K. Kholmurodov et al. / Natural Science 3 (2011) 1011-1021
Copyright © 2011 SciRes. OPEN ACCESS
1020
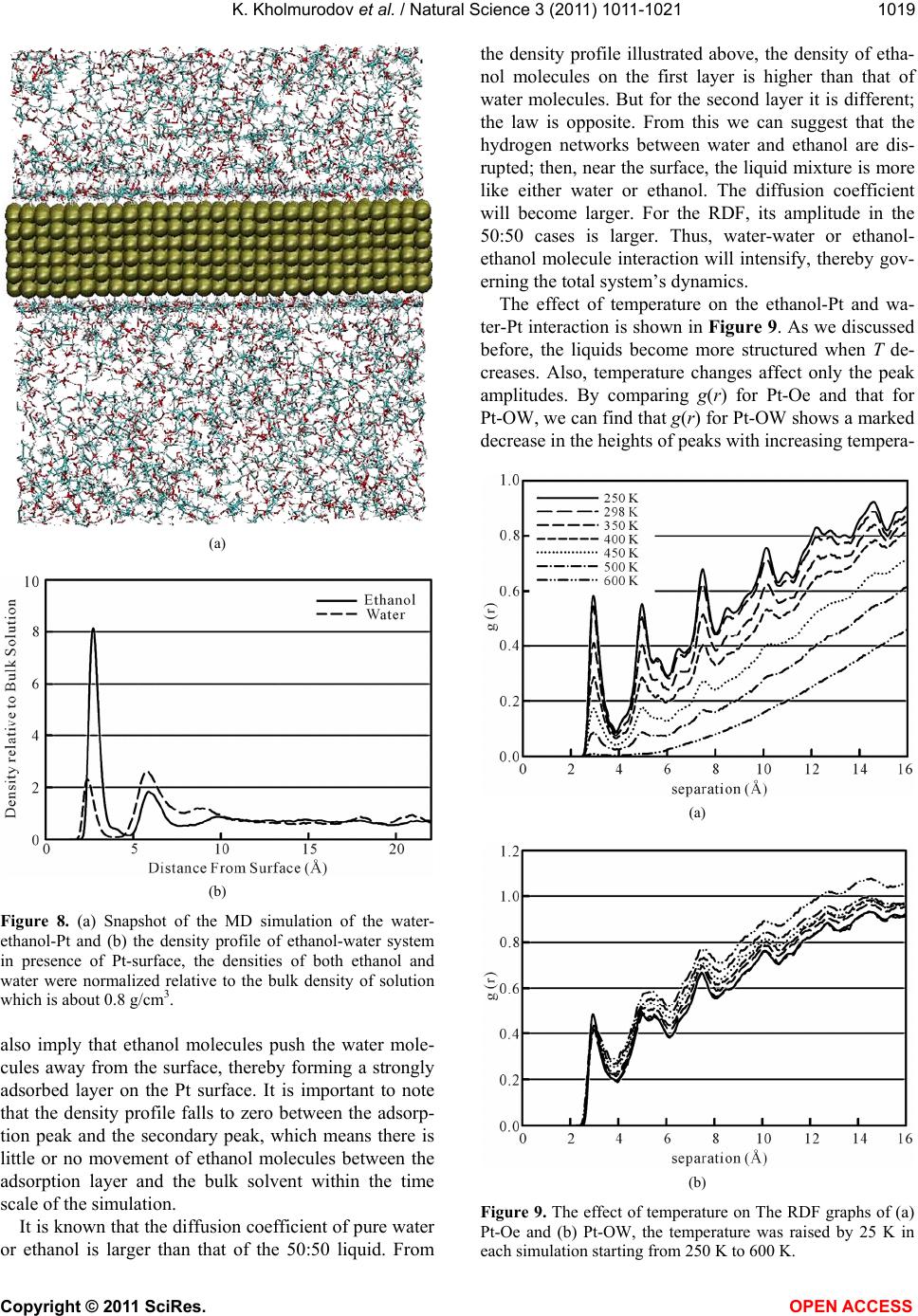
ture; finally, no peaks are observed in its RDF graph at
600 K. In contrast to the RDF of Pt-OW, the heights of
the peaks in the RDF graph of Pt-Oe do not behave so:
only the height of the first peak slightly decreases, and
the other peaks show slight increases in their heights
with increasing temperature. All these results prove that
at high temperatures most of water molecules get re-
moved from the surface by ethanol molecules until only
ethanol molecules are found at the Pt surface at 600 K
(supplementary videos visualizing our system at 600 K
are provided). The height of the first peak in the RDF
graph of Pt-Oe slightly decreases with increasing tem-
perature, which is due to the desorption process that can
occur at higher temperatures.
