Journal of Biomedical Science and Engineering
Vol.6 No.7(2013), Article ID:34772,20 pages DOI:10.4236/jbise.2013.67093
The interrelationship between insulin resistane and Alzheimer development
![]()
Biochemistry Department, Faculty of Science, Alexandria University, Alexandria, Egypt
Email: *d.ghareeb@yahoo.com, doaa.ahmed@alex-sci.edu.eg
Copyright © 2013 Doaa A. Ghareeb et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 8 March 2013; revised 20 April 2013; accepted 10 May 2013
Keywords: Insulin Resistance; B-Amyloid; Oxidative Stress; Brain Inflammation
ABSTRACT
Abnormalities in insulin metabolism, characteristic of T2DM, are among the major factors thought to mechanistically influence the onset of AD. These abnormalities are thought to play a role in AD via their influence on the synthesis and degradation of Aβ and as a consequence of the cascade of neuronal alterations resulting from the effects of danger/alarm signals from oligomeric amyloid species. Additionally, recent studies have indicated that certain signal transduction pathways downstream of the InsR may also promote the generation of Aβ peptides by modulating the cleavage of the parent Aβ precursor protein (AβPP) at the γ-secretase site, a cleavage site necessary for Aβ amyloidogenicity. Glucose homeostasis is critical for energy generation, neuronal maintenance, neurogenesis, neurotransmitter regulation, cell survival and synaptic plasticity. It also plays a key role in cognitive function. In an insulin resistance condition, there is a reduced sensitivity to insulin resulting in hyperinsulinemia; this condition persists for several years before becoming full blown diabetes. Toxic levels of insulin negatively influence neuronal function and survival, and elevation of peripheral insulin concentration acutely increases its cerebrospinal fluid (CSF) concentration. Peripheral hyperinsulinemia correlates with an abnormal removal of the amyloid beta peptide (Aβ) and an increase of tau hyperphosphorylation as a result of augmented cdk5 and GSK3β activities. This leads to cellular cascades that trigger a neurodegenerative phenotype and decline in cognitive function. Chronic peripheral hyperinsulinemia results in a reduction of insulin transport across the BBB and reduced insulin signaling in brain, altering all of insulin’s actions, including its anti-apoptotic effect. However, the increase in brain insulin levels resulting from its peripheral administration at optimal doses has shown a cognition enhancing effect on patient with AD.
1. INTRODUCTION
Insulin Resistance
Clamp studies [1,2] have demonstrated IR in both muscle and adipose tissues. Abnormalities in substrates particularly free fatty acids (FFAs), and the deposition of ectopic fat in muscle [3,4] as well as a number of cytokines such as tumor necrotic factor alpha (TNF-α) and interleukin-6 (IL-6) have been proposed as mediators of IR, although the precise mechanism remains incompletely defined [5,6].
Mechanisms of Insulin Resistance
Glucose transporter-4 (GLUT-4) is stored in intracellular vesicles. Insulin binds to its receptor in the plasma membrane, resulting in phosphorylation of the receptor and activation of insulin-receptor substrates (IRS) molecules. These substrates form complexes with docking proteins such as phosphoinositide-3 kinase (PI3K) at its 85-kDa subunit (p 85) by means of SH2 (Scr homology region 2) domains. Then p 85 is constitutively bound to the catalytic subunit (p 110). Activation of PI3K is a major pathway in the mediation of insulin-stimulating glucose transport and metabolism. It activates phosphoinositidedependent kinases that participate in the activation of protein kinase B (PKB or Akt) and atypical forms of protein kinase C (PKC). This process results in the translocation of the glucose transporter to the plasma membrane. Exercise stimulates glucose transport by pathways that are independent phosphoinositide-3 kinase and that may involve 5'-AMP-activated kinase (AMPK) as shown in Figure 1 [7,8].
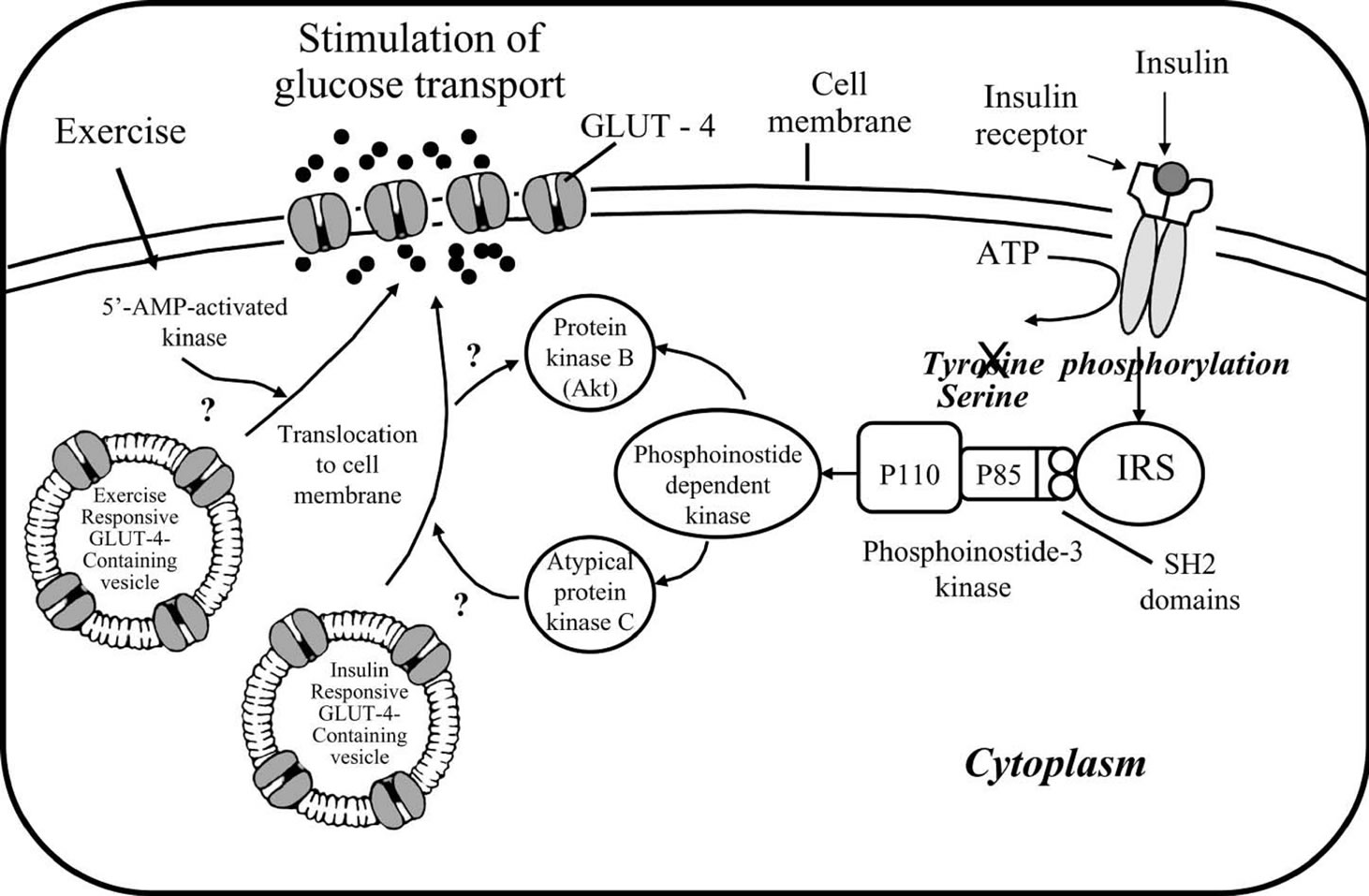
Figure 1. Outline of the insulin pathway and glucose receptors in the skeletal muscle. The primary defect is a substitution of serine for tyrosines in the kinase and prevents the normal cascade of intracellular signaling for glucose entry into the cell. Note that exercise increases glucose uptake via a mechanism independent from the insulin receptor [10].
The abnormalities in glucose uptake into the cell appear to result from defects in intracellular signaling. A number of acquired factors including hyperinsulinemia, hyperglycemia, TNF-α, and FFAs, ceramide and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) have been implicated in altering insulin signaling in patients with obesity and type 2 diabetes [8,9].
The most likely mechanism of IR within the muscle cell is cytokine induced serine rather than tyrosine phosphylation of IRS-1 [11]. A similar abnormality of IRS-2 in the hepatocyte is also likely to be an important mediator of IR with the liver [12,13].
Understanding the intracellular signaling of insulin pathways may lead to new approaches to the treatment of IR and presumably NAFLD. The recent proposal that IR syndrome could be an inflammatory disorder may be particularly relevant [14]. The balance between pro-inflammatory and anti-inflammatory cytokines derived from adipocytes and other tissue may determine the development of IR. Data regarding the mechanism of actions and the beneficial effects of peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists [15,16] and long chain polyunsaturated fatty acids [17] give credence to the inflammatory nature of IR. The recent report that salicylates or disruption of inhibitor of nuclear factor kappa-B kinase subunit beta (IKK-β) reversed obesity and diet-induced insulin resistance gives further support to this concept [18].
Although hyperinsulinemia is a common finding in NAFLD, it is often overlooked in its pathophysiology. However, insulin may injure the liver both directly and indirectly [19,20]. Patients undergoing chronic ambulatory peritoneal dialysis develop hepatic steatosis, but only when insulin is added to the peritoneal fluid dialysate [21,22]. Interestingly, the steatosis only develops at the surface of the liver and in some instances has the histologic appearance of NASH [21]. This direct effect may be due to insulin’s ability to generate oxidative stress [20] or up-regulation of the lipogenic protein, sterol regulatory elemental binding protein (SREBP) [23]. It also appears to have direct profibrogenic effects by stimulating connective tissue growth factor, especially in the presence of hyperglycemia [24].
This latter explanation may explain the observation that NAFLD patients with type 2 diabetes mellitus (T2- DM) have a particularly poor prognosis [25-27]. Evidence [28] also indicates that insulin may be directly involved in causing endoplasmic reticulum stress along with the unfolding protein response and apoptosis. This in itself may exacerbate insulin resistance [29-31] (Figure 2).
2. INSULIN AND BRAIN FUNCTION
2.1. Glucose & Insulin Signaling
2.1.1. Insulin and Insulin Receptor
The effects of insulin on the central nervous system (CNS) are affected by its availability to this separate physiological compartment. The term blood-brain barrier (BBB) in its most restrictive sense refers to the vascular
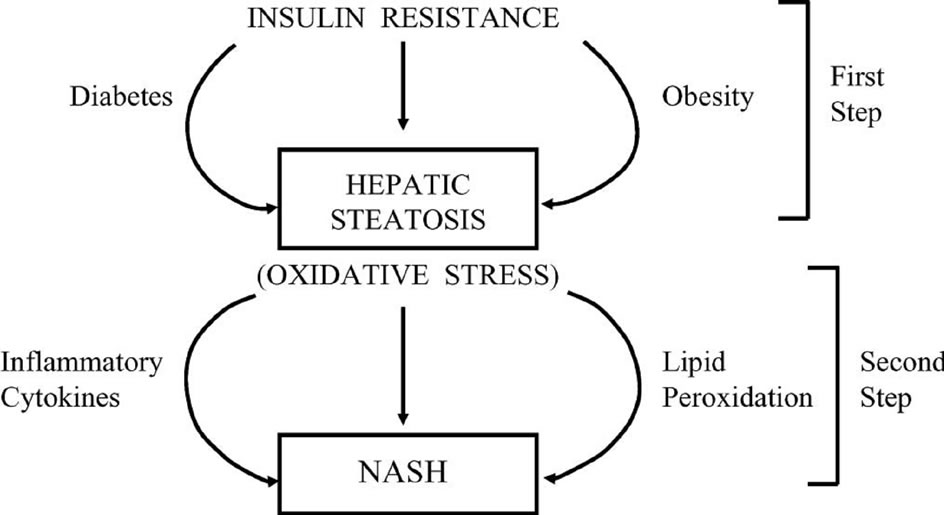
Figure 2. The pathophysiology of NAFLD involving insulin resistance. Insulin resistance causes hepatic steatosis, a process enhanced by obesity and/or T2DM. Once developed, hepatic steatosis may remain in a benign state or progress to NASH [10].
bed of the CNS, which is specially modified to prevent the unrestricted transfer of molecules between the blood and the extracellular fluid of the CNS [32]. The BBB plays a critical role in the transduction of signals between the CNS and peripheral tissues. It does so through several mechanisms, including the direct transport of peptides and regulatory proteins such as insulin (Figure 3) [33].
