Paper Menu >>
Journal Menu >>
 Vol.1, No.3, 152-158 (2009) doi:10.4236/health.2009.13025 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/HEALTH/ Health Difference in regulation mechanisms of ENaC by aldosterone and glucocorticords Chengchun Tang1, Hao Zhang2, Su Wang3, Juyou Wu3, Yuchun Gu3, Jeng Wei3 1Department of Cardiology, The 1st Affiliated Hospital of Southeast University, Nanjing, China 2General Surgery, Hua Shan Hospital, China 3Department of Physiology, University of Birmingham, Edgbaston, UK; Corresponding author: y.gu@bham.ac.uk Received 11 October 2009; revised 15 September 2009; accepted 22 September 2009. ABSTRACT Na+ transport occurs across many epithelial surfaces and plays a key role in regulating salt and water absorption. The molecular pathway underlying this Na+ transport is the epithelial Na channel (ENaC), which is strictly determined by a variety of hormones like aldosterone, ADH and glucocorticoids. In this study, we found that stimulation of either aldosterone or dexameth- asone (Dex) distributed ENaC channel on the apical membrane of mouse cortical collecting duct cells (M1). In the single channel recordings from excised membrane, high density ENaC was found in the cell with a dome shape by the treatment of either dex or aldosterone. However, low active ENaC was revealed in intact cells treated with dex, when compared to cells treated with aldosterone. Only 5.84% of cells treated with dex containing ENaC exhibited ENaC current transition in the cell-attach recording, whereas 40% of cells treated with aldosterone containing ENaC exhibited ENaC current transition. ENaC currents appeared rapid rundown within 5 min- utes since formation of inside-out configuration in cells treated with aldosterone but not with dex. SKF-525A, a general antagonist of CYP, failed to significantly enhance ENaC activity in intact cells treated with dex, but EGTA, which deforming the cells, increased the ENaC activity in the cells treated with dex. PTX, an antagonist of G-protein, reversed the effect of aldosterone on number of active ENaC in intact cells. Based on our obser- vation, we concluded that there are different mechanisms in regulation of ENaC activity be- tween stimulation of aldosterone and glucocor- ticoids. The activation of G-protein is required to maintain the activity of ENaC in the collecting ducts. Keywords: ENaC; Aldosterone; Glucocorticoids; Single Channel Recording 1. INTRODUCTION Active transport of Na+ across epithelial cells plays an important role in maintenance of homeostasis of electro- lytes and water in the body. Hormone sensitive Na+ re- absorption is mainly mediated by epithelial Na+ channels (ENaC), which are distributed in distal and collecting nephron ducts, and alveolar epithelial cells. Disorder of ENaC could lead to Na+ and water retention [1], ‘dry lung’ and even death due to pulmonary edema [2]. Nev- ertheless, ENaC activities are strictly under the control of a variety of hormones like aldosterone, ADH, and glu- cocorticoids. Aldosterone is known to enhance the density of ENaC on apical membrane of epithelial cells by modifying gene expression. Several aldosterone sensitive genes including serum and glucocorticoid (GC) inducible kinase (SGK-1) [3,4], PI3-K [5] have implicated the action mechanism of GC. Previously in vivo and in vitro studies in a number of models show that endogenous and exogenous GC could enhance expression of ENaC and increase the Na+ uptake in airway [6-13], gut [14] and kidney [6,15]. The regula- tion of ENaC by GC determines alveolar fluid clearance and lung development [9]. However, it still remains un- clear of the mechanism in regulation of ENaC by GC, even by aldosterone. A slight difference in the expression of ENaC subunits has been observed in some studies. The expression of α subunit of ENaC is the most pronounced in lung, correlating with the high GC level, whereas ex- pression of α, β and γ subunits of ENaC is gradually in- creased in kidney and colon [9]. Aldosterone is the major regulator in kidney, although GC could also exert the effects on ENaC expression in collecting ducts [6,15]. Till today it is still less clear whether GC and aldosterone exert the same effects on ENaC. In this study, we em- ployed the electrophysiology methods to examine ENaC activity in cells stimulated with either dexamethasone (dex) or aldosterone. 