4. CONCLUSION REMARKS
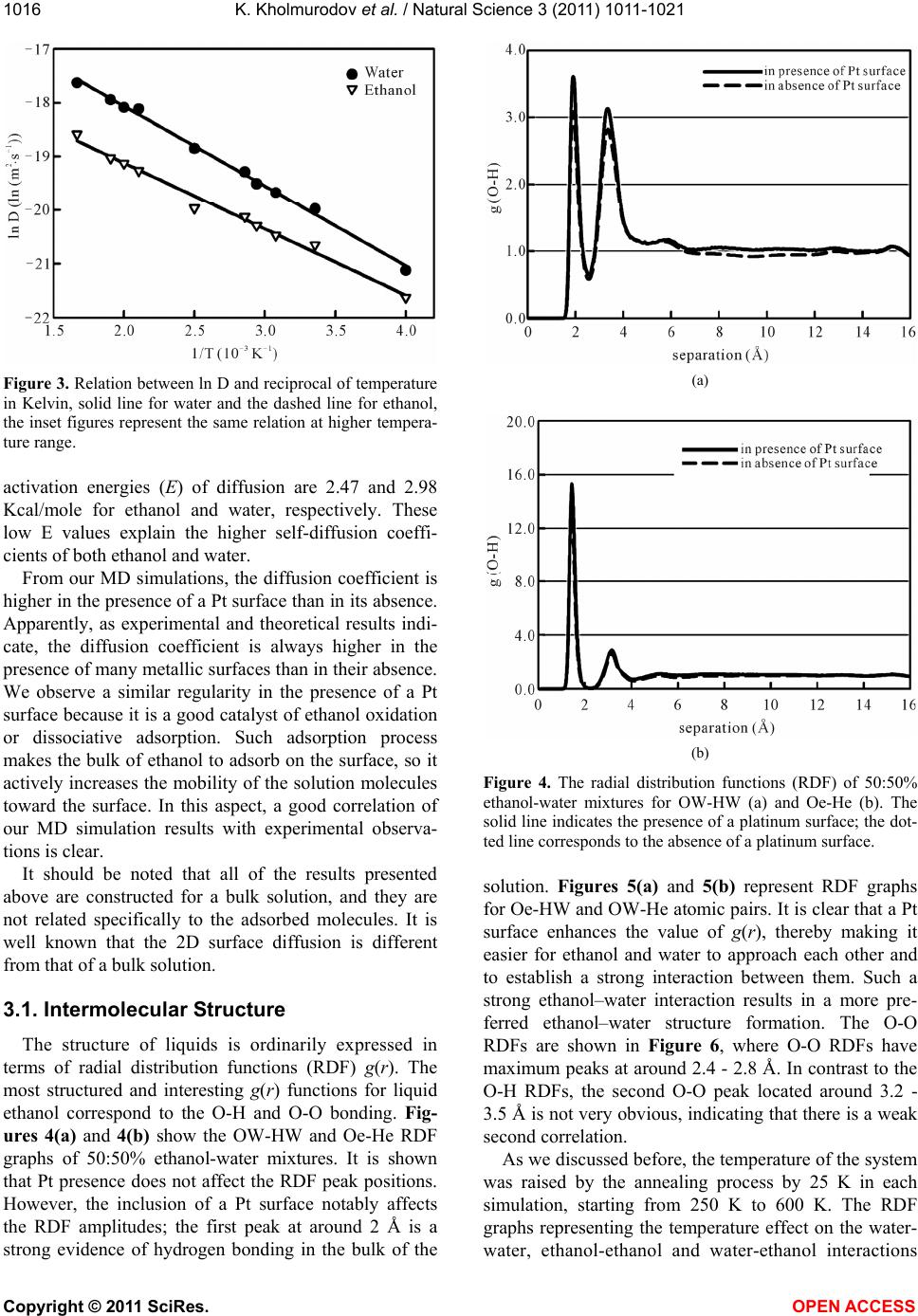
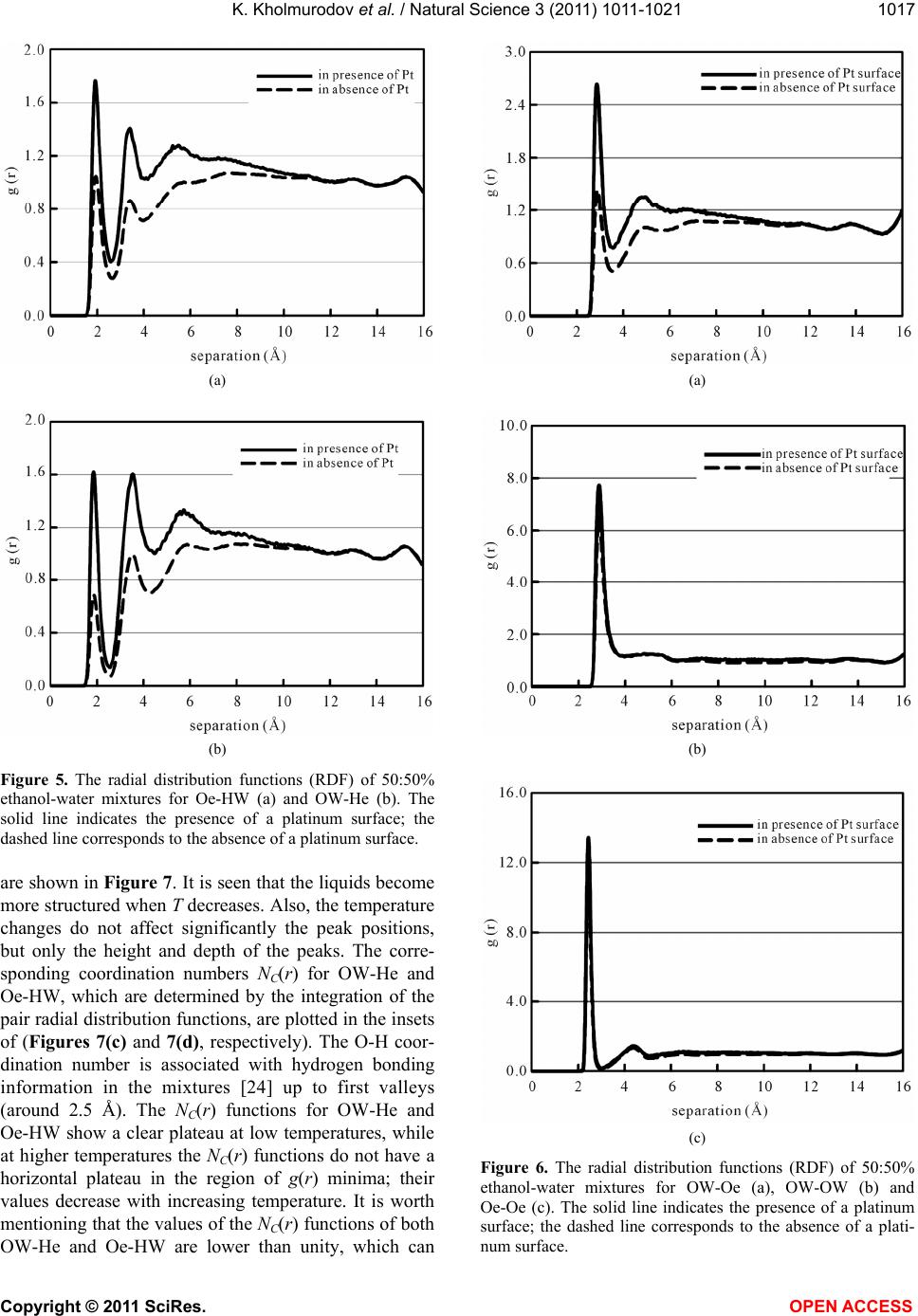
We simulated a water-ethanol mixture in the presence
and absence of a Pt surface using DL_POLY version
2.19. The self-diffusion coefficients of both water and
ethanol in the presence and absence of a Pt surface were
calculated; an excellent agreement with the experimental
results has been found within an error of 7.4%. From our
MD simulations the enhancement of the self-diffusion
coefficients of both water and ethanol molecules related
to the ethanol-water structure have been well-established
in the presence of a Pt surface. As experimental and
theoretical results indicate, the diffusion coefficient is
always higher in the presence of many metallic surfaces
than in their absence. The temperature of the system was
varied using annealing process and its effects on self-
diffusion coefficient and radial distribution functions
(RDF) graphs were illustrated. The RDF graphs in
addition to the density profile have been built, and RDF
correlations with the self-diffusion coefficients of both
ethanol and water molecules are illustrated.
