Journal of Materials Science and Chemical Engineering
Vol.07 No.07(2019), Article ID:93529,6 pages
10.4236/msce.2019.77005
Synthesis of a New Lopinavir Phosphinic Analog as HIV-1 Inhibitor
Ruba Kellow1,2, Magdalini Matziari2
1Institute of Translational Medicine, Faculty of Health & Life Sciences, University of Liverpool, Liverpool, UK
2Department of Chemistry, Xi’an Jiaotong-Liverpool University (XJTLU), Suzhou, China




Received: May 27, 2019; Accepted: July 7, 2019; Published: July 10, 2019
ABSTRACT
HIV/AIDS has been one of the most devastating global diseases. HIV-1 protease proteolytic action is responsible for the manufacture of grown, infectious species, consequently HIV-1 protease has become an attractive goal in the treatment and therapy of HIV. Several HIV-1 protease inhibitors based therapeutic agents are under investigation or currently in the market. Lopinavir (ABT-378) has a great value in this research field. Therefore, different methods have appeared aiming to develop efficient analogs by the utilization of variable techniques, since Lopinavir had showed low bioavailability when being prescribed alone, and various side effects after the combination of Lopinavir with another HIV-1 inhibitors such as Ritonavir, which is available in the markets nowadays under the brand name Kaletra. Replacement of the hydroxyethylene moiety in Lopinavir structure, which is responsible for the monohydroxylated metabolites with the stable to hydrolysis phosphinic group has been considered, since that hydroxyl group in the central core is responsible for the interaction with the carboxylic acid in the catalytic aspartyl residue of HIV-1 by hydrogen bonding and consequently supports the drug affinity to the protease. The small scale processes for the synthetic strategies for the new candidate phosphinic analog of Lopinavir protease inhibitor (PL1) is presented here in along with some preliminary pharmacological data.
Keywords:
HIV-1, Lopinavir, PL1

1. Introduction
Human immunodeficiency virus is considered as a group of retroviruses causing sicknesses characterized by a delay in the onset of symptoms after infection called lentivirus. This virus has two main types, HIV-1 which is mainly responsible for this epidemic, and HIV-2, which is related to the first type but it is widespread in western Africa [1]. HIV-1 protease is classified as an aspartic acid protease which contains 99 amino acid residues that include the active site at the interface between the protein units with a homodimer function, and similar to all dimers, it is symmetrical, with two fold rotational (C2) symmetry [2]. Lopinavir (ABT-378) (Figure 1) is a peptidomimetic HIV-1 protease inhibitor which is considered to interact with the active catalytic site of the protease. Therefore, it has the ability to prevent the transformation process of polyprotein precursors into the mature form, and as a result, it blocks the division and replication operation of the protease inside the virus. It was discovered in 2000 as an improved analog of Ritonavir, and was firstly prescribed to control the interactions between Val 82 active site residues of the HIV-1 protease with the inhibitor, however the residuals of the virus were considerably transformed into drug resistant strains [3].
Phosphinic peptides act as potent and selective inhibitors of metalloproteases (Matrix metalloproteinase, Angiotensin-converting enzyme, Neprilysin, etc.) that are involved in many pathophysiological conditions, such as cancer and hypertension, exhibiting high selectivity and low toxicity properties [4] [5] [6]. Phosphinic peptide inhibitors of aspartic acid proteases have also been reported for serine proteases [7] and serine kinases [8].
2. Results and Discussion
2.1. Retrosynthetic Strategy for PL1
According to the retrosynthetic analysis study, four main fragments are present in PL1, a-d, which come from the disconnections 1 - 3. As shown in Figure 2, fragment a corresponds to the Cbz-(R)-Phe-PO2H2 [9], fragment b come from an acid chloride, fragment c corresponds to (2-nitroallyl) benzene [10], and fragment d to a valine analog [11].
2.2. Synthesis of Fragment a
The synthesis of ((R)-1-(((benzyloxy) carbonyl) amino)-2-phenylethyl) phosphinic acid 7a (precursor of fragment a, Scheme 1) had taken place after several

Figure 1. Lopinavir and Ritonavir structures.

Figure 2. Main disconnections for PL1.