There is solid evidence that insulin can cross the BBB by a saturable transport process mediated by the InsR protein [32,34-40]. This transporter is not uniformly distributed throughout the BBB [33].
There is also evidence of local insulin synthesis in brain [39,40,42,43], although its function has not been elucidated. Therefore, the origin of brain insulin is a subject under investigation. Insulin receptors are present in the CNS, and were localized for the first time by ligand autoradiography and confirmed by immunohistochemistry and autoradiography [44-46]. CNS insulin receptors differ from their peripheral counterparts both in structure, function, and molecular weight [47]. Insulin receptors are widely distributed in the brain with the highest concentration in the olfactory bulb, hypothalamus, cerebral cortex, cerebellum and hippocampus. They are expressed in all regions of brain in both neurons and glial cells. Insulin receptors are distributed throughout the neuronal processes and the cell body, but are concentrated in the synaptic endings [48-52].
The signaling and biological effects of the insulin have been widely studied mainly in the classical insulin target tissues, e.g. liver, fat and skeletal muscle, with respect to glucose uptake, regulation of cell proliferation, gene expression and the suppression of hepatic glucose production [48]. However, insulin plays many roles within the CNS. It has been shown that some of the CNS effects of insulin are the opposite of those exerted in peripheral tissues. In particular, CNS insulin increases glucose and inhibits feeding, whereas serum insulin decreases glucose and increases feeding [40,53]. Thus, to some extent, insulin acts as its own counter regulatory hormone, with CNS insulin producing features of IR [32]. Thus, in the CNS, insulin participates in the regulation of feeding behavior and energy homeostasis, neuronal maintenance, neurogenesis, and neurotransmitter regulation. In addition, it has a role on cognitive functions as supported by neuronal activity, and in the control of aging-related processes [54].
2.1.2. Glucose, Insulin and Cognition
The brain consumes metabolic energy disproportionate to its size. It uses glucose (primary fuel), largely through oxidative metabolism. Glucose deprivation leads to coma, seizures, and produces potentially permanent brain damage. Glucose is continually supplied from cerebral blood flow and must be transported into the brain through the endothelial cells that form the BBB to reach neurons and glial cells by facilitated diffusion. Carriers of glucose are the glucose transport proteins (GLUT), which allow glucose entry into individual cells (neurons and glial cells) [55]. Animal models show that the cerebral blood flux depends on glucose levels in the blood stream; there is a compensatory mechanism to maintain adequate delivery of glucose fuel to the brain [56,57].
Evidence suggests regionally specific effects of insulin on brain glucose metabolism. Insulin does not seem to influence basal cerebral glucose metabolism or transport of glucose into the brain [58-63]. Insulin affects the use of glucose in specific regions of the brain, most likely by selective distribution of insulin-sensitive GLUT isoforms, which overlap with the distribution of insulin and InsR in the brain [64-67]. Insulin, through the InsR localized in the hypothalamus, contributes to the regulation of food intake and energy homeostasis and leads to changes in body weight by anorexigenic or orexigenic effects produced by increasing or decreasing levels of insulin in brain respectively. Moreover, insulin in the CNS modifies peripheral glucose metabolism by increasing insulin sensitivity in peripheral tissue [36,68,69].
In brain, insulin is not a major regulator of glucose metabolism. In vitro studies showed that insulin regulates glucose uptake of glial cells, but did not influence neuronal glucose uptake [70,71]. Insulin can influence neurons directly by mechanisms unrelated to modulation of glucose uptake. Neurotransmitter release, neuronal-outgrowth, tubulin activity, neuronal survival and synaptic plasticity are all directly modulated by insulin [72-75].
Studies in human and animal models have shown that an increase in brain insulin has a cognition-enhancing effect, independently of changes in peripheral glucose [76-80].
Moreover, there is cumulative evidence to support the effects of insulin and InsR on cognition, mediated by a
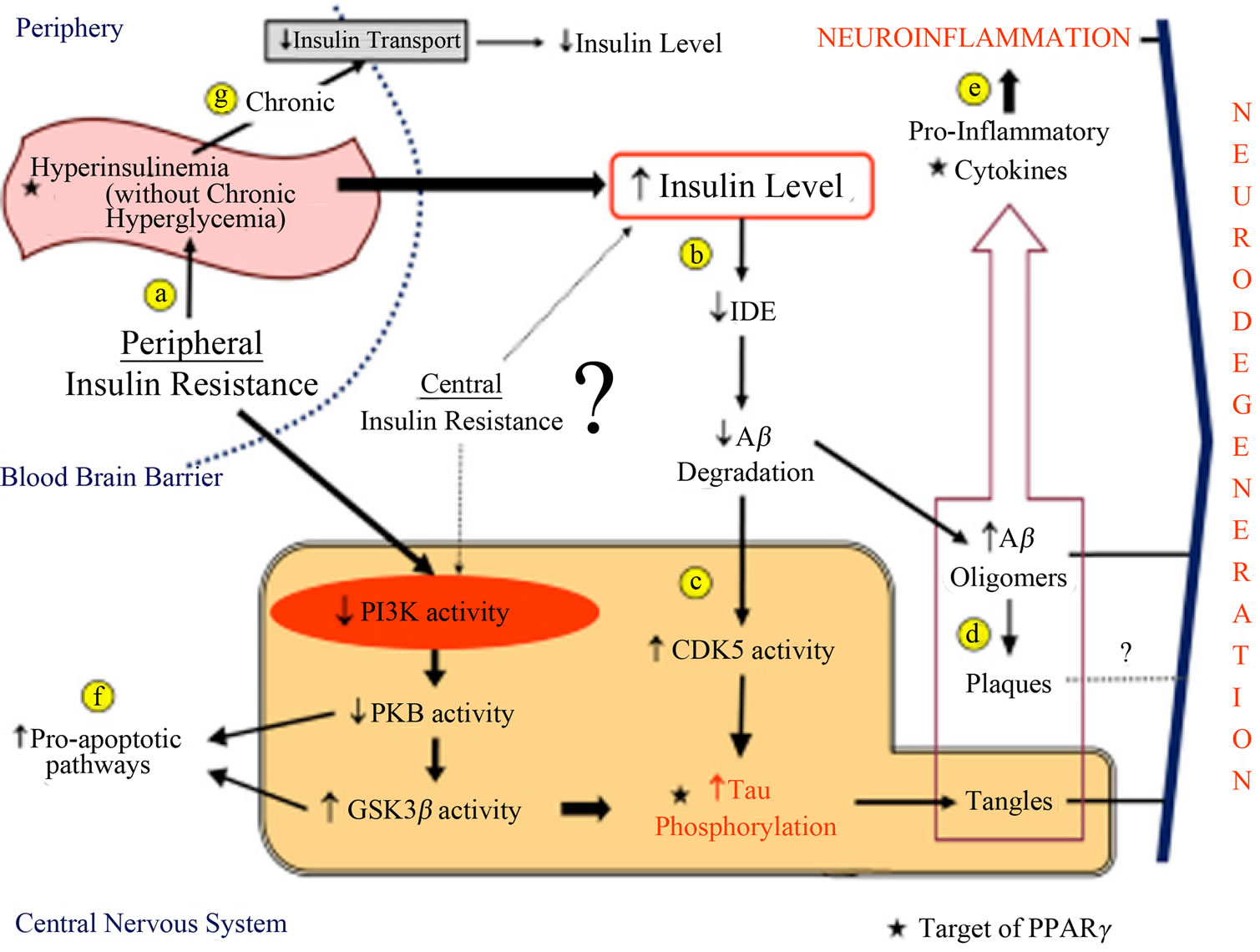
Figure 3. Schematic representation of molecular pathways linking IR and neurodegeneration, and therapeutic target of PPAR-γ ligands. (a) Peripheral IR results in periods of hyperinsulinemia to overcome the hyperglycemia; (b) Hyperinsulinemia, with the possible contribution of central insulin resistance, leads to increased insulin levels in the CNS that kidnaps IDE resulting in a decrease of amyloid-beta peptide degradation, which up regulates cdk5 with consequent tau hyperphosphorylation in the meantime that increases Aβ oligomers; (c) Peripheral insulin resistance is associated with decreased insulin signaling in neuron, which also can be a result of central insulin resistance, with decreased PI3K and PKB activity, leading an increased GSK3 activity. Cdk5 and GSK3 activities increase tau phosphorylation and tangles formation; (d) Oligomers are more toxic than Senile Plaques; they are one of the responsible elements for activation of the innate immune response in the glia; (e) These oligomers are among factors that lead to Neuroinflammation; (f) Down regulation of PKB activity and a high GSK3 activity are also related with pro-apoptotic pathways that result in neurodegeneration; (g) Thus, chronic hyperinsulinemia (without hyperglycemia) results in down-regulation of insulin transporters in the BBB and decreases the levels of insulin in the CNS [41].
modulatory role in learning and memory processes [48, 54,72,81,82]. Insulin also modulates CNS concentration of neurotransmitters associated with important roles in cognition such as acetylcholine (ACh), norepinephine and dopamine [83,84]. It has been shown that, in an early stage of memory formation, an alteration of gene expression of InsR in the rat hippocampus in response to learning experiences occurs [85]. Taken together, all these findings suggest that insulin may influence normal memory function.
The biological basis of learning and memory processes resides in synaptic strength, where insulin signaling plays a modulator role on synaptic long-term potentiation (LTP) and long-term depression (LTD), two opposite forms of activity-dependent synaptic modifications. Insulin signaling modulates synaptic plasticity by: 1) Promoting the recruitment of γ-aminobutyric acid (GABA) receptors on post-synaptic membranes; 2) Influencing N-Methyl-D-aspartate (NMDA) receptor conductance (neuronal calcium ion (Ca2+) influx); and 3) Regulating 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid (AMPA) receptor cycling [86-92].
2.2. Hyperinsulinemia/Hyperglycemia in Brain
Alterations in circulating glucose levels can negatively affect the CNS because neurons have a consistently high glucose demand. Neuronal glucose uptake depends on extracellular glucose concentration, but chronic hyperglycemia results in cellular damage (glucose neurotoxicity). Therefore, a tight metabolic control is very important [93-95]. Studies on hyperglycemic rodents have shown cognitive impairment in addition to functional and structural alterations in the brain [94].
A growing body of evidence suggests that peripheral insulin abnormalities increase the risk for memory loss and neurodegenerative disorders such as Alzheimer’s disease (AD), but acute and chronic hyperinsulinemia have opposing effects on memory performance (Figure 4). A possible explanation for that is discussed below. Chronic hyperinsulinemia has a negative influence on memory, since T2DM has been associated with longterm impairment in cognitive function in humans and animal model studies.
On the other hand, acute increases of peripheral or brain insulin have an enhanced memory performance effect [76,78,96-100]. Prediabetic conditions with hyperinsulinemia but not chronic hyperglycemia may persist for many years before progression to T2DM. Hyperinsulinemia exposes the cells including neurons to unphysiologically high levels of insulin for a long period of time. Studies have shown that high concentrations of insulin affect the function and survival of neurons in culture by sensitizing them to toxin and stress-induced insults [101].