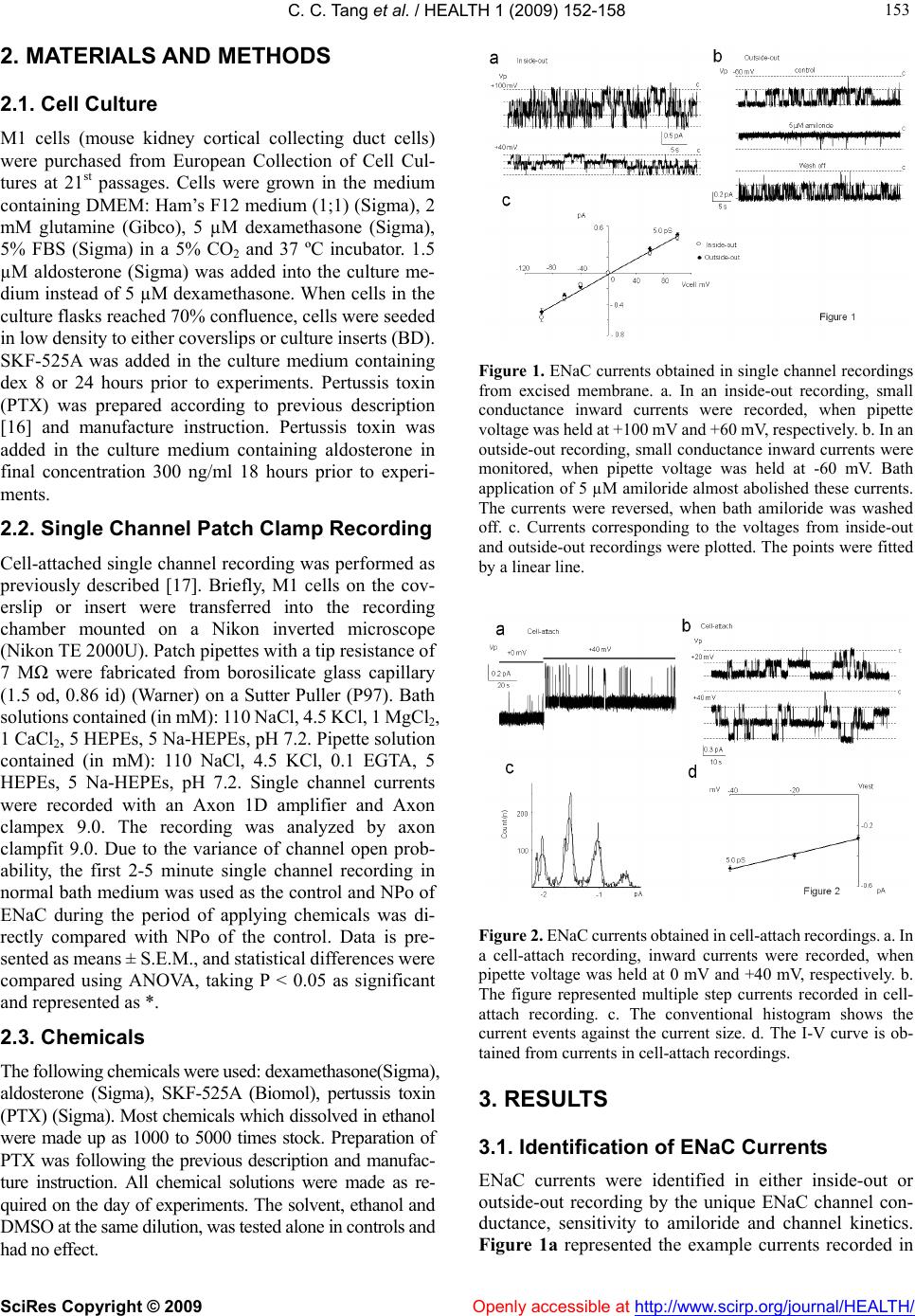 C. C. Tang et al. / HEALTH 1 (2009) 152-158 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/HEALTH/ 153 153 2. MATERIALS AND METHODS 2.1. Cell Culture M1 cells (mouse kidney cortical collecting duct cells) were purchased from European Collection of Cell Cul- tures at 21st passages. Cells were grown in the medium containing DMEM: Ham’s F12 medium (1;1) (Sigma), 2 mM glutamine (Gibco), 5 µM dexamethasone (Sigma), 5% FBS (Sigma) in a 5% CO2 and 37 ºC incubator. 1.5 µM aldosterone (Sigma) was added into the culture me- dium instead of 5 µM dexamethasone. When cells in the culture flasks reached 70% confluence, cells were seeded in low density to either coverslips or culture inserts (BD). SKF-525A was added in the culture medium containing dex 8 or 24 hours prior to experiments. Pertussis toxin (PTX) was prepared according to previous description [16] and manufacture instruction. Pertussis toxin was added in the culture medium containing aldosterone in final concentration 300 ng/ml 18 hours prior to experi- ments. 2.2. Single Channel Patch Clamp Recording Cell-attached single channel recording was performed as previously described [17]. Briefly, M1 cells on the cov- erslip or insert were transferred into the recording chamber mounted on a Nikon inverted microscope (Nikon TE 2000U). Patch pipettes with a tip resistance of 7 MΩ were fabricated from borosilicate glass capillary (1.5 od, 0.86 id) (Warner) on a Sutter Puller (P97). Bath solutions contained (in mM): 110 NaCl, 4.5 KCl, 1 MgCl2, 1 CaCl2, 5 HEPEs, 5 Na-HEPEs, pH 7.2. Pipette solution contained (in mM): 110 NaCl, 4.5 KCl, 0.1 EGTA, 5 HEPEs, 5 Na-HEPEs, pH 7.2. Single channel currents were recorded with an Axon 1D amplifier and Axon clampex 9.0. The recording was analyzed by axon clampfit 9.0. Due to the variance of channel open prob- ability, the first 2-5 minute single channel recording in normal bath medium was used as the control and NPo of ENaC during the period of applying chemicals was di- rectly compared with NPo of the control. Data is pre- sented as means ± S.E.M., and statistical differences were compared using ANOVA, taking P < 0.05 as significant and represented as *. 2.3. Chemicals The following chemicals were used: dexamethasone(Sigma), aldosterone (Sigma), SKF-525A (Biomol), pertussis toxin (PTX) (Sigma). Most chemicals which dissolved in ethanol were made up as 1000 to 5000 times stock. Preparation of PTX was following the previous description and manufac- ture instruction. All chemical solutions were made as re- quired on the day of experiments. The solvent, ethanol and DMSO at the same dilution, was tested alone in controls and had no effect. Figure 1. ENaC currents obtained in single channel recordings from excised membrane. a. In an inside-out recording, small conductance inward currents were recorded, when pipette voltage was held at +100 mV and +60 mV, respectively. b. In an outside-out recording, small conductance inward currents were monitored, when pipette voltage was held at -60 mV. Bath application of 5 µM amiloride almost abolished these currents. The currents were reversed, when bath amiloride was washed off. c. Currents corresponding to the voltages from inside-out and outside-out recordings were plotted. The points were fitted by a linear line. Figure 2. ENaC currents obtained in cell-attach recordings. a. In a cell-attach recording, inward currents were recorded, when pipette voltage was held at 0 mV and +40 mV, respectively. b. The figure represented multiple step currents recorded in cell- attach recording. c. The conventional histogram shows the current events against the current size. d. The I-V curve is ob- tained from currents in cell-attach recordings. 3. RESULTS 3.1. Identification of ENaC Currents ENaC currents were identified in either inside-out or outside-out recording by the unique ENaC channel con- ductance, sensitivity to amiloride and channel kinetics. Figure 1a represented the example currents recorded in  C. C. Tang et al. / HEALTH 1 (2009) 152-158 SciRes Copyright © 2009 http://www.scirp.org/journal/HEALTH/ 154 an inside-out recording. Currents were elicited when pipette voltages (Vp) were held at +100 mV and +60 mV, respectively. In an outside-out recording, the small con- ductance currents possessed conductance similar to those obtained in inside-out recording. These currents were reversely sensitive to amiloride in the range of micro- molar (Figure 1b). I-V curves constructed by currents obtained in either inside-out recording (n=50) or out- side-out recording (n=20) were almost overlapped (Fig- ure 1c) and possessed the same conductance (5.1 pS, fitted by linear line, r2=0.99). Figure 2a and Figure 2b represented the currents obtained in the cell-attach re- cordings. Currents in Figure 2b contained a multiple transition status. I-V curve (Figure 2d) constructed by currents in cell-attach recordings possessed the similar conductance (5.0 pS, fitted by linear line, r2=0.99). Therefore, we concluded that currents detected in single channel recording in M1 cells were mediated by ENaC channel. Figure 3. The rundown of ENaC currents in cells treated with either dex or aldosterone. a represented single channel currents of ENaC in an inside-out recording within 10 minutes since formation of inside-out configuration in a cell treated with dex. b represented single channel currents of ENaC in an inside-out recording in a cell treated with aldosterone. c. Summary plots of NPo (+40 mV in a cell-attach recording) showing a sudden rundown during the period from 3rd to 5th minute in cells treated with aldosterone and a constant NPo in cells treated with dex. 3.2. Different ENaC Activities in M1 Cells Treated with Either Dexmethasone (Dex) or Aldosterone There was no significant difference in cell morphology of M1 cells by different treatments with either dex or al- dosterone. Electrophysiology experiments were per- formed, when cells formed a monolayer. As a standard protocol, cell-attach recording was carried out for 5-10 minutes in each cell prior to single channel recording from the excised membrane including inside-out re- cording and outside-out recording. In cells treated with dex, 325 cells exhibited ENaC currents in either in- side-out or outside-out recording, but only 19 of 325 cells possessed the detectable current transition of ENaC channel during 5-minute of cell-attach recording (Table 1). However, in cells treated with aldosterone, 150 cells exhibited ENaC currents in either inside-out or out- side-out recording, and 60 of 150 cells possessed de- tectable current transition of ENaC channel during 5-minute of cell-attach recording. Based on our observa- tion, a large number of cells contained ENaC channels but they did not mediate Na+ currents, when cells were in the intact condition. About 5.84% of cells treated with dex could mediate Na+ currents, whereas 40% of cells treated with aldosterone could mediate Na+ currents, although these cells contained the ENaC in apical side of mem- brane. However, cells possessed low active ENaC (p<0.05, in comparing with aldosterone treatment group) on the apical membrane when cells were incubated with aldosterone and PTX (300 ng/ml) 18 hours prior to ex- periments. Figure 4. The morphology of M1 cells in monolayer selected for the patch clamping study. a, b, c, d represented the morphology of M1 cells in monolayer. Numbers 1 to 7 represented the cell to be studied. e and f represented the cell morphology after 10 minutes of treatment of EGTA. Figure 5. M1 cells with certain patterns possessed high density of ENaC currents. 3.3. Rundown of ENaC Currents in Cells Treated with Either Dex or Aldosterone A6 [18,19] were stable for the initial 4 minutes and ex hibited a sudden rundown during the period from the 5th to 10th minute. In cells treated with dex, ENaC currents Previous studies have demonstrated that ENaC currents in Openly accessible at 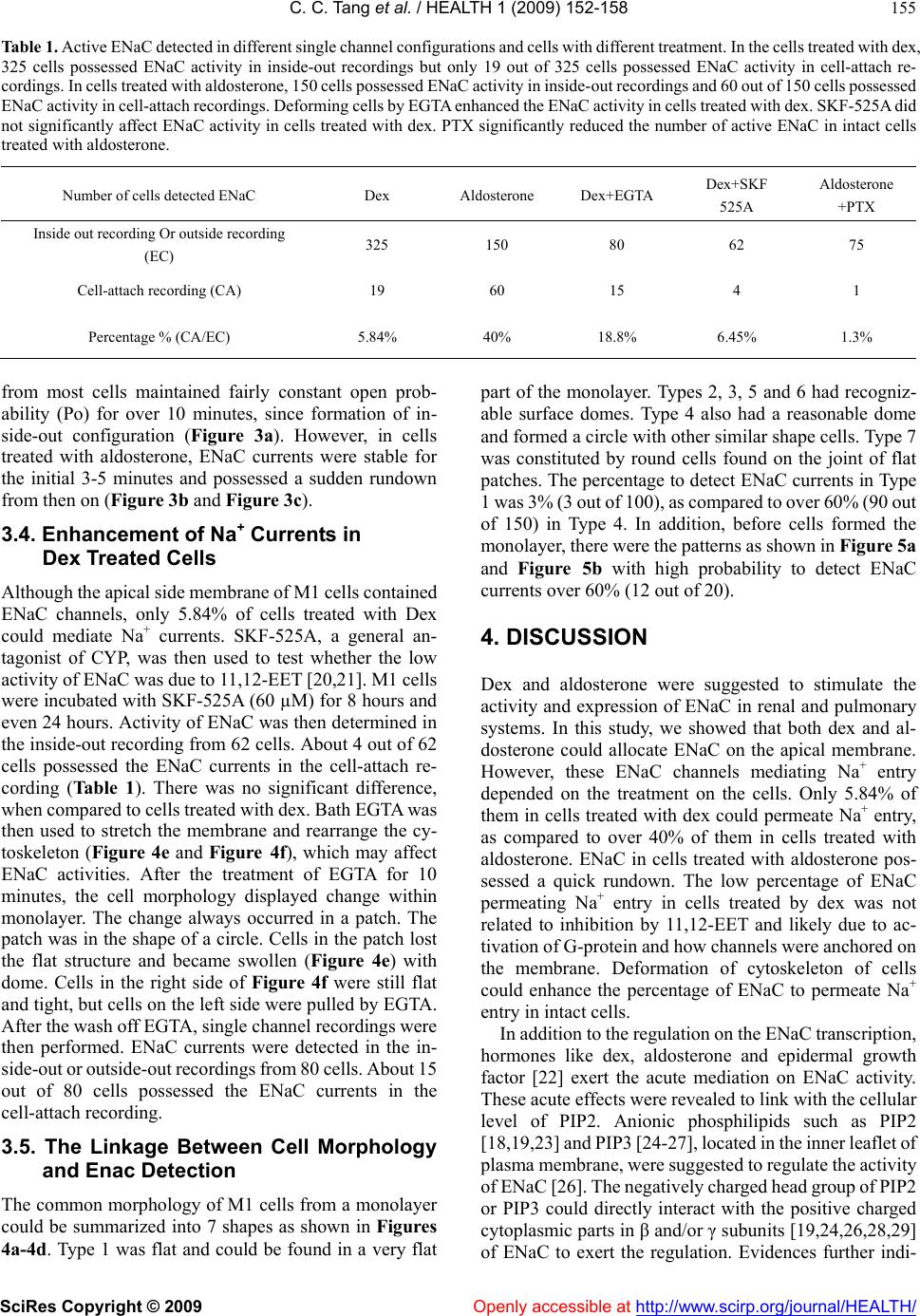 C. C. Tang et al. / HEALTH 1 (2009) 152-158 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/HEALTH/ 155 Table 1. Active ENaC detected in different single channel configurations and cells with different treatment. In the cells treated with dex, 325 cells possessed ENaC activity in inside-out recordings but only 19 out of 325 cells possessed ENaC activity in cell-attach re- cordings. In cells treated with aldosterone, 150 cells possessed ENaC activity in inside-out recordings and 60 out of 150 cells possessed ENaC activity in cell-attach recordings. Deforming cells by EGTA enhanced the ENaC activity in cells treated with dex. SKF-525A did not significantly affect ENaC activity in cells treated with dex. PTX significantly reduced the number of active ENaC in intact cells treated with aldosterone. Number of cells detected ENaC Dex Aldosterone Dex+EGTA Dex+SKF 525A Aldosterone +PTX Inside out recording Or outside recording (EC) 325 150 80 62 75 Cell-attach recording (CA) 19 60 15 4 1 Percentage % (CA/EC) 5.84% 40% 18.8% 6.45% 1.3% from most cells maintained fairly constant open prob- ability (Po) for over 10 minutes, since formation of in- side-out configuration (Figure 3a). However, in cells treated with aldosterone, ENaC currents were stable for the initial 3-5 minutes and possessed a sudden rundown from then on (Figure 3b and Figure 3c). 3.4. Enhancement of Na+ Currents in Dex Treated Cells Although the apical side membrane of M1 cells contained ENaC channels, only 5.84% of cells treated with Dex could mediate Na+ currents. SKF-525A, a general an- tagonist of CYP, was then used to test whether the low activity of ENaC was due to 11,12-EET [20,21]. M1 cells were incubated with SKF-525A (60 µM) for 8 hours and even 24 hours. Activity of ENaC was then determined in the inside-out recording from 62 cells. About 4 out of 62 cells possessed the ENaC currents in the cell-attach re- cording (Table 1). There was no significant difference, when compared to cells treated with dex. Bath EGTA was then used to stretch the membrane and rearrange the cy- toskeleton (Figure 4e and Figure 4f), which may affect ENaC activities. After the treatment of EGTA for 10 minutes, the cell morphology displayed change within monolayer. The change always occurred in a patch. The patch was in the shape of a circle. Cells in the patch lost the flat structure and became swollen (Figure 4e) with dome. Cells in the right side of Figure 4f were still flat and tight, but cells on the left side were pulled by EGTA. After the wash off EGTA, single channel recordings were then performed. ENaC currents were detected in the in- side-out or outside-out recordings from 80 cells. About 15 out of 80 cells possessed the ENaC currents in the cell-attach recording. 3.5. The Linkage Between Cell Morphology and Enac Detection The common morphology of M1 cells from a monolayer could be summarized into 7 shapes as shown in Figures 4a-4d. Type 1 was flat and could be found in a very flat part of the monolayer. Types 2, 3, 5 and 6 had recogniz- able surface domes. Type 4 also had a reasonable dome and formed a circle with other similar shape cells. Type 7 was constituted by round cells found on the joint of flat patches. The percentage to detect ENaC currents in Type 1 was 3% (3 out of 100), as compared to over 60% (90 out of 150) in Type 4. In addition, before cells formed the monolayer, there were the patterns as shown in Figure 5a and Figure 5b with high probability to detect ENaC currents over 60% (12 out of 20). 4. DISCUSSION Dex and aldosterone were suggested to stimulate the activity and expression of ENaC in renal and pulmonary systems. In this study, we showed that both dex and al- dosterone could allocate ENaC on the apical membrane. However, these ENaC channels mediating Na+ entry depended on the treatment on the cells. Only 5.84% of them in cells treated with dex could permeate Na+ entry, as compared to over 40% of them in cells treated with aldosterone. ENaC in cells treated with aldosterone pos- sessed a quick rundown. The low percentage of ENaC permeating Na+ entry in cells treated by dex was not related to inhibition by 11,12-EET and likely due to ac- tivation of G-protein and how channels were anchored on the membrane. Deformation of cytoskeleton of cells could enhance the percentage of ENaC to permeate Na+ entry in intact cells. In addition to the regulation on the ENaC transcription, hormones like dex, aldosterone and epidermal growth factor [22] exert the acute mediation on ENaC activity. These acute effects were revealed to link with the cellular level of PIP2. Anionic phosphilipids such as PIP2 [18,19,23] and PIP3 [24-27], located in the inner leaflet of plasma membrane, were suggested to regulate the activity of ENaC [26]. The negatively charged head group of PIP2 or PIP3 could directly interact with the positive charged cytoplasmic parts in β and/or γ subunits [19,24,26,28,29] of ENaC to exert the regulation. Evidences further indi-  C. C. Tang et al. / HEALTH 1 (2009) 152-158 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/HEALTH/ 156 cated that aldosterone elevates the cellular concentration of PIP3 by activating PI-3-kinase to lock the cytoplasmic termini of ENaC to the inner surface of plasma membrane [3] and/or pull the channel to activate ENaC [25]. Al- dosterone-induced protein K-Ras localizes PI3-K near ENaC. The spatial organization of phosphatidylinositides and specific phospholipid precursors [30,31] within cel- lular-membrane might determine the turnover of ENaC to allow Na+ currents [32]. Therefore, the activation of PI3-K and spatial organization of phosphatidylinositides on the apical membrane by aldosterone might explain the active ENaC in the cells treated with aldosterone, but not dex, suggesting the presence of another mechanism. Degeneration of PIP2 by PLC [33] could lead to unlocking of cytoplasmic termini of ENaC [25,33], re- sulting in decreased ENaC activity. It was manifested as the rundown of ENaC in excise membrane recording. Rundown of ENaC currents in excise membrane re- cordings reflected the simultaneous/continuous activation of PLC in a platform interacting with PIP2 and ENaC. Such platforms were revealed in G protein activated in- ward rectifying K+ channel [34], trp channel [35] and suggested to locate in β and γ subunits of ENaC [19]. Moreover, PLC-γ via receptor typrosine kinase [22] and PLC-β via Gq/11 [33,36] were demonstrated to mediate the degeneration of PIP2 in epithelial cells. However, the effects of PIP2 on ENaC activity are suggested to be permissive rather than regulatory [26]. Declines in PIP2 levels are parallel to the loss of ENaC activity, whereas addition of exogenous PIP2 rarely causes ENaC open probability to exceed the control level in excise mem- brane recording [36]. Increase in ENaC open probability by addition of PIP2 with GTP in inside-out recordings [19] implicates that the activation of G-protein plays an im- portant role in regulating ENaC activity. Decrease in ENaC open probability by dialysis of intracellular GTP, which is not dependent on PI-3 K [37], further supported this hypothesis. We, therefore, proposed that activation of G-protein in cells treated with aldosterone might be the key to keep active ENaC on the membrane. Activation of G-protein could activate PLC, resulting in PIP2 degen- eration. The rapid rundown of ENaC in cells treated with aldosterone might reflect activation of G-protein. In intact cells, the degeneration and resynthesis of phosphatidy- linositides reach a balance to maintain the lipid compo- nent of the membrane structure [38,39], but the lack of resynthesis of PIP2 pathway in excised membrane re- cording causes the decline of PIP2, resulting in rundown of ENaC. Like aldosterone, dex too distributes ENaC on the apical membrane via a similar mechanism as aldos- terone via SGK-1 [3,4,7,40,41]. It is demonstrated by the ENaC activity detected in inside-out recordings in cells treated with dex. The low active ENaC in intact cells treated with dex and constant ENaC open probability in inside-out recording might be attributed to the low activ- ity of G-protein. The linkage between aldosterone and activation of G-protein could be implicated by experi- ments that prevention of ATP release due to stimulation of aldosterone abolishes the aldosterone action [17]. Our observation that PTX incubation significantly reverses the effect of aldosterone on active ENaC in intact cells is consistent to previous report [42] and further supports our hypothesis. 11,12 - EET, a metabolite of AA via CYP epoxygenase, was demonstrated to mediate the direct inhibition of ENaC [20]. The evidence that EET mediates the inhibi- tion effect of adenosine [21] on ENaC indicates a pro- found pathway to regulate the cellular EET level. The other rational explanation to low active ENaC in cells treated with dex is due to presence of cellular inhibitor of ENaC. We, therefore, applied SKF-525A, a general an- tagonist for CYP, to reduce the cellular EET level. There was no significant difference in the ENaC activity in intact cells, suggesting that EET is not the case to respond to low active ENaC. It is widely accepted that ENaC activity is strongly regulated by cytoskeleton elements [43-45]. Recent evi- dence that activation of P2 receptors in epithelial baso- lateral membrane could deform the cells and eventually activate ENaC addressed the classic observation that aldosterone action on Na+ reabsorption relies on ATP production. Cell cytoskeletons could be deformed by many methods like ATP stimulation and EGTA. Our pre- vious work demonstrated that elevation in [Ca2+]i could reduce ENaC open probability, but there was no signifi- cant difference in ENaC activity, when 500 nM and 0 nM Ca2+ were in cytoplasmic medium. EGTA was, therefore, used to deform the cell. A significant increase in ENaC activity in cells treated with dex demonstrated that ele- ments of cytoskeleton regulate ENaC activity. It also explains that ENaC activity is commonly observed in the excised membrane recording. However, the mechanism remains unclear. Consistent to previous observation [11], ENaC channels are in high density and distributed in the cells containing splitting pattern, suggesting that ENaC might involve cell differentiation and proliferation. The recognized dome in some cells, which contain ENaC on apical membrane, might be formed due to Na+ entry via ENaC with subsequent water entry. In conclusion, aldosterone and glucocorticords could increase the expression of ENaC and distribute ENaC on the apical membrane of cortical collecting duct cells. Low active ENaC is found in cells treated with dex. Activation of G-protein is required to keep the channel active. Al- dosterone could lead to activation of G-protein to there- fore possess high ENaC activity in intact cells. REFERENCES [1] Y. Guan, C. Hao, D. R. Cha, R. Rao, W. Lu, D. E. Kohan, M. A. Magnuson, R. Redha, Y. Zhang, M. D. Breyer, (2005) Thiazolidinediones expand body fluid volume  C. C. Tang et al. / HEALTH 1 (2009) 152-158 SciRes Copyright © 2009 Openly accessible at http://www.scirp.org/journal/HEALTH/ 157 157 through PPARgamma stimulation of ENaC-mediated re- nal salt absorption. Nat Med, 11, 861-866. [2] P. Factor, G. M. Mutlu, L. Chen, J. Mohameed, A. T. Akhmedov, F. J. Meng, T. Jilling, E. R. Lewis, M. D. Johnson, A. Xu, D. Kass, J. M. Martino, A. Bellmeyer, J. S. Albazi, C. Emala, H. T. Lee, L. G. Dobbs, S. Matalon, (2007) Adenosine regulation of alveolar fluid clearance. Proc Natl Acad Sci U S A, 104, 4083-4088. [3] J. D. Stockand, (2002) New ideas about aldosterone sig- naling in epithelia. Am J Physiol Renal Physiol, 282, F559-576. [4] C. Boyd, A. Naray-Fejes-Toth, (2005) Gene regulation of ENaC subunits by serum- and glucocorticoid-inducible kinase-1. Am