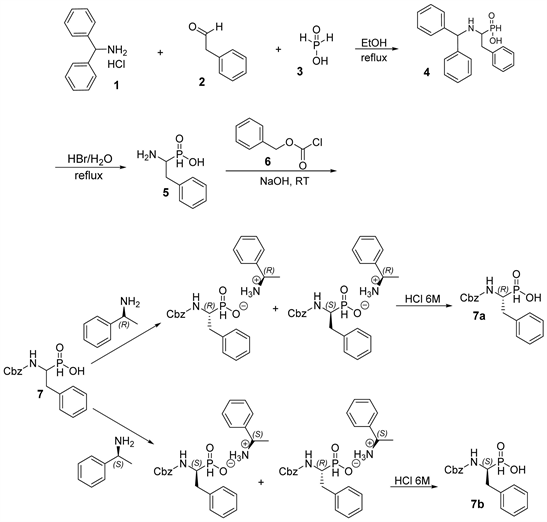
Scheme 1. Synthesis of fragment a.
steps, started from the addition of 3 to the in situ formed imine of 1 and 2 (Scheme 1). Removal of the diphenyl group by refluxing in strong acid followed by the protection of the amino group of the obtained product with benzyl carbonochloridate 6 to produce the product 7 (precursor of fragment a) in the form of racemic mixture (Scheme 1). Then, the resolution through the formation of diastereomeric salts using (R)-phenylethyl amine led to the desired (R)-isomer 7a [9]. As shown in Scheme 1, the mixture of enantiomers of 7 was converted to salts with the chiral amine (R)-phenylethyl amine. The salt containing a mixture of two diastereomers, which was separated by crystallization. The procedure was repeated with (S)-phenylethyl amine to produce the (S)-isomer 7b. The enantiomeric purity of the 7a and 7b was determined by optical rotation measurements.
2.3. Synthesis of the Precursor for Fragment c
The most high yielding attempt to obtain 9 was the reduction of (E)-(2-nitrovinyl) benzene 8 as shown in Scheme 2, followed by olefination to get product 10.
2.4. Synthesis of Fragment d
As shown in Scheme 3, the synthesis of 13 (fragment d) was accomplished in high yield through two steps beginning from the conversion of L-valine 11 to N-phenoxycarbonyl-L-valine and then transformed to the derivative 13 [11].
2.5. Assembly of PL1
PL1 was synthesized according to the series of reactions shown in Scheme 4, started from the assembly of the precursor for fragment c (2-nitroallyl) benzene (10) with fragment a (7a) through Michael addition reaction to afford 14 in a good yield. Then, the reduction reaction of 14, which was achieved under very mild conditions leading to conversion of the nitro (−NO2) group to amine (−NH2) in a high yield without affecting the Cbz-protecting group to produce compound 15 as shown in Scheme 4.The next step was to assembly the resulted 15 with valine urea (fragment d) (13), which affording16 in a good yield. Removal of carboxybenzyl (Cbz-) group of16 was produced product 17 as shown in Scheme 4.The final step to obtain PL1 was the coupling of 17 with fragment b, which was accomplished in good yield (Scheme 4).
The synthesized PL1was tested against HIV-1 to test its potency as phosphinic inhibitor using HIV-1 Protease Inhibitor Assay Kit (Fluorometric) (ab211106) by Abcam. The result displayed that PL1 efficiently inhibits HIV-1 with an IC50 in the low nanomolar range (IC50 = 50 nm).
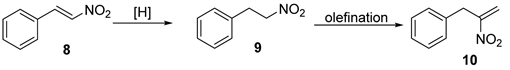
Scheme 2. Synthesis of the precursor for fragment c.
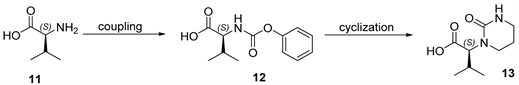
Scheme 3. Synthesis of fragment d.
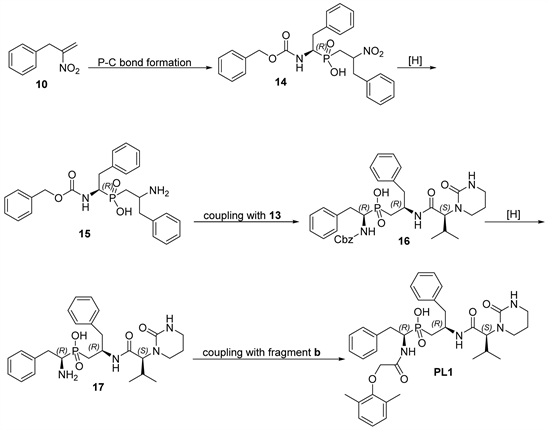
Scheme 4. Assembly of PL1.
3. Conclusion
HIV/AIDS has become a devastating disease and resulted in an enormous human suffering. Lopinavir played a major role in the era of boosted protease inhibitor therapy, and in offering a good option to patients affected with this virus. Considering those findings, PL1, a new potent Lopinavir phosphinic analog as HIV-1 inhibitor was designed and synthesized. Its potency was assessed toward HIV-1 inhibitory and pharmacokinetic activities.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Kellow, R. and Matziari, M. (2019) Synthesis of a New Lopinavir Phosphinic Analog as HIV-1 Inhibitor. Journal of Materials Science and Chemical Engineering, 7, 36-41. https://doi.org/10.4236/msce.2019.77005
References
- 1. Goodman, L., Gilman, A., Brunton, L., Lazo, J. and Parker, K. (2006) Goodman & Gilman’s the Pharmacological Basis of Therapeutics. 11th Edition, McGraw-Hill, New York.
- 2. Lee, T., Laco, G., Torbett, B., Fox, H., Lerner, D., Elder, J. and Wong, C. (1998) Analysis of the S3 and S3’ Subsite Specificities of Feline Immunodeficiency Virus (FIV) Protease: Development of a Broad-Based Protease Inhibitor Efficacious against FIV, SIV, and HIV In Vitro and Ex Vivo. Proceeding of the National Academy of Science, 95, 939-944. https://doi.org/10.1073/pnas.95.3.939
- 3. Prabu-Jeyabalan, M., Nalivaika, E., King, N. and Schiffer, C. (2003) Viability of a Drug-Resistant HIV-1 Protease Mutant: Structural Insights for Better Antiviral Therapy. Journal of Virology, 77, 1306-1315. https://doi.org/10.1128/JVI.77.2.1306-1315.2003
- 4. Georgiadis, D. and Dive, V. (2015) Phosphinic Peptides as Potent Inhibitors of Zinc-Metalloproteases. Topics in Current Chemistry, 360, 1-38. https://doi.org/10.1007/128_2014_571
- 5. Mucha, A., Kafarski, P. and Berlicki, L. (2011) Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. Journal of Medicinal Chemistry, 54, 5955-5980. https://doi.org/10.1021/jm200587f
- 6. Matziari, M., Beau, F., Cuniasse, P., Dive, V. and Yiotakis, A. (2004) Evaluation of P1’-Diversified Phosphinic Peptides Leads to the Development of Highly Selective Inhibitors of MMP-11. Journal of Medicinal Chemistry, 47, 325-336. https://doi.org/10.1021/jm0308491
- 7. Sieńczyk, M. and Oleksyszyn, J. (2009) Irreversible Inhibition of Serine Proteases-Design and In Vivo Activity of Diaryl α-Aminophosphonate Derivatives. Current Medicinal Chemistry, 16, 1673-1687. https://doi.org/10.2174/092986709788186246
- 8. Németh, G., Greff, Z., Sipos, A., Varga, Z., Székely, R., Sebestyén, M., Jászay, Z., Béni, S., Nemes, Z., Pirat, J., Volle, J., Virieux, D., Gyuris, á., Kelemenics, K., áy, é., Minarovits, J., Szathmary, S., Kéri, G. and Örfi, L. (2014) Synthesis and Evaluation of Phosphorus Containing, Specific CDK9/CycT1 Inhibitors. Journal of Medicinal Chemistry, 57, 3939-3965. https://doi.org/10.1021/jm401742r
- 9. Baylis, E.K., Campbell, C.D. and Dingwall, J.D. (1984) 1-Aminoalkylphosphonous Acids. Part 1. Isosteres of the Protein Amino Acids. Journal of the Chemical Society, Perkin Transactions, 1, 1-2853. https://doi.org/10.1039/p19840002845
- 10. Han, Y., Zheng, B. and Peng, Y. (2015) Construction of Chiral 2-Substituted Octahydroindoles from Cyclic Ketones and Nitroolefins Bearing only One α-Substituent. Advanced Synthesis & Catalysis, 357, 1136-1142. https://doi.org/10.1002/adsc.201400851
- 11. Stoner, E.J., Cooper, A.J., Dickman, D.A., Kolaczkowski, L., Lallaman, J.E., Liu, J., Oliver-Shaffer, P.A., Patel, K.M., Paterson, J.B., Plata, D.J., Riley, D.A., Sham, H.L., Stengel, P.J. and Tien, J. (2000) Synthesis of HIV Protease Inhibitor ABT-378 (Lopinavir). Organic Process Research & Development, 4, 264-269. https://doi.org/10.1021/op990202j