Moreover, a relationship between glycosylation of tauproteins, its aggregation ability and the neurodegenerative phenotype has been revealed [102]. Peripheral injection of high doses of insulin in mice caused a rapid and dose-dependent increase in tau phosphorylation in the CNS [103]. In vitro studies indicate that insulin modulates the levels of amyloid-beta (Aβ) peptide by promoting the release of intracellular Aβ. Physiological insulin levels promote Aβ clearance by peptide degradation, a mechanism that involves insulin degrading enzyme (IDE) activity [104-106]. A low level of insulin in brain reduces Aβ release from intracellular to extracellular compartments and high levels reduce Aβ degradation in the extracellular compartment [107-111].
Impaired verbal memory has been reported in individuals with hyperinsulinemia and not chronic hyperglycemia [112]. Epidemiological studies have indicated that hyperinsulinemia, independent of glucose levels, constitutes one of the risk factors for dementia in the T2DM population as well as in the non-diabetic population, since the raising of peripheral insulin concentration acutely increases its concentration in the cerebrospinal fluid (CSF) and brain [96,97,113,114]. Prolonged peripheral hyperinsulinemia down-regulates BBB functions and the InsR activity and reduces insulin transport into the brain [114,115]. Thus, hyperinsulinemia during the development of T2DM is neurotoxic. Development of these complications depends on the duration of a diabetic condition, up regulation of circulating glucose, glycosylated proteins, etc., and the quality of metabolic control [93].
IR, hyperinsulinemia and T2DM are associated with elevated inflammatory markers and increased risk for AD [97,113,116-118] (Figure 3). In adults with T2DM and impaired glucose tolerance, an abnormal level of soluble
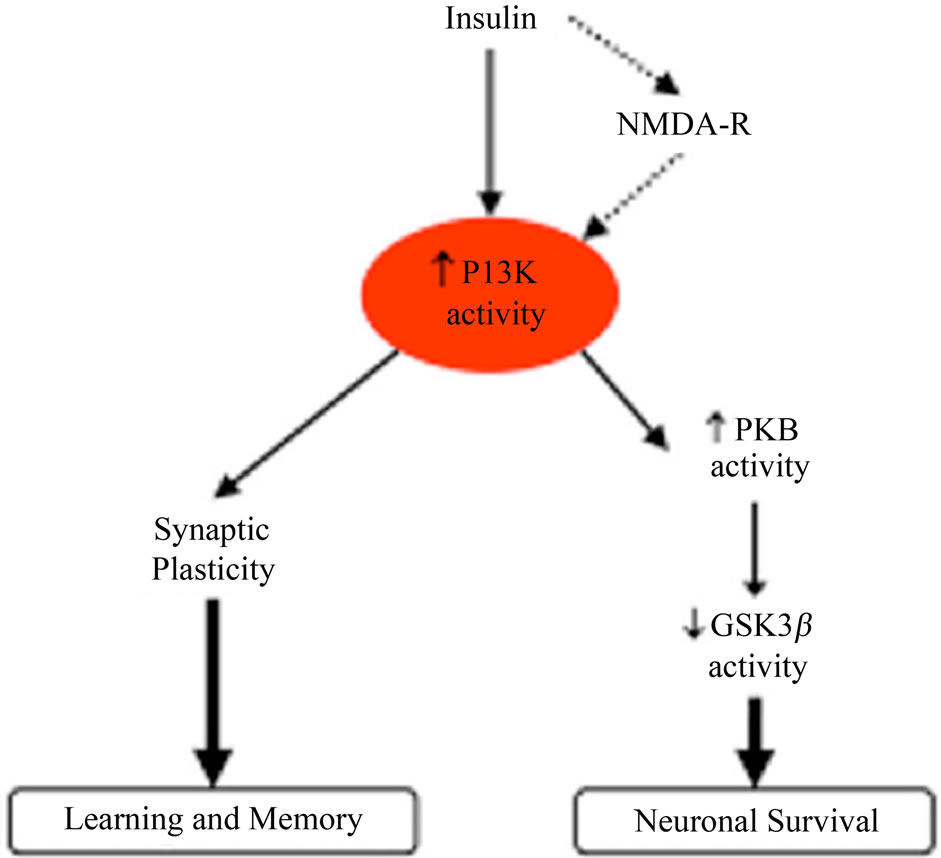
Figure 4. Schematic representation of the relationships between insulin signaling, learning, memory and neuronal survival [41].
tumor necrotic factor receptor-1 (TNF-R1) (death receptor domain) and tumor necrotic factor receptor-2 (TNFR2) (cell survival domain) has been reported. Increased levels of TNF-R1 and decreased levels of TNF-R2 have also been observed in AD brain [119-121].
2.3. Impaired Glucose Uptake and Metabolism in the AD Brain
Studies using positron emission tomography (PET) have demonstrated that glucose metabolism is reduced markedly in the cerebral cortex in early stage AD and in the mild cognitive impairment (MCI) that is believed to be a precursor of AD, and that the reduction parallels the worsening of dementia symptoms, establishing that glucose uptake and metabolism are impaired in AD brain [122]. However, the causes of the impairment of glucose uptake/metabolism in AD brain are not well understood. Several findings suggest that this impairment is a cause, rather than a consequence, of neurodegeneration. The impaired cerebral glucose consumption is more severe than the impaired oxygen consumption in AD, suggesting that the former is not the result of the latter.
The brain is highly dependent upon glucose as a source of energy. The impaired glucose uptake/metabolism might lead to deficient synaptic activity and cellular homeostasis, as these are very sensitive to energy deficiency. Impaired glucose uptake/metabolism also causes reduced formation of acetyl-CoA and, consequently, affects the synthesis of Ach [123]. Deficient activity of the cholinergic system is one of the most significant biochemical deficiencies in AD pathology, discovered decades ago [124], and is the basis of the first generation of AD drugs, the cholinesterase (ChE) inhibitors donepecil, galantamine and rivastigmine. The reduced cellular availability of acetyl-CoA may also cause decreased formation of both intracellular cholesterol and neurosteroids, which are the main lipid components of cell membranes. Membrane abnormalities have been observed in AD brains.
The Aβ peptide (or Aβ oligomers) has been shown to cause decreased glucose uptake/metabolism [125]. Furthermore, apolipoprotein E4 (ApoE4) and diabetes mellitus are known to be major risk factors for AD [126]. Individuals with ApoE4 alleles have lower brain glucose metabolism than those carrying ApoE2 or ApoE3 alleles. Diabetic patients are characterized by deficient glucose uptake/metabolism. Hence, it can be speculated that ApoE4 genotype together with diabetes may increase the risk for AD by impairment in brain glucose uptake/metabolism.
3. LINK BETWEEN IR AND AD
3.1. Relationships between IR and AD Pathogenesis
Abnormalities in insulin metabolism, characteristic of T2DM, are among the major factors thought to mechanistically influence the onset of AD. These abnormalities are thought to play a role in AD via their influence on the synthesis and degradation of Aβ and as a consequence of the cascade of neuronal alterations resulting from the effects of danger/alarm signals from oligomeric amyloid species (Figure 3) [127]. Additionally, recent studies have indicated that certain signal transduction pathways downstream of the InsR may also promote the generation of Aβ peptides by modulating the cleavage of the parent Aβ precursor protein (AβPP) at the γ-secretase site, a cleavage site necessary for Aβ amyloidogenicity [128].
Although this evidence tentatively suggests that T2DM might play an important role in AD through mechanisms that involve Aβ peptide generation, alternative studies suggest that insulin may also provoke amyloid accumulation by limiting Aβ degradation via direct competition for the insulin degrading enzyme (IDE). Another major substrate of IDE is the Aβ peptide. The IDE degrades monomeric but not the oligomeric Aβ peptide [104,105]. IDE is a zinc-metallopeptidase that preferentially cleaves proteins with a propensity to form pleated sheet-rich amyloid fibrils, such as Aβ peptides. This relationship of IDE with Aβ is supported by recent evidence indicating that IDE activity in the brain is negatively correlated with Aβ content, and that IDE expression is decreased in the AD brain [110]. Insulin regulates the levels of IDE. Hyperinsulinemia reduces Aβ degradation by reduction of IDE levels and binding competition (Figure 3) [107- 111].
It has been reported that Aβ-40 and Aβ-42 reduce insulin binding and InsR autophosphorylation. The reduction in this binding seems to be caused by a decrease in the affinity of insulin to the InsR. This suggests that Aβ is a direct competitive inhibitor of insulin binding and action [129], an aspect that demands further investigation.
The strikingly reduced CNS expression of genes encoding insulin, insulin-like growth factor-I (IGF-I), and insulin-like growth factor-II (IGF-II), as well as the insulin and IGF-I receptors in AD led to some authors to suggest that AD may represent a neuroendocrine disorder that resembles diabetes mellitus [130]. In addition, the same researchers demonstrated that alterations insulin levels and IGF expression and deterioration of insulin function with the course of AD progression, were linked with an ACh decrease in the brain [131]. There is evidence supporting the notion that high plasma insulin levels and peripheral IR affect Aβ-42 levels, inflammation in the CNS and cognitive performance of individuals [132]. From such evidence, a model can be constructed describing how this metabolic profile contributes to the pathogenesis of AD. There are several etiological factors leading to the final common expression in the AD pathology [131].
Insulin plays an important role in memory and brain function in general. Peripheral hyperinsulinemia and IR induce a number of deleterious effects in the CNS that interfere with these functions, in a manner that is exacerbated by obesity and aging. In particular, effects on Aβ regulation and neuroinflammation are potential culprits in promoting aging-related memory impairment in some cases of AD (Figure 3). This possibility has obvious relevance for adults with T2DM. However, it is worth noting that hyperinsulinemia and IR afflict many nondiabetic adults with conditions such as obesity, impaired glucose tolerance, cardiovascular disease, and hypertension [133-135].
Indeed, in recent years, cumulative evidence has been gained on the involvement of alteration in neuronal lipoproteins activity, as well as a role of cholesterol and other lipids in the pathogenesis of this neurodegenerative disorder. In relation to hypercholesterolemia, several reports have shown that elevated serum cholesterol levels and elevated levels of Aβ are linked with AD risk. Cholesterol influences the activity of the enzymes involved in the metabolism of the amyloid precursor protein (APP) and in the production of Aβ, but the mechanism by which cholesterol affects Aβ production and metabolism is not fully understood [136].
3.2. Metabolomics of Insulin in the Context of Neuronal Survival and AD Onset
To understand the link between insulin, IR, neuronal survival and AD onset, it is important identify the link between the key molecules involved in the intracellular pathways utilized by insulin to exert its effect. IR in the periphery produces acute episodes of hyperinsulinemia without chronic hyperglycemia. High levels of insulin in plasma are correlated with high levels of insulin in brain [114] leading to neurotoxic effects [101].
Studies in transgenic mouse models of AD have shown that diet-induced peripheral IR promotes amyloidosis suggesting that peripheral IR can influence Aβ production in the brain [111,137]. These findings in association of a reduced basal signaling of insulin in cortex with an increased degree of AD neuropathology argue that peripheral insulin resistance promotes neuronal IR when genetic background predisposes to AD. However, further studies are necessary to clarify this.
Insulin binding to the InsR leads to autophosphorylation of the InsR which initiates several signaling cascades. One of these is the lipid kinase PI3K cascade, which is associated with almost all of the metabolic action of insulin. PI3K activation leads to the activation of PKB and glycogen synthase kinase 3 (GSK3). Activation of PKB inactivates GSK3 by phosphorylation [138,139]. The neuroprotective effect of IGF-I results from activation of PKB [140]. Expression of PKB protects neurons against toxin-induced death and protects PC12 cells against Aβ peptide-induced death [141-143].
IR in the periphery produces hyperinsulinemia, while in the brain it decreases IDE activity. The effects of IDE include abnormal Aβ removal and plaque formation, increased cell division protein kinase 5 (cdk5) activity, and increased activity of GSK3. Dysregulation of cdk5 is a major molecular event in the pathway to neurodegeneration [144-146]. GSK3 activity has been implicated in the pathology of AD in different ways. AD brain exhibits a dysregulated expression of this kinase as well as changes in its activity [147,148], leading to hyperphosphorylation of tau and tangle formation. Several studies have shown that GSK3 activity is required for induction of neuronal apoptosis, while inhibition of GSK3 promotes neuronal survival [72]. Insulin signaling induces the phosphorylation and inhibition of GSK3 [138].
All of the negative effects of hyperinsulinemia in the CNS and their associated functions could be exacerbated or similarly produced by a central IR in conjunction with the peripheral IR or secondary to it. This is supported by the evidence of central molecular IR in a mouse model of AD with induced peripheral IR, where a reduced basal signaling (reduced phosphorylation of IR, PKB and GSK3, as well as decreased PI3K activity) was shown in the cerebral cortex [111,137].
Studies of insulin action in brain are focused on the basic effects of insulin signaling. Insulin acts like a neurotrophic factor, since it promotes neuronal survival [75,149]. Studies in vitro of intracellular pathways utilized by insulin for synaptic plasticity have identified a link to neuronal protection against cell death [75,149]. Intracellular pathways utilized by insulin to influence synaptic plasticity and neuronal survival converge on the PI3K pathway [81]. The increased catalytic activity of PI3K results in the phosphorylation and activation of antiapoptotic substrates [81]. PKB is of major importance in mediating the effects of PI3K in neuronal survival, and in vitro and in vivo studies have shown that activated PKB protects against apoptosis.
4. ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is a progressive and fatal neurodegenerative disorder manifested by cognitive and memory deterioration, progressive impairment of activities of daily living, and a variety of neuropsychiatric symptoms and behavioral disturbances. Prevalence studies suggest that in 2000 the number of persons with AD in the United States was 4.5 million [150]. The percentage of persons with AD increases by a factor of two with approximately every five years of age, meaning that 1% of 60-year-olds and about 30 % of 85-year-olds have the disease [151]. Without advances in therapy, the number of symptomatic cases in the United States is predicted to rise to 13.2 million by 2050 [150].
The amyloid hypothesis states that formation of β- amyloid (Aβ) peptides by neurons is the prime trigger of the pathogenesis of AD, and during years Aβ deposition was considered to be the cause of the disease. Evidence supporting the amyloid hypothesis has been recently reviewed [152]. Aβ protofibrils activate microglia, inciting an inflammatory response and release of neurotoxins or neurotoxic cytokines [153].
4.1. Diagnosis
AD is the most common form of dementia in the elderly. Dementia is commonly recognized with use of the criteria of the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV) [154,155]. The diagnosis of AD is most often based on the criteria developed by the National Institute of Neurologic and Communicative Disorders and Stroke-AD and Related Disorders Association (NINCDS-ADRDA) [156], according to which the diagnosis is classified as definite (clinical diagnosis with histologic confirmation), probable (typical clinical syndrome without histologic confirmation), or possible (typical clinical features but no alternative diagnosis apparent; no histologic confirmation). Typical sensitivity and specificity values for the diagnosis of probable AD with the use of these criteria are 0.65 and 0.75, respectively [157].
The classic clinical features of AD are an amnesic type of memory impairment [158,159], deterioration of language [160], and visuospatial deficits [161,162]. Motor and sensory abnormalities, gait disturbances, and seizures are uncommon until the late phases of the disease [156].
Functional and behavioral disturbances are characteristic of the disease. Patients progress from the loss of higher-level activities of daily living, such as check writing and the use of public transportation, through abnormalities of basic activities of daily living, such as eating, grooming, and using the toilet, as the disease enters advanced phases [163]. Behavioral disturbances also progress over the course of the illness [155]. Mood change and apathy commonly develop early and continue for the duration of the disease. Psychosis and agitation are characteristic of the middle and later phases of the disease [164].
As part of the assessment of dementia, laboratory studies are necessary to identify causes of dementia and coexisting conditions that are common in the elderly. Thyroid function tests and measurement of the serum vitamin B12 level are required to identify specific alternative causes of dementia. A complete blood count; measurement of blood urea nitrogen (BUN), serum electrolyte, and blood glucose levels; and liver-function tests should be performed [165]. Specialized laboratory studies such as a serologic test for syphilis, the erythrocyte sedimentation rate (ESR), a test for human immunodeficiency virus antibody, or screening for heavy metals are indicated when historical features or clinical circumstances suggest that infections, inflammatory diseases, or exposure to toxins may be contributing to the dementia.
Neuroimaging plays an important role in the diagnosis of AD and is particularly helpful in excluding alternative causes of dementia. It is recommended that patients undergo structural imaging of the brain with computed tomography (CT) or magnetic resonance imaging (Figure 5) at least once in the course of their dementia [165]. Functional imaging with PET (Figure 5) or single-photon-emission CT may be helpful in the differential diagnosis of disorders associated with dementia [166].
4.2. Pathophysiology
There is increasing consensus that the production and accumulation of Aβ peptide are central to the pathogenesis of AD [168]. Evidence supporting a pivotal role for Aβ includes the following: mutations in the gene encoding APP lead to early-onset AD; all currently known mutations associated with AD increase the production of Aβ; in patients with trisomy 21 (Down’s syndrome) and three copies of the gene for APP, neuropathological characteristics of AD develop by midlife; Aβ is neurotoxic in vitro and leads to cell death; over expression of human APP in


Figure 5. Scans of patients with probable AD. A magnetic resonance image shows cortical atrophy and scan shows reduced glucose metabolism in the parietal lobes bilaterally (blue-green) as compared with more normal metabolism in other cortical areas (yellow) [167].
transgenic mouse models of AD results in neuritic plaques similar to those seen in humans with AD; transgenic mice over expressing the human APP have evidence of learning and memory deficits, in concert with the accumulation of amyloid; the ApoE ε4 genotype, a major risk factor for AD, leads to accelerated deposition of amyloid; and the generation of antiamyloid antibodies in humans with AD seems to ameliorate the disease process [169-173] Formation of neurofibrillary tangles, oxidation and lipid peroxidation, glutamatergic excitotoxicity, inflammation, and activation of the cascade of apoptotic cell death are considered secondary consequences of the generation and deposition of Aβ [168] (Figure 6). This hypothesized amyloid cascade underlies attempts to modify the onset and course of AD through identification of antiamyloid agents, antioxidants, antiinflammatory drugs, compounds that limit the phosphorylation of tau-protein, antiapoptotic agents, and glutamatergic N-methyld-aspartate receptor antagonists.
Cell dysfunction and cell death in nuclear groups of neurons responsible for maintenance of specific transmitter systems lead to deficits in acetylcholine, norepinephrine, and serotonin [174,175]. Alternate hypotheses regarding the pathophysiology of AD place greater emphasis on the potential role of tau-protein abnormalities, heavy metals, vascular factors, or viral infections.
4.3. Oxidative Damage and AD
4.3.1. Inflammation and AD
It has been speculated that this inflammatory response associated with the presence of neuritic plaques is secondary to Aβ accumulation and could be involved in neuronal damage and with the progression of the disease. Activated microglia and reactive astrocytes surrounding extracellular deposits of Aβ initiate an inflammatory re-
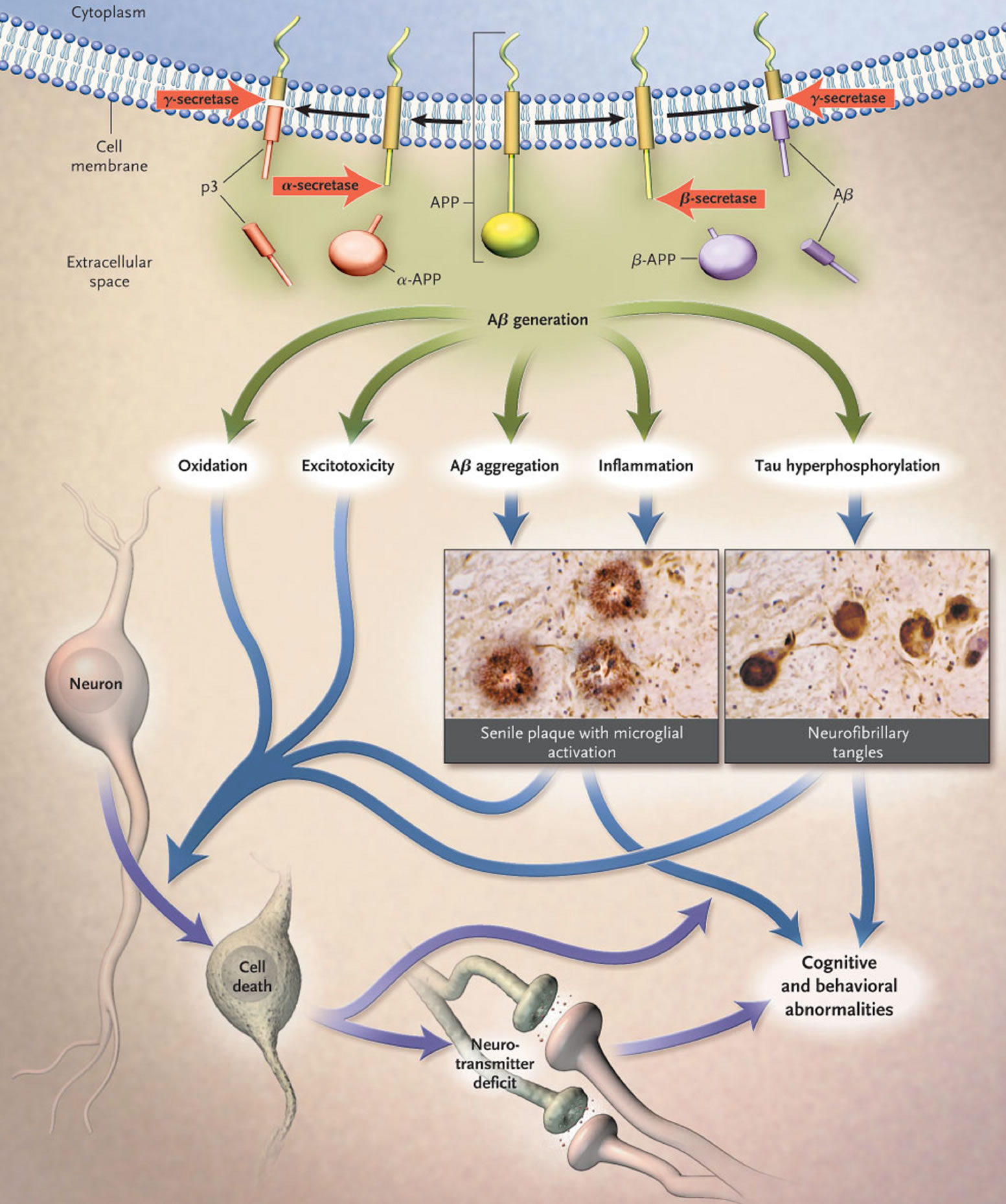
Figure 6. Putative amyloid cascade. This hypothesis of the amyloid cascade, which progresses from the generation of the Aβ peptide from APP, through multiple secondary steps, to cell death, forms the foundation for current and emerging options for the treatment of AD [167].
sponse characterised by a local cytokine-mediated acute phase response, activation of the complement cascade and subsequent further cell damage [176-178]. The main inflammatory players in AD are:
1) Microglia Microglia constitute around 10% of the cells in the nervous system. They represent the first line of defense against invading pathogens or other types of brain tissue injury. Under pathological situations, such as neurodegenerative disease, stroke and tumor invasion, these cells become activated, surround damaged and dead cells and clear cellular debris from the area, like phagocytic macrophages of the immune system [179]. Activated microglia up-regulate a variety of surface receptors, including the major histocompatibility complex and complement receptors [180]. They also experience dramatic morphological changes from resting ramified cells to motile activated amoeboid microglia [181]. Once immunostimulated in response to neurodegenerative events, these microglia cells release a variety of proinflammatory mediators such as cytokines, ROS, complement factors, neurotoxic secretory products, free radical species and nitric oxide (NO) which all can contribute to both neuronal dysfunction and cell death, ultimately creating a vicious cycle [182].
Microglia has been suggested to be preferentially associated with certain amyloid plaque types [183]. Aβ peptides and APP are potent glial activators [184,185] and disruption of APP gene and its proteolytic products delay and decrease microglial activation [186]. The extend of astrocytosis and microglial activation is directly dependent on the amyloid load, and treatment with β- sheet breakers peptide results in reduced brain inflammation [187]. Aβ is able to stimulate NF-β-dependent pathway that is required for cytokine production [188]. The subsequent activation of extracellular signal-regulated kinase (ERK) and MAPK pathways by Aβ binding to the microglial cell surface induces proinflammatory gene expression and leads to the production of cytokines and chemokines [189]. Not only Aβ, but also the C-terminal 100 amino acids of β-APP, which are present also in senile plaques, can induce astrocytosis and neuronal death. The C-terminal 100 exposure results in activation of MAPK pathways as well as NF-κB [190]. On the other hand, other proteins involved in APP processing have been implicated in the inflammatory response. Loss of presenilin function in presenilin conditional knockout mice leads to differential up-regulation of inflammatory markers in the cerebral cortex, such as strong microglial activation, and elevated levels of glial fibrillary acidic protein (GFAP), complement component C1q, and cathepsin S [191]. This effect might not be directly related to APP metabolism, since presenilin has been also shown to interact with β-catenin and to be implicated in the cleavage of other type I transmembrane proteins, such as Notch, CD44, Ecadherin, receptor tyrosine-protein kinase erbB-4 (ErbB-4) and the Notch ligands [192]. In some situations the role of microglia has been found to be beneficial, since activated microglia can reduce Aβ accumulation by increasing its phagocytosis, clearance and degradation [105,193]. Secreted Aβ1-40 and Aβ1-42 peptides are constitutively degraded by neprilysin and IDE, a metalloprotease released by microglia and other neural cells, whose enzymatic activity is enhanced by inflammatory events, such as lipopolysaccharide (LPS) stimulation [104]. Microglia can also secrete a number of soluble factors, such as the glia-derived neurotrophic factor (GDNF), which are potentially beneficial to the survival of neurons [180].
2) Astrocytes Astrocytes are known to be important for Aβ clearance and degradation, for providing trophic support to neurons, and for forming a protective barrier between Aβ deposits and neurons [194,195]. The presence of large numbers of astrocytes associated with Aβ deposits in AD suggests that these lesions generate chemotactic molecules that mediate astrocyte recruitment. Astrocytes throughout the entorhinal cortex of AD patients gradually accumulate Aβ-42 positive material and the amount of this material correlates positively with the extent of local AD pathology. Aβ-42 within astrocytes appears to be of neuronal origin, most likely accumulated via phagocytosis of local degenerated dendrites and synapses, especially in the cortical molecular layer [196] Figure 7.
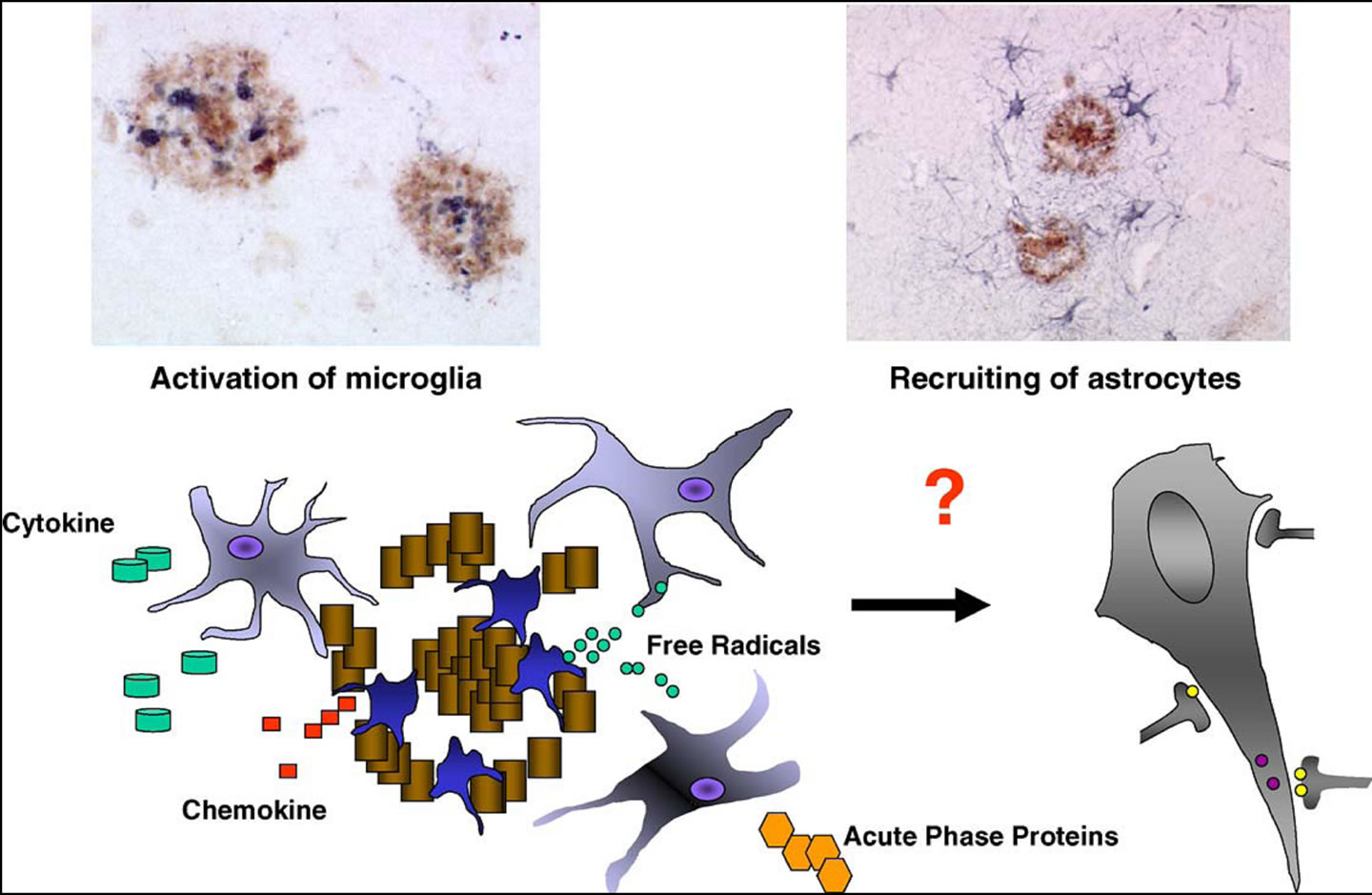
Figure 7. Representation of the local inflammatory response by amyloid-beta. Staining of activated microglia and the recruitment of astrocytes in a neuritic plaque from the brain of an AD patient. Schematic representation of Aβ deposition, which promotes microglial activation and the recruitment of astrocytes, release of cytokines and cytotoxic molecules that are harmful for neurons. At the same time, microglia and astrocytes are able to phagocyte Aβ and to secrete IDE, which contribute to the clearance of Aβ [211].
However, under certain conditions related to chronic stress, the role of astrocytes may not be beneficial. Recruited astrocytes assemble at the Aβ plaque site most likely prolonging neuroinflammation and contributing to NO-mediated neurotoxicity by expressing the inducible nitric oxide synthase (iNOS) and the L-arginine-supplying enzyme argininosuccinate synthetase [197]. A recent report suggests that astrocytes could be also a source for Aβ, because they over express beta-site APP cleaving enzyme 1 (BACE1 or β-secretase) in response to chronic stress [195]. Oxidative damage could be already an early event in the disease, even when there is no Aβ deposition [198]. In this line, a report has shown microglial activation in early stages of AD, comparing PET and volumetric magnetic resonance imaging (MRI) in patients with mild to moderate dementia with healthy individuals [199].
4.3.2. Inflammatory Cytokines and Chemokines@NolistTemp# Nearly all the cytokines and chemokines that have been studied in AD, including IL-1β, IL-6, TNF-α, IL-8, transforming growth factor-β (TGF-β) and macrophage inflammatory protein-1α (MIP-1α) seem to be up-regulated in AD compared with control individuals [200]. An association of AD with polymorphisms in a number of proinflammatory genes has been described, including IL-1 [201], IL-6 [202], TNF-α [203,204], and 1-antichymotrypsin, an acute phase protein [205]. Animal models of AD, such as Tg2567 over expressing APP carrying the Swedish mutation, also show enhanced levels for TNF-α, IL-1a, IL-1β, chemoattractant protein-1, cyclooxigenase-2 (COX-2) and complement component 1q [206-209]. In addition, an increased immunoreactivity in IL-1 and COX-2 in mice over expressing the mutant human P301 tau-proteins have been reported [210]. The production of interleukins and other cytokines and chemokines may also lead to microglial activation, astrogliosis, and further secretion of proinflammatory molecules and amyloid, thus perpetuating the cascade [189].REFERENCES
- Marchesini, G., Brizi, M., Bianchi, G., Tomassetti, S., Bugianesi, E., Lenzi, M., McCullough, A.J., Natale, S., Forlani, G. and Melchionda, N. (2001) Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes, 50, 1844-1850. doi:10.2337/diabetes.50.8.1844
- Sanyal, A.J., Campbell-Sargent, C., Mirshahi, F., Rizzo, W.B., Contos, M.J., Sterling, R.K., Luketic, V.A., Shiffman, M.L. and Clore, J.N. (2001) Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology, 120, 1183- 1192. doi:10.1053/gast.2001.23256
- Petersen, K.F., Befroy, D., Dufour, S., Dziura, J., Arijan, C., Rothman, D.L., DiPietro, L., Cline, G.W. and Shulman, G.I. (2003) Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science, 300, 1140- 1142. doi:10.1126/science.1082889
- Iossa, S., Mollica, M.P., Lionetti, L., Crescenzo, R., Tasso, R. and Liverini, G. (2004) A possible link between skeletal muscle mitochondrial efficiency and age-induced Insulin Resistance. Diabetes, 53, 2861-2866. doi:10.2337/diabetes.53.11.2861
- Saltiel, A.R. and Kahn, C.R. (2001) Insulin signaling and the regulation of glucose and lipid metabolism. Nature, 414, 799-806. doi:10.1038/414799a
- Samuel, V.T. and Shulman, G.I. (2005) Insulin resistance in NAFLD: Potential mechanisms and therapies. In: Farrell, G.C., George, J., Hall, P., et al., Eds. Fatty Liver Disease—NASH and Related Disorders, Oxford Publishing Limited, Blackwell, 38-54.
- Shepherd, P.R. and Kahn, B.B. (1999) Glucose transporters and insulin action—implications for insulin resistance and diabetes mellitus. New England Journal of Medicine, 341, 248-257. doi:10.1056/NEJM199907223410406
- Virkamaki, A., Ueki, K. and Kahn, C.R. (1999) Proteininteraction in insulin signaling and the molecular mechanisms of insulin resistance. Journal of Clinical Investigation, 103, 931-943. doi:10.1172/JCI6609
- Combettes-Souverain, M. and Issad, T. (1998) Molecular basis on insulin action. Diabetes Metabolism, 24, 477- 489.
- McCullough, A.J. (2006) Pathophysiology of nonalcoholic steatohepatitis. Journal of Clinical Gastroenterology, 40, S17-S29.
- Synkiotis, G.P. and Papavassiliou, AG. (2001) Serine phosphorylation of insulin receptor substrates-1: A novel target for the reversal of insulin resistance. Molecular Endocrinology, 15, 1864-1869.
- Kido, Y., Burks, D.J., Withers, D., Bruning, J.C., Kahn, C.R., White, M.F. and Accili, D. (2000) Tissue-specific insulin resistance in mice with combined mutations of insulin receptor, IRS-1 and IRS-2. Journal of Clinical Investigation, 105, 199-205. doi:10.1172/JCI7917
- Previs, S.F., Withers, D.J., Ren, J.M., White, M.F. and Shulman, G.I. (2000) Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. Journal of Biological Chemistry, 275, 38990- 38994. doi:10.1074/jbc.M006490200
- Das, U.N. (2001) Is obesity an inflammatory condition? Nutrition, 17, 953-966. doi:10.1016/S0899-9007(01)00672-4
- Promrat, K., Lutchman, G., Uwaifo, G.I., Freedman, R.J., Soza, A., Heller, T., Doo, E., Ghany, M., Premkumar, A., Park, Y., Liang, T.J., Yanovski, J.A., Kleiner, D.E. and Hoofnagle, J.H. (2004) A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology, 39, 188-196. doi:10.1002/hep.20012
- Neuschwander-Tetri, B.A., Brunt, E.M., Wehmeier, K.R., Oliver, D. and Bacon, B.R. (2003) Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology, 38, 1008- 1017.
- Sekiya, M., Yahagi, N., Matsuzaka, T., Najima, Y., Nakakuki, M., Nagai, R., Ishibashi, S., Osuga, J., Yamada, N. and Shimano, H. (2003) Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology, 38, 1529-1539.
- Yuan, M., Konstantopoulos, N., Lee, J., Hansen, L., Li, ZW., Karin, M. and Shoelson, S.E. (2001) Reversal of obesity and diet-induced insulin resistance with salicylates on targeted disruption of IKK-β. Science, 293, 1673- 1677. doi:10.1126/science.1061620
- Parola, M. and Robino, G. (2001) Oxidative stress-related molecules and liver fibrosis. Journal of Hepatology, 35, 297-306. doi:10.1016/S0168-8278(01)00142-8
- Goldstein, B.J., Mahadev, K. and Wu, X. (2005) Insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes, 54, 311-321. doi:10.2337/diabetes.54.2.311
- Wanless, I.R., Bargman, J.M., Oreopoullos, D.G. and Vas, S.I. (1989) Subcapsular steatonecrosis in response to peritoneal insulin deliver: A clue to the pathogenesis of steatonecrosis in obesity. Modern Pathology, 2, 69-74.
- Khalili, K., Lan, F.P., Hanbidge, A.E., Muradali, D., Oreopoulos, D.G. and Wanless, I.R. (2003) Hepatic subcapsular steatosis in response to intraperitoneal insulin delivery: CT findings and prevalence. American Journal of Roentgenology, 180, 1601-1604. doi:10.2214/ajr.180.6.1801601
- Li, X.L., Man, K., Ng, K.T., Lee, T.K., Lo, C.M. and Fan, S.T. (2004) Insulin in UW solution exacerbates hepatic ischemia/reperfusion injury by energy depletion through the IRS-2/SREBP-1c pathway. Liver Transplantation, 10, 1172-1182. doi:10.1002/lt.20240
- Paradis, V., Perlemuter, G., Bonvoust, F., Dargere, D., Parfait, B., Vidaud, M., Conti, M., Huet, S., Ba, N., Buffet, C. and Bedossa, P. (2001) High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology, 34, 738-744.
- Matteoni, C.A., Younossi, Z.M., Gramlich, T., Boparai, N., Liu, Y.C. and McCullough, A.J. (1999) Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology, 116, 1413-1419. doi:10.1016/S0016-5085(99)70506-8
- Younossi, Z.M., Gramlich, T., Matteoni, C.A., Boparai, N. and McCullough, A.J. (2004) Nonalcoholic fatty liver disease in patients with type II diabetes. Clinical Gastroenterology and Hepatology, 2, 262-265. doi:10.1016/S1542-3565(04)00014-X
- El-Serag, H.B., Tran, T. and Everhart, J.E. (2004) Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology, 126, 460-468. doi:10.1053/j.gastro.2003.10.065
- Ozcan, U., Cao, Q., Yilmaz, E., Lee, A.H., Iwakoshi, N.N., Ozdelen, E., Tuncman, G., Görgün, C., Glimcher, L.H. and Hotamisligil, G.S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type II diabetes. Science, 306, 457-461. doi:10.1126/science.1103160
- Muoio, D.M. (2004) Insulin resistance takes a trip through the ER. Science, 306, 4285-4426. doi:10.1126/science.1104680
- Hasegawa, T., Yoneda, M., Nakamura, K., Makino, I. and Terano, A. (2001) Plasma transforming growth factorbeta1 level and efficacy of α-tocopherol in patients with non-alcoholic steatohepatitis: A pilot study. Journal of Alimentary Pharmacology & Therapeutics, 15, 1667-1672. doi:10.1046/j.1365-2036.2001.01083.x
- Robertson, G., Leclercq, I. and Farrell, G.C. (2001) Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. American Journal of Physiology—Gastrointestinal and Liver Physiology, 281, G1135-G1139.
- Banks, W.A. (2004) The source of cerebral insulin. European Journal of Pharmacology, 490, 5-12. doi:10.1016/j.ejphar.2004.02.040
- Banks, W.A. (2006) Blood-brain barrier and energy balance. Obesity, 14, 234S-237S. doi:10.1038/oby.2006.315
- Margolis, R.U. and Altszuler, N. (1967) Insulin in the cerebrospinal fluid. Nature, 215, 1375-1376. doi:10.1038/2151375a0
- Steffens, A.B., Scheurink, A.J., Porte Jr., D. and Woods, S.C. (1988) Penetration of peripheral glucose and insulin into cerebrospinal fluid in rat. American Journal of Physiology, 255, R200-R204.
- Schwartz, M.W., Figlewicz, D.P., Baskin, D.G., Woods, S.C. and Porte Jr., D. (1992) Insulin in the brain: A hormonal regulator of energy balance. Endocrine Reviews, 13, 387-414.
- Baura, G.D., Foster, D.M., Porter Jr., D., Kahn, S.E., Bergman, R.N., Cobelli, C. and Schwartz, M.W. (1993) Saturable transport of insulin from plasma into the central nervous system of dogs in vivo: A mechanism for regulated insulin delivery to the brain. Journal of Clinical Investigation, 92, 1824-1830. doi:10.1172/JCI116773
- Banks, W.A., Jaspan, J.B. and Kastin, A.J. (1997) Selective, physiological transport of insulin across the bloodbrain barrier: Novel demonstration by species-specific radioimmunoassay. Peptides, 18, 1257-1262. doi:10.1016/S0196-9781(97)00198-8
- Banks, W.A., Jaspan, J.B., Huang, W. and Kastin, A.J. (1997) Transport of insulin across the blood-brain barrier: Saturability at euglycemic doses of insulin. Peptides, 18, 1423-1429. doi:10.1016/S0196-9781(97)00231-3
- Woods, S.C., Seeley, R.J., Baskin, D.G. and Schwartz, M.W. (2003) Insulin and blood-brain barrier. Current Pharmaceutical Design, Vol. 9, 795-800. doi:10.2174/1381612033455323
- Neumann, K.F., Rojo, L., Navarrete, L.P., Farías, G., Reyes, P. and Maccioni, R.B. (2008) Insulin resistance and Alzheimer’s disease: Molecular links & clinical implications. Current Alzheimer Research, 5, 438-447. doi:10.2174/156720508785908919
- Devaskar, S.U., Giddings, S.J., Rajakumar, P.A., Carnaghi, L.R., Menon, R.K. and Zahm, D.S. (1994) Insulin gene expression and insulin synthesis in mammalian neuronal cells. Journal of Biological Chemistry, 269, 8445-8454.
- Rulifson, E.J., Kim, S.K. and Nusse, R. (2002) Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science, 296, 1118-1120. doi:10.1126/science.1070058
- Havrankova, J., Roth, J. and Brownstein, M. (1978) Insulin receptors are widely distributed in central nervoussystem of rat. Nature, 272, 827-829. doi:10.1038/272827a0
- Van Houten, M., Posner, B.I., Kopriwa B.M. and Brawer, J.R. (1979) Insulin binding sites in the rat brain: In vivo localization to the circumventricular organs by quantitative radioautography. Endocrinology, 105, 666-673. doi:10.1210/endo-105-3-666
- Baskin, D.G., Woods, S.C., West, D.B., Van Houten, M., Posner, B.I., Dorsa, D.M. and Porte Jr., D. (1983) Immunocytochemical detection of insulin in rat hypothalamus and its possible uptake from cerebrospinal fluid. Endocrinology, 113, 1818-1825. doi:10.1210/endo-113-5-1818
- Heidenreich, K.A. Zahniser, N.R. Berhanu, P. Brandenburg D. and Olefsky, J.M. (1983) Structural differences between insulin receptors in the brain and peripheral target tissues. Journal of Biological Chemistry, 258, 8527- 8530.
- Plum, L., Schubert, M. and Brüning, J.C. (2005) The role of insulin receptor signaling in the brain. Trends in Endocrinology and Metabolism, 16, 59-65. doi:10.1016/j.tem.2005.01.008
- Havrankova, J., Brownstein, M. and Roth, J. (1981) Insulin and insulin receptors in rodent brain. Diabetologia, 20, 268-273. doi:10.1007/BF00254492
- Wozniak, M., Rydzewski, B., Baker, S.P. and Raizadai, M. (1993) The cellular and physiological actions of insulin in the central nervous system. Neurochemistry International, 22, 1-10. doi:10.1016/0197-0186(93)90062-A
- Unger, J.W. and Betz, M. (1998) Insulin receptors and signal transduction proteins in the hypothalamo-hypophyseal system: A review on morphological findings and functional implications. Histology and Histopathology, 13, 1215-1224.
- Abbott, M.A., Wells, D.G. and Fallon, J.R. (1999) The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. Journal of Neuroscience, 19, 7300-7308.
- Florant, G.L., Singer, L., Scheurink, A.J., Park, C.R., Richardson, R.D. and Woods, S.C. (1991) Intraventicular insulin reduces food intake and body weight of marmots during the summer feeding period. Physiology and Behavior, 49, 335-338. doi:10.1016/0031-9384(91)90053-Q
- Craft, S. and Watson, G.S. (2004) Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurology, 3, 169-178. doi:10.1016/S1474-4422(04)00681-7
- Clarke, D.D. and Sokoloff, L. (1999) Circulation and energy metabolism of the brain. In: Siegel, G.J., Agranoff, B.W., Albers, R.W., Fisher, S.K. and Uhler, M.D., Eds., Basic Neurochemistry, 6th Edition, Lippincott-Ratven, Philadelphia, 637-669.
- Duckrow, R.B., Beard, D.C. and Brennan, R.W. (1987) Regional cerebral blood flow decreases during chronic and acute hyperglycemia. Stroke, 18, 52-58. doi:10.1161/01.STR.18.1.52
- Pelligrino, D.A. Segil, L.J. and Albrecht, R.F. (1990) Brain glucose utilization and transport and cortical function in chronic vs. acute hypoglycemia. American Journal of Physiology, 259, E729-E735.
- Lucignani, G., Namba, H., Nehlig, A., Porrino, L.J., Kennedy, C. and Sokoloff, L. (1987) Effects of insulin on local cerebral glucose utilization in the rat. Journal of Cerebral Blood Flow & Metabolism, 7, 309-314. doi:10.1038/jcbfm.1987.68
- Marfaing, P., Penicaud, L., Broer, Y., Mraovitch, S., Calando, Y. and Picon, L. (1990) Effects of hyper insulinemia on local cerebral insulin binding and glucose utilizetion in normoglycemic awake rats. Neuroscience Letters, 115, 279-285.
- Doyle, P. Cusin, I., Rohner-Jeanrenaud, F. and Jeanrenaud, B. (1995) Four day hyperinsulinemia in euglycemic conditions alters local cerebral glucose utilization in specific brain nuclei of freely moving rats. Brain Research, 684, 47-55. doi:10.1016/0006-8993(95)00402-C
- Fanelli, C.G., Dence, C.S., Markham, J., Videen, T.O., Paramore, D.S., Cryer, P.E. and Powers, W.J. (1998) Blood-to-brain glucose transport and cerebral glucose metabolism are not reduced in poorly controlled type 1 diabetes. Diabetes, 47, 1444-1450. doi:10.2337/diabetes.47.9.1444
- Hasselbalch, S.G., Knudsen, G.M., Videback, C., Pinborg, L.H., Schmidt, J.F., Holm, S., and Paulson, O.B. (1999) No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes, 48, 1915-1921. doi:10.2337/diabetes.48.10.1915
- Bingham, E.M., Hopkins, D., Smith, D., Pernet, A., Hallett, W., Reed, L., Marsden, P.K. and Amiel, S.A. (2002) The role of insulin in human brain glucose metabolism: An 18-fluoro-deoxyglucose positron emission tomogramphy study. Diabetes, 51, 3384-3390. doi:10.2337/diabetes.51.12.3384
- Brant, A.M., Jess, T.J., Milligan, G., Brown, C.M. and Gould, G.W. (1993) Immunological analysis of glucose transporters expressed in different regions of the rat brain and central nervous system. Biochemical and Biophysical Research Communications, 192, 1297-1302. doi:10.1006/bbrc.1993.1557
- El Messari, S., Leloup, C., Quignon, M., Brisorgueil, M.J., Penicaud, L. and Arluison, M. (1998) Immunocytochemical localization of the insulin-responsive glucose transporter 4 (Glut4) in the rat central nervous system. Journal of Comparative Neurology, 399, 492-512. doi:10.1002/(SICI)1096-9861(19981005)399:4<492::AID-CNE4>3.0.CO;2-X
- Ibberson, M., Uldry, M. and Thorens, B. (2000) GLUTX1, a novel mammalian glucose transporter expressed in the central nervous system and insulin sensitive tissues. Journal of Biological Chemistry, 275, 4607-4612. doi:10.1074/jbc.275.7.4607
- Reagan, L.P., Gorovits, N., Hoskin, E.K., Alves, S.E., Katz, E.B., Grillo, C.A., Piroli, G.G., McEwen, B.S. and Charron, M.J. (2001) Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proceedings of the National Academy of Sciences of the United States of America, 98, 2820-2825. doi:10.1073/pnas.051629798
- Obici, S., Zhang, B.B., Karkanias G. and Rossetti, L. (2002) Hypothalamic insulin signaling is required for inhibition of glucose production. Nature Medicine, 8, 1376- 1382. doi:10.1038/nm1202-798
- Niswender, K.D. and Schwartz, M.W. (2003) Insulin and leptin revisited: Adiposity signals with overlapping physiological and intracellular signaling capabilities. Frontiers in Neuroendocrinology, 24, 1-10. doi:10.1016/S0091-3022(02)00105-X
- Clarke, D.W., Boyd Jr., F.T., Kappy, M.S. and Raizada, M.K. (1984) Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. Journal of Biological Chemistry, 259, 11672-11675.
- Schubert, M., Gautam, D., Surjo, D., Ueki, K., Baudler, S., Schubert, D, Kondo, T, Alber, J, Galldiks, N, Küstermann, E., Arndt, S., Jacobs, A.H., Krone, W., Kahn, C.R. and Brüning, J.C. (2004) Role for neuronal Insulin Resistance in neurodegenerative disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 3100-3105. doi:10.1073/pnas.0308724101
- Cole, A.R., Astell, A., Green, C. and Sutherland, C. (2007) Molecular connexions between dementia and diabetes. Neuroscience & Biobehavioral Reviews, 31, N 1046-1063. doi:10.1016/j.neubiorev.2007.04.004
- Mill, J.F., Chao, M.V. and Ishii, D.N. (1985) Insulin, insulin-like growth factor II, and nerve growth factor effects on tubulin mRNA levels and neurite formation. Proceedings of the National Academy of Sciences of the United States of America, 82, 7126-7130. doi:10.1073/pnas.82.20.7126
- Wang, C., Li, Y., Wible, B., Angelides, K.J. and Ishii, D.N. (1992) Effects of insulin and insulin-like growth factors on neurofilament mRNA and tubulin mRNA content in human neuroblastoma SH-SY5Y cells. Molecular Brain Research, 13, 289-300. doi:10.1016/0169-328X(92)90212-T
- Tanaka, M., Sawada, M., Yoshida, S., Hanaoka F. and Marunouchi, T. (1995) Insulin prevents apoptosis of external granular layer neurons in rat cerebellar slice cultures. Neuroscience Letters, 199, 37-40. doi:10.1016/0304-3940(95)12009-S
- Craft, S., Asthana, S., Newcomer, J.W., Wilkinson, C.W., Matos, I.T., Baker, L.D., Cherrier, M., Lofgreen, C., Latendresse, S., Petrova, A., Plymate, S., Raskind, M., Grimwood, K. and Veith, R.C. (1999) Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Archives of General Psychiatry, 56, 1135-1140. doi:10.1001/archpsyc.56.12.1135
- Kern, W., Born, J., Schreiber, H. and Fehm, H.L. (1999) Central nervous system effects of intranasally administered insulin during euglycemia in men. Diabetes, 48, 557-563. doi:10.2337/diabetes.48.3.557
- Park, C.R., Seeley, R.J., Craft, S. and Woods, S.C. (2000) Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiology & Behavior, 68, 509- 514. doi:10.1016/S0031-9384(99)00220-6
- Kern, W., Peters, A., Fruehwald-Schultes, B., Deininger, E., Born, J. and Fehm, H.L. (2001) Improving influence of insulin on cognitive functions in humans. Neuroendocrinology, 74, 270-280. doi:10.1159/000054694
- Craft, S., Asthana, S., Cook, D.G., Baker, L.D., Cherrier, M., Purganan, K., Wait, C., Petrova, A., Latendresse, S., Watson, G.S., Newcomer, J.W., Schellenberg, G.D. and Krohn, A.J. (2003) Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology, 28, 809-822. doi:10.1016/S0306-4530(02)00087-2
- Van Der Heide, L.P., Ramakers, G.M. and Smidt, M.P. (2006) Insulin signaling in the central nervous system: Learning to survive. Progress in Neurobiology, 79, 205- 221. doi:10.1016/j.pneurobio.2006.06.003
- Dou, J.T., Chen, M., Dufour, F., Alkon, D.L. and Zhao, W.Q. (2005) Insulin receptor signaling in long-term memory consolidation following spatial learning. Learning & Memory, 12, 646-655. doi:10.1101/lm.88005
- Figlewicz, D.P., Szot, P., Israel, P.A., Payne C., and Dorsa, D.M. (1993) Insulin reduces norepinephrine transporter mRNA in vivo in rat locus coeruleus. Brain Research, 602, 161-164. doi:10.1016/0006-8993(93)90258-O
- Kopf, S. and Baratti, C. (1999) Effects of posttraining administration of insulin on retention of a habituation response in mice: Participation of a central cholinergic mechanism. Neurobiology of Learning and Memory, 71, 50-61. doi:10.1006/nlme.1998.3831
- Zhao, W., Chen, H., Xu, H., Moore, E., Meiri, N., Quon, M.J. and Alkon, D.L. (1999) Brain insulin receptors and spatial memory: Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. Journal of Biological Chemistry, 274, 34893-34902. doi:10.1074/jbc.274.49.34893
- Wang, Y.T. and Salter, M.W. (1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature, 369, 233-235. doi:10.1038/369233a0
- Wan, Q., Xiong, Z.G., Man, H.Y., Ackerley, C.A., Braunton, J., Lu, W.Y., Becker, L.E., MacDonald, J.F. and Wang, Y.T. (1997) Recruitment of functional GABA (A) receptors to postsynaptic domains by insulin. Nature, 388, 686-690. doi:10.1038/41792
- Man, H.Y., Lin, J.W., Ju, W.H., Ahmadian, G., Liu, L., Becker, L.E., Sheng, M. and Wang, Y.T. (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron, 25, 649-662. doi:10.1016/S0896-6273(00)81067-3
- Kneussel, M. (2002) Dynamic regulation of GABA (A) receptors at synaptic sites. Brain Research Reviews, 39, 74-83. doi:10.1016/S0165-0173(02)00159-5
- Malenka, R.C. (2003) Synaptic plasticity and AMPA receptor trafficking. Annals of the New York Academy of Sciences, 1003, 1-11. doi:10.1196/annals.1300.001
- Huang, C.C. Lee, C.C. and Hsu, K.S. (2004) An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. Journal of Neurochemistry, 89, 217-231. doi:10.1111/j.1471-4159.2003.02307.x
- Van Der Heide, L.P., Kamal, A., Artola, A., Gispen, W.H. and Ramakers, G.M. (2005) Insulin modulates hippocampal activity-dependent synaptic plasticity in N-methyld-aspartate receptor and phosphatidylinositol-3-kinasedependent manner. Journal of Neurochemistry, 94, 1158- 1166. doi:10.1111/j.1471-4159.2005.03269.x
- Pirart, J. (1977) Diabetes Mellitus and its degenerative complications: Prospective study of 4400 patients observed between 1947 and 1973. Diabetes & Metabolism, 3, 245-256.
- Gispen, W.H. and Biessels, G.I. (2000) Cognition and synaptic plasticity in Diabetes Mellitus. Trends in Neurosciences, 23, 542-549. doi:10.1016/S0166-2236(00)01656-8
- Tomlinson, D.R. and Gardiner, N.J. (2008) Glucose neurotoxicity. Nature Reviews Neuroscience, 9, 36-45. doi:10.1038/nrn2294
- Ott, A., Stolk, R.P., Hofman, A., Van Harskamp, F., Grobbee, D.E. and Breteler, M.M. (1996) Association of diabetes mellitus and dementia: The Rotterdam study. Diabetologia, 39, 1392-1397. doi:10.1007/s001250050588
- Ott, A., Stolk, R.P., Van Harskamp, F., Pols, H.A., Hofman, A. and Breteler, M.M. (1999) Diabetes mellitus and the risk of dementia: The Rotterdam study. Neurology, 53, 1937-1942. doi:10.1212/WNL.53.9.1937
- Leibson, C.L., Rocca, W.A., Hanson, V.A., Cha, R., Kokmen, E., O’Brien, P.C. and Palumbo, P.J. (1997) Risk of dementia among persons with diabetes mellitus: A population-based cohort study. American Journal of Epidemiology, 145, 301-308. doi:10.1093/oxfordjournals.aje.a009106
- Fehm, H.L., Perras, B., Smolnik, R., Kern, W. and Born, J. (2000) Manipulating neuropeptidergic pathways in humans: A novel approach to neuropharmacology. European Journal of Pharmacology, 405, 43-54.
- Reger, M.A., Watson, G.S., Green, P.S., Wilkinson, C.W., Baker, L.D., Cholerton, B., Fishel, M.A., Plymate, S.R., Breitner, J.C., DeGroodt, W., Mehta, P. and Craft, S. (2008) Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology, 70, 440-448. doi:10.1212/01.WNL.0000265401.62434.36
- Schafer, M. and Erdo, S.L., (1991) Development of glutamate neurotoxicity in cortical cultures: Induction of vulnerability by insulin. Developmental Brain Research, 62, 293-296. doi:10.1016/0165-3806(91)90179-M
- González, C., Farías, G. and Maccioni, R.B. (1998) Modification of tau to an Alzheimer’s type protein interferes with its interaction with microtubules. Cell and Molecular Biology (Noisy-le-grand), 44, 1117-1127.
- Freude, S., Plum, L., Schnitker, J., Leeser, U., Udelhoven, M., Krone, W., Bruning, J.C. and Schubert, M. (2005) Peripheral hyperinsulinemia promotes tau phosphorrylation in vivo. Diabetes, 54, 3343-3348. doi:10.2337/diabetes.54.12.3343
- Qiu, W.Q., Ye, Z., Kholodenko, D., Seubert, P. and Selkoe, D.J. (1997) Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. Journal of Biological Chemistry, 272, 6641-6646. doi:10.1074/jbc.272.10.6641
- Qiu, W.Q., Walsh, D.M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M.B., Rosner, M.R., Safavi, A., Hersh, L.B. and Selkoe, D.J. (1998) Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. Journal of Biological Chemistry, 273, 32730- 32738. doi:10.1074/jbc.273.49.32730
- Gasparini, L., Gouras, G.K., Wang, R., Gross, R.S., Beal, M.F., Greengard, P. and Xu, H. (2001) Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogenactivated protein kinase signaling. Journal of Neuroscience, 21, 2561-2570.
- Perez, A., Morelli, L., Cresto, J.C. and Castano, E.M. (2000) Degradation of soluble amyloid beta peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochemical Research, 25, 247-255.
- Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E.A., Frosch, M.P., Eckman, C.B., Tanzi, R.E., Selkoe, D.J. and Guenette, S. (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid betaprotein, and the beta-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences of the United States of America, 100, 4162-4167. doi:10.1073/pnas.0230450100
- Mayeux, R., Honig, L.S., Tang, M.X., Manly, J., Stern, Y. Schupf, N. and Mehta, P.D. (2003) Plasma Aβ40 and Aβ42 and Alzheimer’s disease: Relation to age, mortality, and risk. Neurology, 61, 1185-1190. doi:10.1212/01.WNL.0000091890.32140.8F
- Cook, D.G., Leverenz, J.B., McMillan, P.J., Kulstad, J.J., Ericksen, S., Roth, R.A., Schellenberg, G.D., Jin, L.W., Kovacina, K.S. and Craft, S. (2003) Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. American Journal of Patholology, 162, 313-319. doi:10.1016/S0002-9440(10)63822-9
- Ho, L., Qin, W., Pompl, P.N., Xiang, Z., Wang, J., Zhao, Z., Peng, Y., Cambareri, G., Rocher, A., Mobbs, C.V., Hof, P.R. and Pasinetti, G.M. (2004) Diet-induced Insulin Resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. Federation of American Societies for Experimental Biology, 18, 902-904.
- Vanhanen, M., Koivisto, K., Kuusisto, J., Mykkänen, L., Helkala, E.L., Hänninen, T., Riekkinen Sr., P., Soininen, H. and Laakso, M. (1998) Cognitive function in an elderly population with persistent impaired glucose tolerance. Diabetes Care, 21, 398-402. doi:10.2337/diacare.21.3.398
- Luchsinger, J.A. Tang, M.X., Shea, S. and Mayeux, R. (2004) Hyperinsulinemia and risk of Alzheimer disease. Neurology, 63, 1187-1192. doi:10.1212/01.WNL.0000140292.04932.87
- Wallum, B.J. Taborsky Jr. G.I., Porte Jr., D., Figlewicz, D.P., Jacobson, L., Beard, J.C., Ward, W.K. and Dorsa, D. (1987) Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. Journal of Clinical Endocrinology and Metabolism, 64, 190-194. doi:10.1210/jcem-64-1-190
- Kaiyala, K.J., Prigeon, R.L., Kahn, S.E., Woods, S.C. and Schwartz, M.W. (2000) Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes, 49, 1525-1533.
- Strachan, M.W., Deary, I.J., Ewing, F.M. and Frier, B.M. (1997) Is type II diabetes associated with an increased risk of cognitive dysfunction? A critical review of published studies. Diabetes Care, 20, 438-445. doi:10.2337/diacare.20.3.438
- Caballero, A.E. (2004) Endothelial dysfunction, inflammation, and insulin resistance: A focus on subjects at risk for type II diabetes. Current Diabetes Reports, 4, 237-246. doi:10.1007/s11892-004-0074-9
- Rojo, L.E., Fernández, J.A., Maccioni, A.A., Jimenez, J.M. and Maccioni, R.B. (2008) Neuroinflammation: Implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Archives of Medical Research, 39, 1-16. doi:10.1016/j.arcmed.2007.10.001
- Aggarwal, S., Gollapudi, S. and Gupta, S. (1999) Increased TNF-α-induced apoptosis in lymphocytes from aged humans: Changes in TNF-α receptor expression and activation of caspases. Journal of Immunology, 162, 2154-2161.
- Dzienis-Straczkowska, S., Straczkowski, M., Szelachowska, M., Stepien, A., Kowalska, I. and Kinalska, I. (2003) Soluble tumor necrosis factor-alpha receptors in young obese subjects with normal and impaired glucose tolerance. Diabetes Care, 26, 875-880. doi:10.2337/diacare.26.3.875
- Zhao, M., Cribbs, D.H., Anderson, A.J., Cummings, B.J., Su, J.H., Wasserman, A.J. and Cotman, C.W. (2003) The induction of the TNF-α death domain signaling pathway in Alzheimer’s disease brain. Neurochemical Research, 28, 307-318. doi:10.1023/A:1022337519035
- Jagust, W.J., Seab, J.P., Huesman, R.H., Valk, P.E., Mathis, C.A., Reed, B.R., Coxson, P.G. and Budinger, T.F. (1991) Diminished glucose transport in Alzheimer’s disease: Dynamic PET studies. Journal of Cerebral Blood Flow and Metabolism, 11, 323-330. doi:10.1038/jcbfm.1991.65
- Gibson, G.E. Petersen, C. and Jenden, D.J. (1981) Brain acetylcholine synthesis decline with senescence. Science, 213, 674-676. doi:10.1126/science.7256270
- Bigl, V., Arendt, T., Fischer, S., Werner, M. and Arendt, A. (1987) The cholinergic system in aging. Gerontology, 33, 172-180.
- Prapong, T., Buss, J., Hsu, W.H., Heine, P., West Greenlee, H. and Uemura, E. (2002) Amyloid beta-peptide decreases neuronal glucose uptake despite causing increase in GLUT3 mRNA transcription and GLUT3 translocation to the plasma membrane. Experimental Neurology, 174, 253-258. doi:10.1006/exnr.2001.7861
- Zhu, X., Perry, G. and Smith, M.A. (2005) Insulin signaling, Diabetes Mellitus and risk of Alzheimer disease. Journal of Alzheimer’s Disease, 7, 81-84.
- Fernández, J., Rojo, L., Kuljiš, R.O. and Maccioni, R.B. (2008) The damage signals hypothesis of Alzheimer’s disease. Journal of Alzheimer’s Disease, 14, 329-333.
- Moreira, P.I. Santos, M.S. Seica, R. and Oliveira, C.R. (2007) Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. Journal of Neurological Sciences, 257, 206-214.
- Xie, L., Helmerhorst, E., Taddei, K., Plewright, B., Van Bronswijk, W. and Martins, R. (2002) Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. Journal of Neuroscience, 22, Article ID: RC221.
- Steen, E., Terry, B.M., Rivera, E.J., Cannon, J.L., Neely, T.R., Tavares, R., Xu, X.J., Wands, J.R. and de la Monte, S.M. (2005) Impaired insulin and insulin-like growth factor expression and signalling mechanisms in Alzheimer’s disease: Is this type 3 diabetes? Journal of Alzheimer’s Disease, 7, 63-80.
- Rivera, E.J., Goldin, A., Fulmer, N., Tavares, R., Wands J.R. and de la Monte, S.M. (2005) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. Journal of Alzheimer’s Disease, 8, 247-268.
- Steven, E.S., Jongsoon, L. and Allison, B.G. (2006) Inflammation and Insulin Resistance. Journal of Clinical Investigation, 116, 1793-1801. doi:10.1172/JCI29069
- Röder, H.M. and Ingram, V.M. (1991) Two novel kinases phosphorylate tau and the KSP site of heavy neurofilament subunits in high stoichiometric ratios. Journal of Neuroscience, 11, 3325-3343.
- Bush, M.L., Miyashiro, J.S. and Ingram, V.M. (1995) Activation of a neurofilament kinase, a tau kinase, and a tau phosphatase by decreased ATP levels in nerve growth factor-differentiated PC12 cells. Proceedings of the National Academy of Sciences of the United States of America, 92, 1861-1865. doi:10.1073/pnas.92.6.1861
- Hong, M. and Lee, V.M. (1997) Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. Journal of Biological Chemistry, 272, 19547-19553. doi:10.1074/jbc.272.31.19547
- Rojo, L., Sjöberg, M.K., Hernández, P., Zambrano, C. and Maccioni, R.B. (2006) Roles of cholesterol and lipids in the etiopathogenesis of Alzheimer’s disease. Journal of Biomedicine & Biotechnology, 2006, Article ID: 73976.
- Gasparini, L., Netzer, W.J. Greengard, P. and Xu, H. (2002) Does insulin dysfunction play a role in Alzheimer’s disease? Trends in Pharmacological Sciences, 23, 288-293. doi:10.1016/S0165-6147(02)02037-0
- Cross, D.A., Alessi, D.R., Cohen, P., Andjelkovich, M. and Hemmings, B.A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785-789. doi:10.1038/378785a0
- Fisher, T.L. and White, M.F. (2004) Signaling pathways: The benefits of good communication. Current Biology, 14, R1005-R1007. doi:10.1016/j.cub.2004.11.024
- Dudek, H., Datta, S.R., Franke, T.F., Birnbaum, M.J., Yao, R., Cooper, G.M., Segal, R.A., Kaplan, D.R. and Greenberg, M.E. (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science, 275, 661-665. doi:10.1126/science.275.5300.661
- Zhou, H., Li, X.M., Meinkoth, J. and Pittman, R.N. (2000) Akt regulates cell survival and apoptosis at a postmitochondrial level. Journal of Cell Biology, 151, 483-494. doi:10.1083/jcb.151.3.483
- Zhou, H., Summers, S.A., Birnbaum, M.J. and Pittman, R.N. (1998) Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. Journal of Cell Biology, 273, 16568-16575.
- Martin, D., Salinas, M., Lopez-Valdaliso, R., Serrano, E., Recuero, M. and Cuadrado, A. (2001) Effect of the Alzheimer amyloid fragment Aβ (25-35) on Akt/PKB kinase and survival of PC12 cells. Journal of Neurochemistry, 78, 1000-1008. doi:10.1046/j.1471-4159.2001.00472.x
- Maccioni, R.B., Otth, C., Concha, I.I. and Muñoz, J.P. (2001) The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer’s pathology. European Journal of Biochemistry, 268, 1518-1527. doi:10.1046/j.1432-1327.2001.02024.x
- Alvarez, A., Muñoz, J.P. and Maccioni, R.B. (2001) A Cdk5-p35 Stable Complex is involved in the β-Amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Experimental Cell Research, 264, 266-274. doi:10.1006/excr.2001.5152
- Alvarez, A., Toro, R., Caceres, A. and Maccioni, R.B. (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid induced neuronal death. Federation of European Biochemical Societies Letters, 459, 421-426. doi:10.1016/S0014-5793(99)01279-X
- Shiurba, R.A., Ishiguro, K., Takahashi, M., Sato, K., Spooner, E.T., Mercken, M., Yoshida, R., Wheelock, T.R., Yanagawa, H., Imahori, K. and Nixon, R.A. (1996) Immunocytochemistry of tau phosphoserine 413 and tau protein kinase I in Alzheimer pathology. Brain Research, 737, 119-132.
- Leroy, K., Boutajangout, A., Authelet, M., Woodgett, J.R., Anderton, B.H. and Brion, J.P. (2002) The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer’s disease. Acta Neuropathologica, 103, 91-99. doi:10.1007/s004010100435
- