T. ABURJAI ET AL.
Copyright © 2011 SciRes. AJAC
937
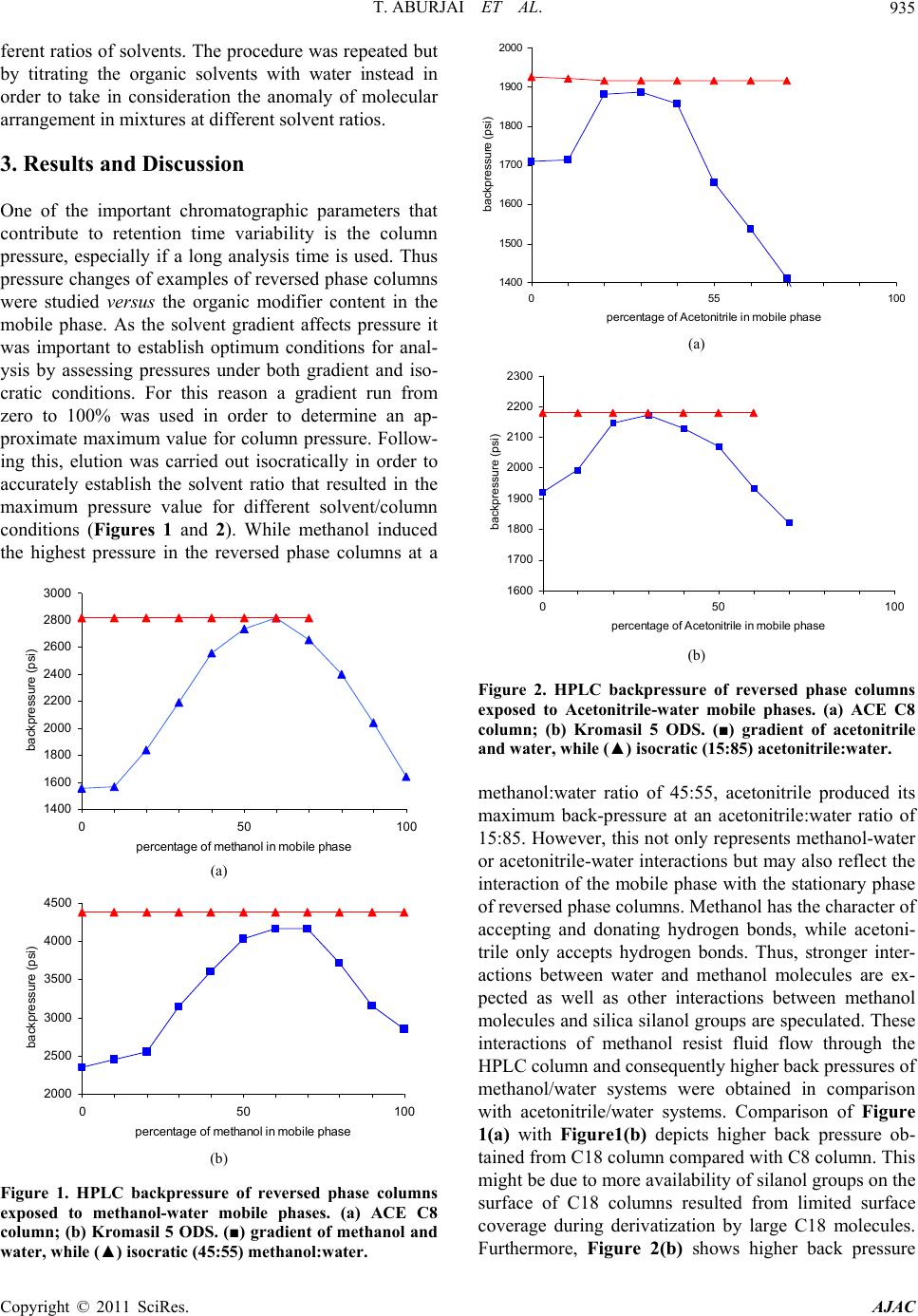
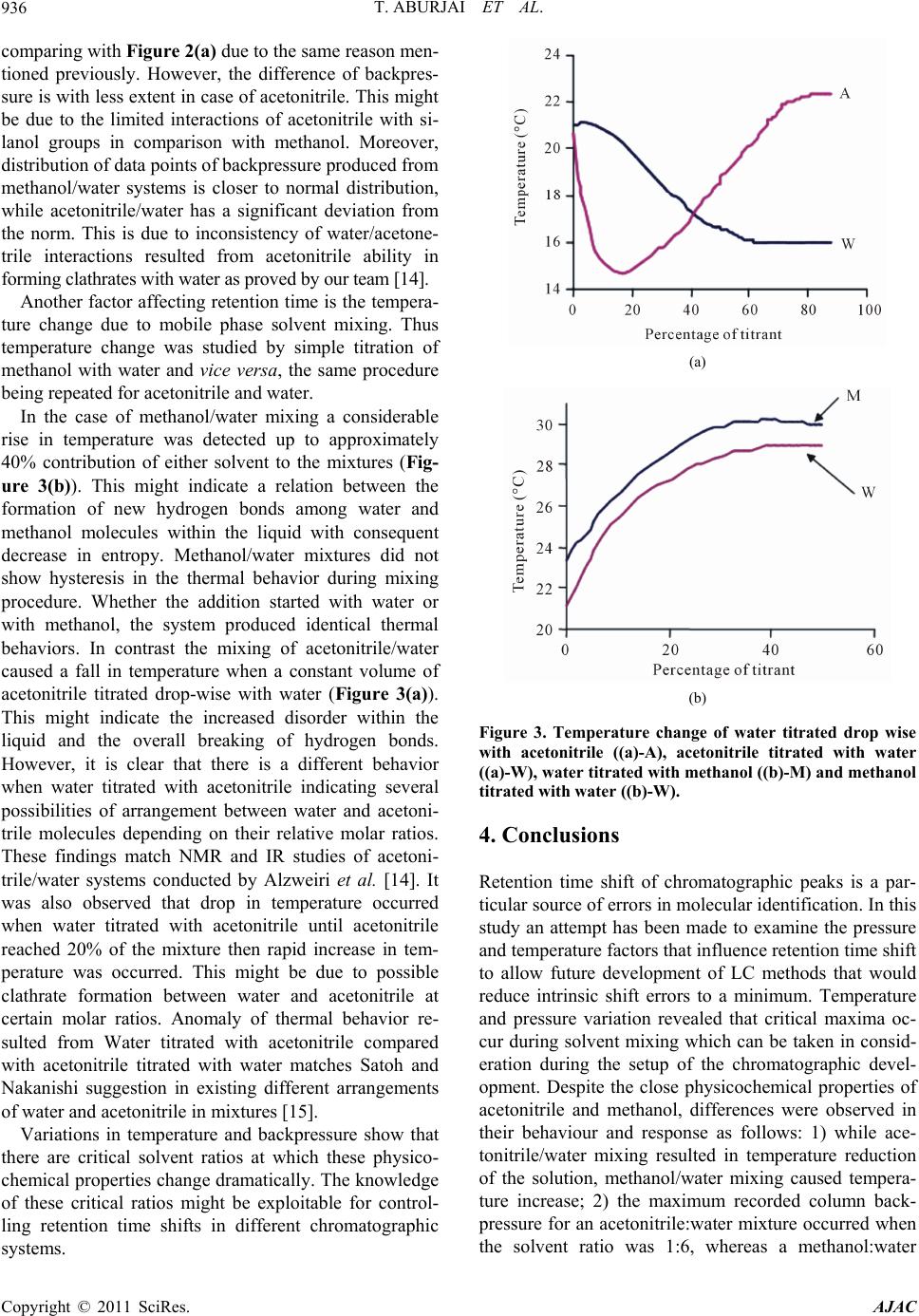
mixture resulted in a maximum backpressure at a solvent
ratio of 1:1.
5. References
[1] E. Bulemela, P. Tremaine and S.-I. Ikawa, “Volumetric
Behavior of Water-Methanol Mixtures in the Vicinity of
the Critical Region,” Fluid Phase Equilibria, Vol. 245,
2006, p. 125.
[2] K. F. Ita McStravick, J. Lambert, N. Teahan and W. Earle
Waghorne, “Enthalpy of Transfer of -CH2- between Wa-
ter and Organic Solvents or Mixed Aqueous Organic
Solvents or Mixed Aquoues Organic Solvent Systems,”
Journal of Molecular Liquids, Vol. 94, 2001, pp. 145-
153.
[3] G. R. Behbehani, S. Ghammamy and W. E. Waghorne,
“Enthalpies of Transfer of Acetonitrile from Water to
Aqueous Methanol, Ethanol and Dimethylsulphoxide
Mixtures at 298.15 K,” Thermochimica Acta, Vol. 448,
No. 1, 2006, pp. 37-40. doi:10.1016/j.tca.2006.06.021
[4] M. Vosough and A. Salemi, “Second-Order Standard
Addition for Deconvolution and Quantification of Fatty
Acids of Fish Oil Using GC-MS,” Talanta, Vol. 73, No. 1,
2007, pp. 30-36. doi:10.1016/j.talanta.2007.02.025
[5] M. Bechtold, A. Felinger, M. Held and S. Panke, “Ad-
sorption Behavior of a Teicoplanin Aglycone Bonded
Stationary Phase under Harsh Overload Conditions,”
Journal of Chromatography A, Vol. 1154, No. 1-2, 2007,
pp. 277-286. doi:10.1016/j.chroma.2007.03.103
[6] D. Bylund, R. Danielsson, G. Malmquist and K. E.
Markides, “Chromatographic Alignment by Warping and
Dynamic Programming as a Pre-Processing Tool for
PARAFAC Modelling of Liquid Chromatography-Mass
Spectrometry Data,” Journal of Chromatography A, Vol.
961, No. 2, 2002, pp. 237-244.
doi:10.1016/S0021-9673(02)00588-5
[7] K. M. Pierce, B. W. Wright and R. E. Synovec, “Unsu-
pervised Parameter Optimization for Automated Reten-
tion Time Alignment of Severely Shifted Gas Chroma-
tographic Data Using the Piecewise Alignment Algo-
rithm,” Journal of Chromatography A, Vol. 1141, No. 1,
2007, pp. 106-116.
doi:10.1016/j.chroma.2006.11.101
[8] F. Gong, Y.-Z. Liang, Y.-S. Fung and F.-T. Chau, “Cor-
rection of Retention Time Shifts for Chromatographic
Fingerprints of Herbal Medicines,” Journal of Chroma-
tography A, Vol. 1029, No. 1-2, 2004, pp. 173-183.
doi:10.1016/j.chroma.2003.12.049
[9] F. O. Andersson, R. Kaiser and S. P. Jacobsson, “Data
Preprocessing by Wavelets and Genetic Algorithms for
Enhanced Multivariate Analysis of LC Peptide Map-
ping,” Journal of Pharmaceutical and Biomedical Analy-
sis, Vol. 34, No. 3, 2004, pp. 531-541.
doi:10.1016/S0731-7085(03)00583-1
[10] K. J. Johnson, B. W. Wright, K. H. Jarman and R. E.
Synovec, “High-Speed Peak Matching Algorithm for Re-
tention Time Alignment of Gas Chromatographic Data
for Chemometric Analysis,” Journal of Chromatography
A, Vol. 996, No. 1-2, 2003, pp. 141-155.
doi:10.1016/S0021-9673(03)00616-2
[11] A. M. van Nederkassel, M. Daszykowski, P. H. C. Eilers
and Y. V. Heyden, “A Comparison of Three Algorithms
for Chromatograms Alignment,” Journal of Chromatog-
raphy A, Vol. 1118, No. 2, 2006, pp. 199-210.
doi:10.1016/j.chroma.2006.03.114
[12] M. Makela and L. Pyy, “Effect of Temperature on Reten-
tion Time Reproducibility and on the Use of Programma-
ble Fluorescence Detection of Fifteen Polycyclic Aro-
matic Hydrocarbons,” Journal of Chromatography A, Vol.
699, No. 1-2, 1995, pp. 49-57.
doi:10.1016/0021-9673(95)00120-C
[13] W. S. Gardner, H. A. Bootsma, C. Evans and P. A. S.
John, “Improved Chromatographic Analysis of 15N:14N
Ratios in Ammonium or Nitrate for Isotope Addition Ex-
periments,” Marine Chemistry, Vol. 48, No. 3-4, 1995, pp.
271-282. doi:10.1016/0304-4203(94)00060-Q
[14] M. Alzweiri, J. Parkinson, D. Watson and S. Steer, “Mi-
croscopic Trends in Methanol/Water and Acetonitrile/
Water Systems,” Jordan Journal of Pharmaceutical Sci-
ences, Vol. 4, 2011, pp. 20-28.
[15] Y. Satoh and K. Nakanishi, “Theoretical Studies of Ace-
tonitrile-Water Mixtures/Monte Carlo Simulation,” Fluid
Phase Equilibria, Vol. 104, 1995, pp. 41-55.
doi:10.1016/0378-3812(94)02638-H