5. ACKNOWLEDGEMENTS
This work has been performed in the framework of joint collabora-
tive agreement Arab Republic of Egypt (ARE)—Joint Institute for
Nuclear Research (JINR) (project #302 “Molecular Dynamics Re-
search of Radiobiological Problems”). This work was supported in part
by the Grant in Aid for the Global Center of Excellence Program of the
Center for Education and Research of Symbiotic, Safe and Secure
System Design from Japan’s Ministry of Education, Culture, Sport,
and Technology.
Supporting Information Available: A visualization video of our
system, ethanol-water in presence of a Pt surface, at 600 K, this video
was created using VMD program. This material is available free of
charge via the internet at http://pubs.acs.org
REFERENCES
[1] Carlos, A.C. and Óscar, J.S. (2007) Fuel ethanol produc-
tion: process design trends and integration opportunities.
Bioresource Technology, 98, 2415-2457.
doi:10.1016/j.biortech.2007.01.002
[2] Carlos, A.C. and Óscar, J.S. (2008) Trends in biotechno-
logical production of fuel ethanol from different feed-
stocks. Bioresource Technology, 99, 5270-5295.
doi:10.1016/j.biortech.2007.11.013
[3] Shen, S.Y., Zhao, T.S. and Xu, J.B. (2010) Carbon sup-
ported PtRh catalysts for ethanol oxidation in alkaline
direct ethanol fuel cell. International Journal of Hydro-
gen Energy, 35, 12911-12917.
doi:10.1016/j.ijhydene.2010.08.107
[4] Vasudevan, V.N. and Leland, M.V. (2007) High perme-
ability membranes for the dehydration of low water con-
tent ethanol by pervaporation. Journal of Membrane
Science, 306, 209-215.
doi:10.1016/j.memsci.2007.08.050
[5] Song, S.Q. and Tsiakaras, P. (2006) Recent progress in
direct ethanol proton exchange membrane fuel cells (DE-
PEMFCs). Applied Catalysis B, 63, 187-193.
doi:10.1016/j.apcatb.2005.09.018
[6] Song, S.Q., Zhou, W.J., Zhou, Z.H., Jiang, L.H., Sun,
G.Q., Xin, Q., Leontidis, V., Kontou, S. and Tsiakaras, P.
(2005) Direct ethanol PEM fuel cells: The case of plati-
num based anodes. International Journal of Hydrogen
Energy, 30, 995-1001.
doi:10.1016/j.ijhydene.2004.11.006
[7] Yao-Chun, W., Chaun, C. and Shin-Pon, J. (2008) Ad-
sorption mechanism and dynamic behavior of water and
ethanol molecules inside Au nanotubes. Chinese Journal
of Catalysis, 29, 1099-1106.
doi:10.1016/S1872-2067(09)60008-5
[8] Kousksou, T., Jamil, A., Zeraouli, Y. and Dumas, J.P.
(2007) Equilibrium liquidus temperatures of binary mix-
tures from differential scanning calorimetry. Chemical
Engineering Science, 62, 6516-6523.
doi:10.1016/j.ces.2007.07.008
[9] Zhang, C., and Yang, X. (2005) Molecular dynamics
simulation of ethanol/water mixtures for structure and
diffusion properties. Fluid Phase Equilibria, 231, 1-10.
doi:10.1016/j.fluid.2005.03.018
[10] Kusalik, P.G., Lyubarts, A.P., Bergman, D.L. and Laak-
sonen, A. (2000) Computer simulation study of tertbutyl
alcohol. Structure in aqueous solution. Journal of Physi-
cal Chemistry B, 104, 9533-9539.
doi:10.1021/jp001887o
[11] Soper, A.K. and Finney, J.L. (1993) Hydration of metha-
nol in aqueous solution. Physical Review Letters, 71,
4346-4350. doi:10.1103/PhysRevLett.71.4346
[12] Sachtler, W.M.H. and Ichikawa, M. (1986) Catalytic site
requirements for elementary steps in syngas conversion
to oxygenates over promoted rhodium. Journal of Physi-
cla Chemistry, 90, 4758-4764.
doi:10.1021/j100411a009
[13] Ahmed, G., Atta, N.F., Darwesh, S.A. and Ali S.M. (2008)
Electrodeposited metals at conducting polymer elec-
trodes. II: Study of the oxidation of methanol at poly
(3-methylthiophene) modified with Pt-Pd Co-catalyst.
Topics in Catalysis, 47, 73-83.
[14] Kholmurodov, Kh., Puzynin, I., Smith, W., Yasuoka, K.
and Ebisuzaki, T. (2001) MD simulation of cluster-sur-
face impacts for metallic phases: soft landing, droplet