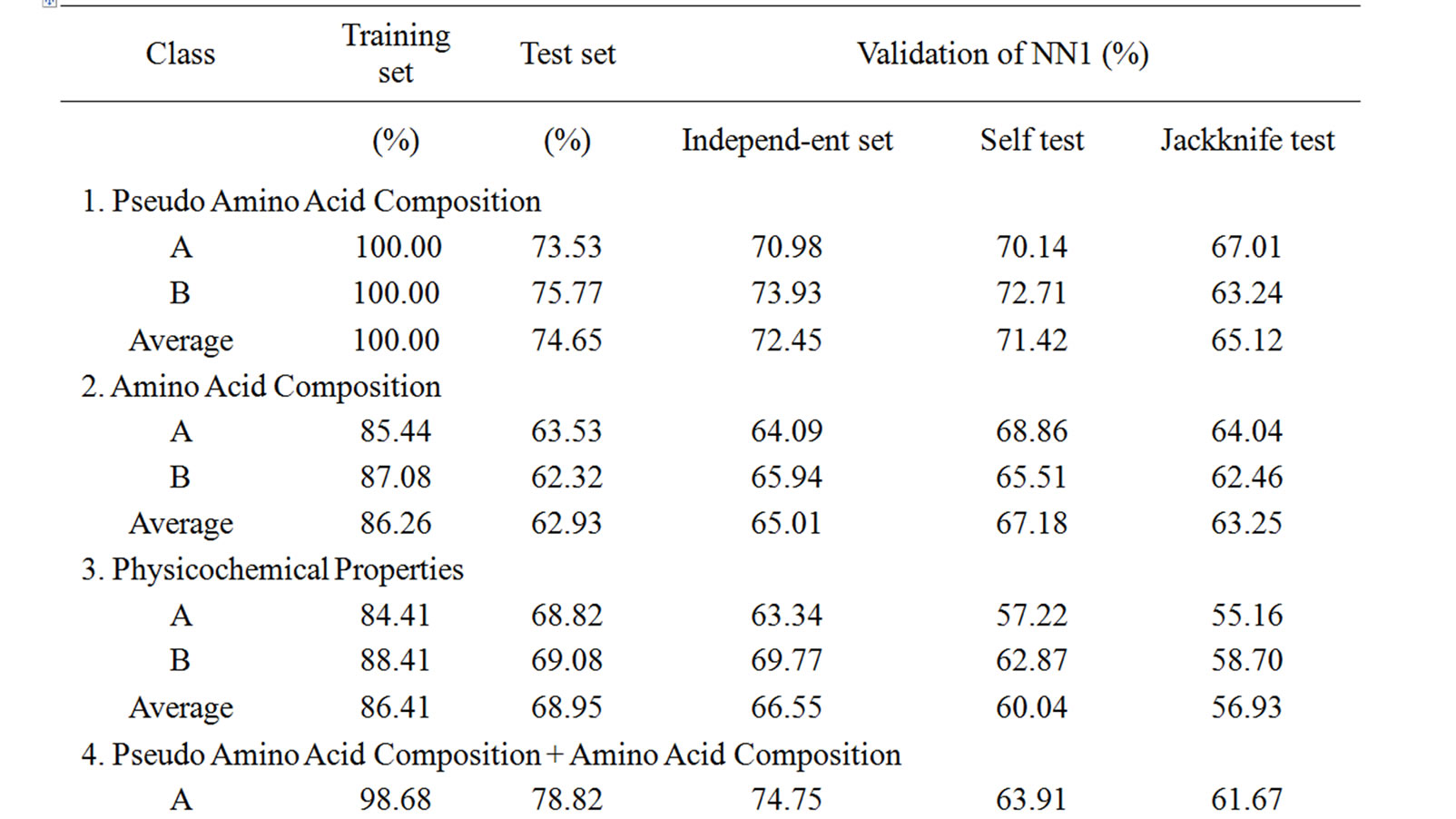

Supplementary Table 1. The summary of the performance accuracy and validation results of 1st layer (NN1) of MetalloPred based on different sequence derived features.
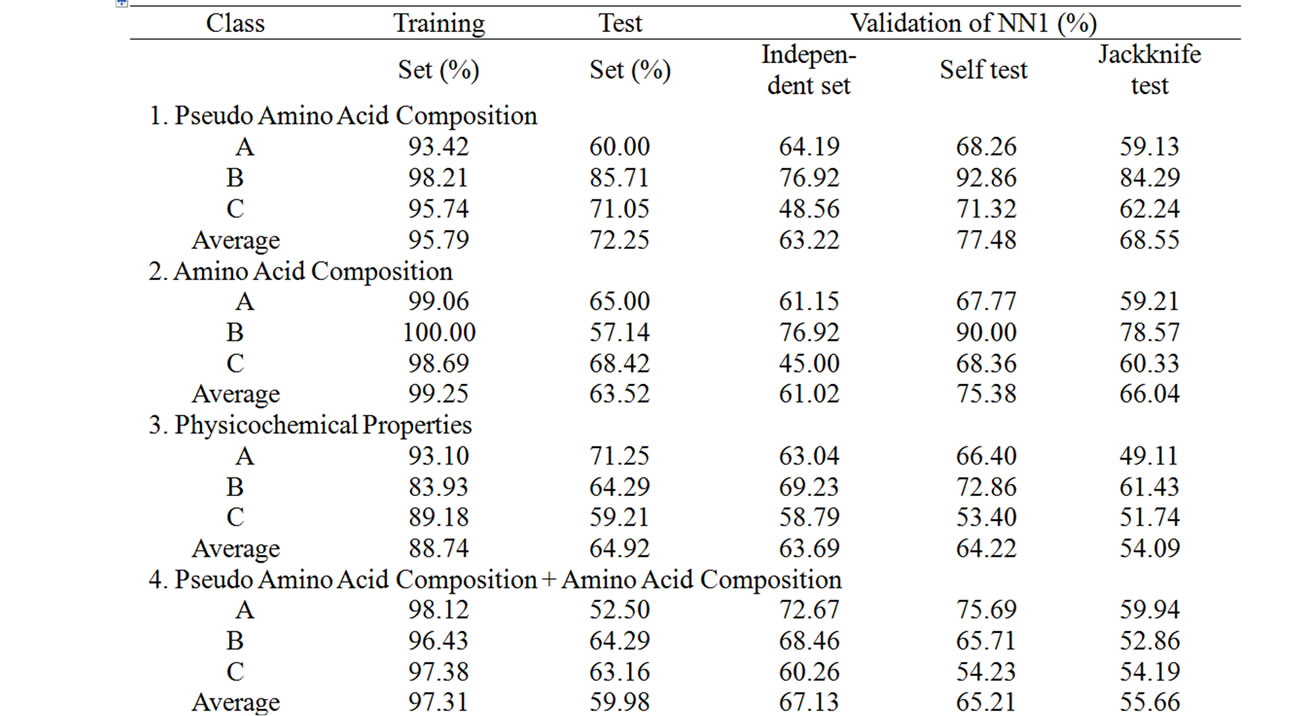
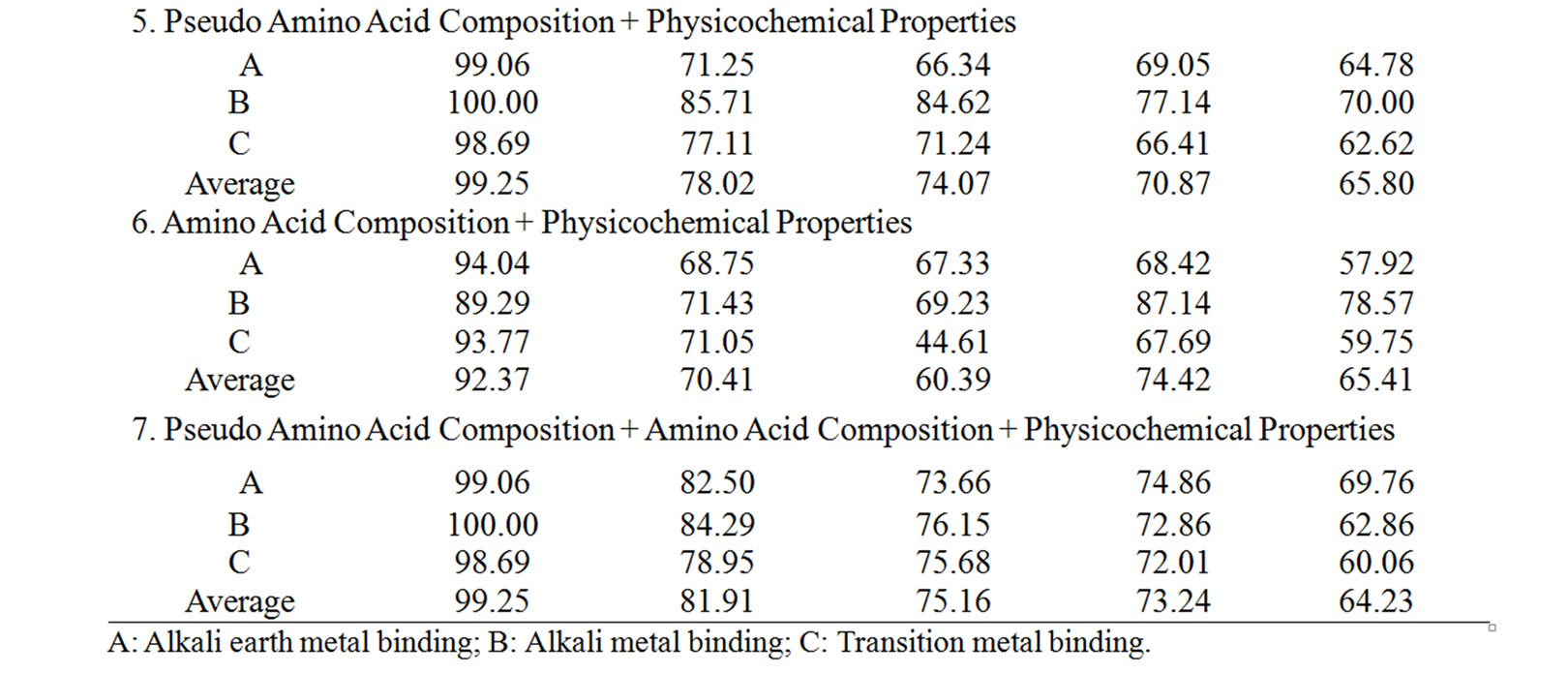
Supplementary Table 2. The summary of the performance accuracy and validation results of 2nd layer (NN2) of MetalloPred based on different sequence derived features.

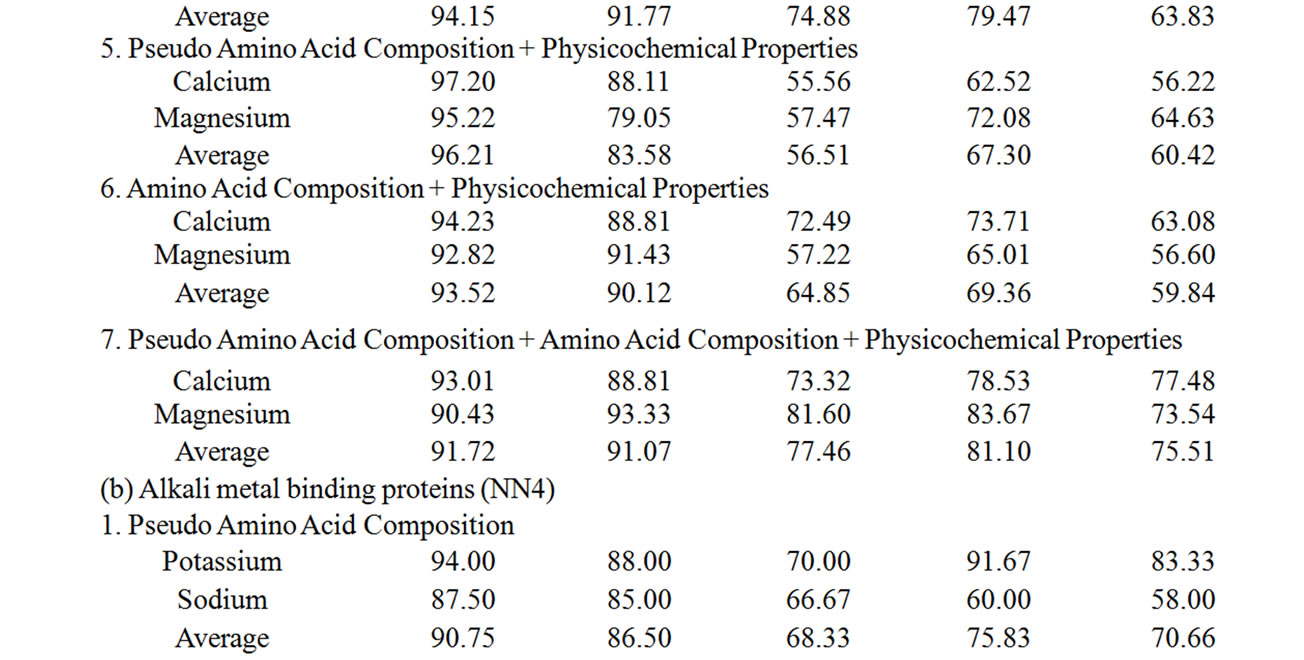
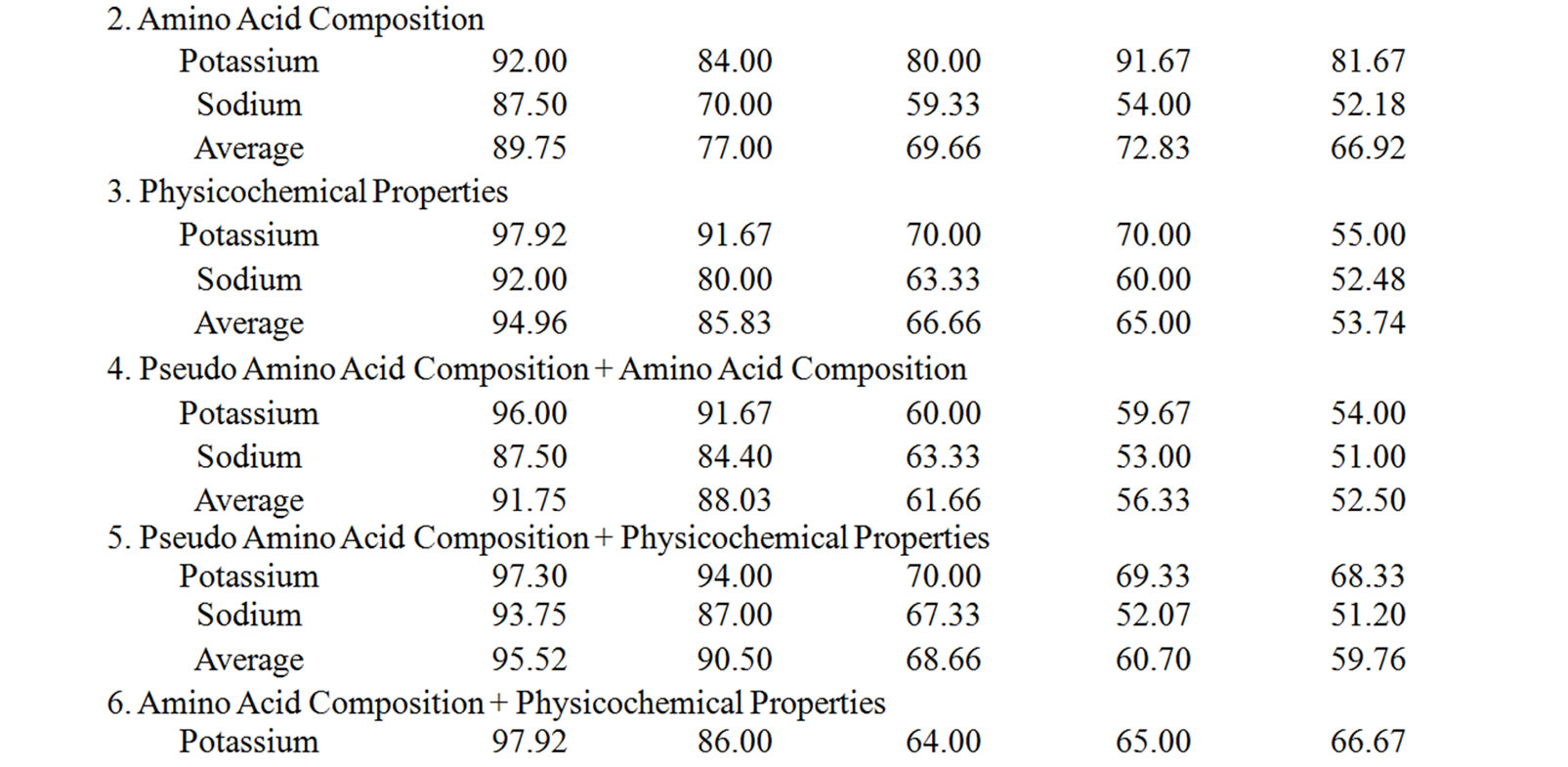

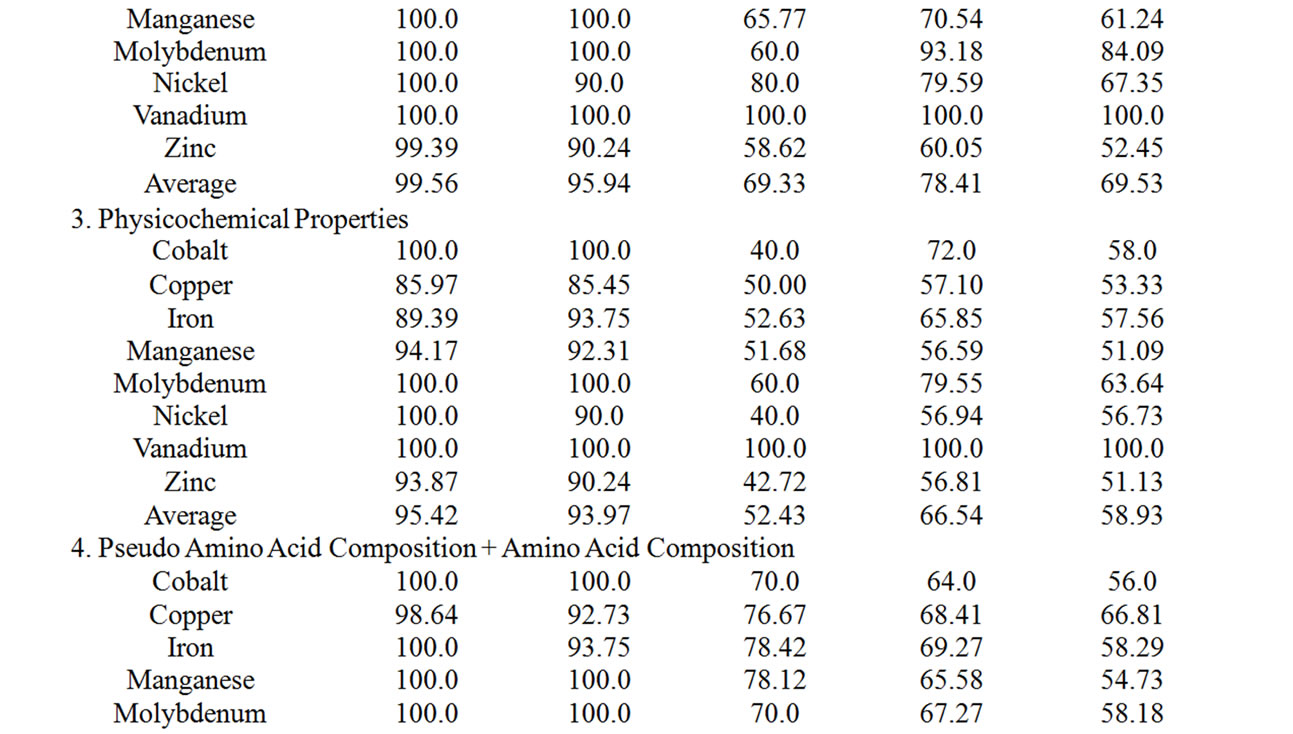

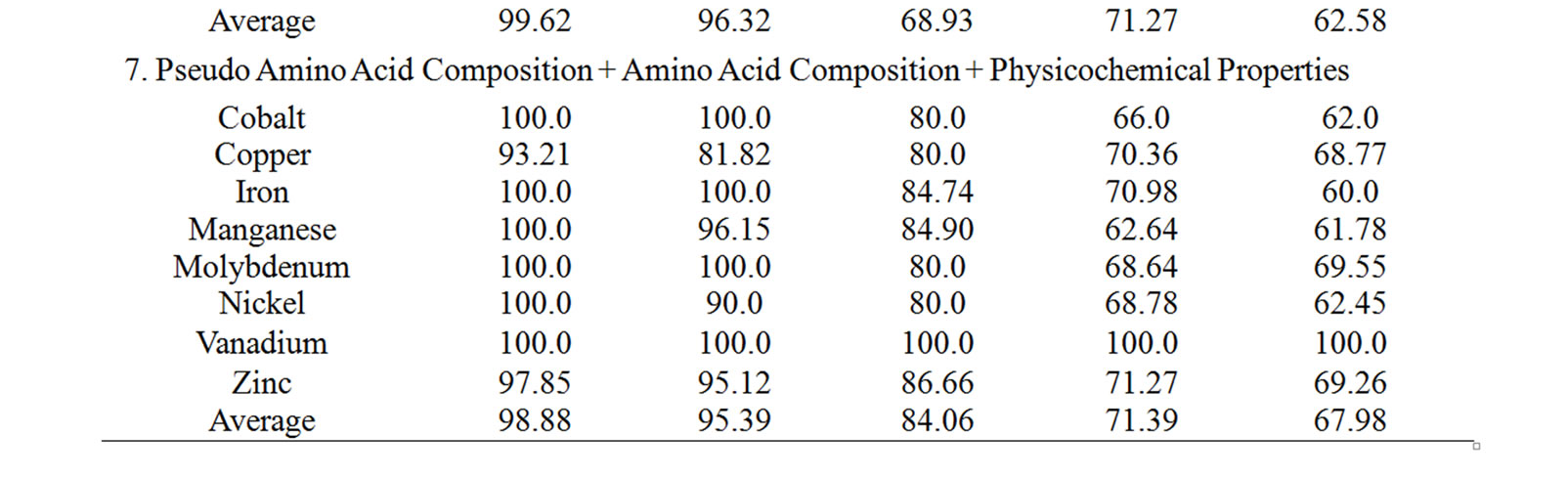
Supplementary Table 3. The summary of the performance accuracy and validation results of 3rd layer of MetalloPred based on different sequence derived features.