J Physiol Renal Physiol, 288, F505-512. [5] J. Wang, P. Barbry, A. C. Maiyar, D. J. Rozansky, A. Bhargava, M. Leong, G. L. Firestone, D. Pearce, (2001) SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am J Physiol Renal Physiol, 280, F303-313. [6] P. MacDonald, S. MacKenzie, L. E. Ramage, J. R. Seckl, R. W. Brown, (2000) Corticosteroid regulation of amilo- ride-sensitive sodium-channel subunit mRNA expression in mouse kidney. J Endocrinol, 165, 25-37. [7] O. A. Itani, S. D. Auerbach, R. F. Husted, K. A. Volk, S. Ageloff, M. A. Knepper, J. B. Stokes, C. P. Thomas, (2002a) Glucocorticoid-stimulated lung epithelial Na(+) transport is associated with regulated ENaC and sgk1 ex- pression. Am J Physiol Lung Cell Mol Physiol, 282, L631-641. [8] O. A. Itani, K. Z. Liu, K. L. Cornish, J. R. Campbell, C. P. Thomas, (2002b) Glucocorticoids stimulate human sgk1 gene expression by activation of a GRE in its 5'-flanking region. Am J Physiol Endocrinol Metab, 283, E971-979. [9] K. Nakamura, J. B. Stokes, P. B. McCray Jr, (2002) En- dogenous and exogenous glucocorticoid regulation of ENaC mRNA expression in developing kidney and lung. Am J Physiol Cell Physiol, 283, C762-772. [10] S. R. Pondugula, J. D. Sanneman, P. Wangemann, P. G. Milhaud, D. C. Marcus, (2004) Glucocorticoids stimulate cation absorption by semicircular canal duct epithelium via epithelial sodium channel. Am J Physiol Renal Physiol, 286, F1127-1135. [11] V. Shlyonsky, A. Goolaerts, R. Van Beneden, S. Sari- ban-Sohraby, (2005) Differentiation of epithelial Na+ channel function: An in vitro model. J Biol Chem, 280, 24181-24187. [12] S. R. Pondugula, N. N. Raveendran, Z. Ergonul, Y. Deng, J Chen, J. D. Sanneman, L. G. Palmer, D. C. Marcus, (2006) Glucocorticoid regulation of genes in the amiloride-sensitive sodium transport pathway by semicircular canal duct epithe- lium of neonatal rat. Physiol Genomics, 24, 114-123. [13] R. R. Quesnell, X. Han, B. D. Schultz, (2007) Glucocor- ticoids stimulate ENaC upregulation in bovine mammary epithelium. Am J Physiol Cell Physiol. [14] M. Horster, (2000) Embryonic epithelial membrane trans- porters. Am J Physiol Renal Physiol, 279, F982-996. [15] M. T. Bens, V. Vallet, F. Cluzeaud, L. Pascual-Letallec, A. Kahn, M. E. Rafestin-Oblin, B. C. Rossier, A. Vandewalle, (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol, 10, 923-934. [16] H. R. Kaslow, L. K. Lim, J. Moss, D. D. Lesikar, (1987) Structure-activity analysis of the activation of pertussis toxin. Biochemistry, 26, 123-127. [17] J. Gorelik, Y. Zhang, D. Sanchez, A. Shevchuk, G. Frolenkov, M. Lab, D. Klenerman, C. Edwards, Y. Kor- chev, (2005) Aldosterone acts via an ATP autocrine/ paracrine system: the Edelman ATP hypothesis revisited. Proc Natl Acad Sci U S A, 102, 15000-15005. [18] H. P. Ma, S. Saxena, D. G. Warnock, (2002b) Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC). J Biol Chem, 277, 7641-7644. [19] G. Yue, B. Malik, G. Yue, D. C. Eaton, (2002) Phosphati- dylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J. Biol Chem, 277, 11965-11969. [20] Y. Wei, D. H. Lin, R. Kemp, G. S. Yaddanapudi, A. Nas- jletti, J. R. Falck, W. H. Wang, (2004) Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol, 124, 719-727. [21] Y. Wei, P. Sun, Z. Wang, B. Yang, M. A. Carroll, W. H. Wang, (2006) Adenosine inhibits ENaC via cytochrome P-450 epoxygenase-dependent metabolites of arachidonic acid. Am J Physiol Renal Physiol, 290, F1163-1168. [22] Q. Tong, J. D. Stockand, (2005) Receptor tyrosine kinases mediate epithelial Na(+) channel inhibition by epidermal growth factor. Am J Physiol Renal Physiol, 288, F150- 161. [23] D. W. Hilgemann, S. Feng, C. Nasuhoglu, (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE, RE19. [24] M. N. Helms, L. Liu, Y. Y. Liang, O. Al-Khalili, A. Vandewalle, S. Saxena, D. C. Eaton, H. P. Ma, (2005) Phosphatidylinositol 3,4,5-trisphosphate mediates aldos- terone stimulation of epithelial sodium channel (ENaC) and interacts with gamma-ENaC. J Biol Chem, 280, 40885-40891. [25] H. P. Ma, D. C. Eaton, (2005) Acute regulation of epithe- lial sodium channel by anionic phospholipids. J Am Soc Nephrol, 16, 3182-3187. [26] O. Pochynyuk, Q. Tong, J. Medina, A. Vandewalle, A. Staruschenko, V. Bugaj, J. D. Stockand, (2007) Molecu lar determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J Gen Physiol, 130, 399-413. [27] K. M. Weixel, R. S. Edinger, L. Kester, C. J. Guerriero, H. Wang, L. Fang, T. R. Kleyman, P. A. Welling, O. A. Weisz, J. P. Johnson, (2007) Phosphatidylinositol 4-phosphate 5-kinase reduces cell surface expression of the epithelial sodium channel (ENaC) in cultured collecting duct cells. J. Biol Chem. [28] Q. Tong, N. Gamper, J. L. Medina, M. S. Shapiro, J. D. Stockand, (2004b) Direct activation of the epithelial Na(+) channel by phosphatidylinositol 3,4,5-trisphosphate and phosphatidylinositol 3,4-bisphosphate produced by phos- phoinositide 3-OH kinase. J Biol Chem, 279, 22654- 22663. [29] O. Pochynyuk, Q. Tong, J. Medina, A. Vandewalle, A. Staruschenko, V. Bugaj, J. D. Stockand, (2007) Molecu lar determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J Gen Physiol, 130, 399-413. [30] R. L. Doughman, A. J Firestone, R. A. Anderson, (2003) Phosphatidylinositol phosphate kinases put PI4,5P(2) in its place. J Membr Biol, 194, 77-89.  C. C. Tang et al. / HEALTH 1 (2009) 152-158 SciRes Copyright © 2009 http://www.scirp.org/journal/HEALTH/Openly accessible at 158 [31] P. A. Oude Weernink, M. Schmidt, K. H. Jakobs, (2004) Regulation and cellular roles of phosphoinositide 5- kinases. Eur J Pharmacol, 500, 87-99. [32] A. Staruschenko, O. M. Pochynyuk, Q. Tong, J. D. Stockand, (2005) Ras couples phosphoinositide 3-OH kinase to the epithelial Na+ channel. Biochim Biophys Acta, 1669, 108-115. [33] K. Kunzelmann, T. Bachhuber, R. Regeer, D. Markovich, J. Sun, R. Schreiber, (2005) Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. Faseb J, 19, 142-143. [34] T. Rohacs, J. Chen, G. D. Prestwich, D. E. Logothetis, (1999) Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J Biol Chem, 274, 36065- 36072. [35] R. C. Hardie, (2003a) Regulation of TRP channels via lipid second messengers. Annu Rev Physiol, 65, 735-759. [36] H. P. Ma, L. Li, Z. H. Zhou, D. C. Eaton, D. G. Warnock, (2002a) ATP masks stretch activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol, 282, F501-505. [37] O. Pochynyuk, A. Staruschenko, Q. Tong, J. Medina, J. D. Stockand, (2005) Identification of a functional phos- phatidylinositol 3,4,5-trisphosphate binding site in the epithelial Na+ channel. J Biol Chem, 280, 37565-37571. [38] R. C. Hardie, (2003b) TRP channels in Drosophila photore- ceptors: the lipid connection. Cell Calcium, 33, 385- 393. [39] R. C. Hardie, (2004) Regulation of Drosophila TRP channels by lipid messengers. Novartis Found Symp, 258, 160-167; discussion 167-171, 263-166. [40] D. Pearce, (2003) SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem, 13, 13-20. [41] Q. Tong, R. E. Booth, R. T. Worrell, J. D. Stockand, (2004a) Regulation of Na+ transport by aldosterone: sig- naling convergence and cross talk between the PI3-K and MAPK1/2 cascades. Am J Physiol Renal Physiol, 286, F1232-1238. [42] L. Gambling, R. E. Olver, G. K. Fyfe, P. J. Kemp, D. L. Baines, (1998) Differential regulation of Na+ and Cl- conductances by PTX-sensitive G proteins in fetal lung apical membrane vesicles. Biochim Biophys Acta, 1372, 187-197. [43] B. K. Berdiev, A. G. Prat, H. F. Cantiello, D. A. Ausiello, C. M. Fuller, B. Jovov, D. J Benos, I. I. Ismailov, (1996) Regulation of epithelial sodium channels by short actin filaments. J Biol Chem, 271, 17704-17710. [44] J. B. Zuckerman, X. Chen, J. D. Jacobs, B. Hu, T. R. Kleyman, P. R. Smith, (1999) Association of the epithelial sodium channel with Apx and alpha-spectrin in A6 renal epithelial cells. J Biol Chem, 274, 23286-23295. [45] C. Mazzochi, J. K. Bubien, P. R. Smith, D. J. Benos, (2006) The carboxyl terminus of the alpha-subunit of the amilo- ride-sensitive epithelial sodium channel binds to F-actin. J Biol Chem, 281, 6528-6538. |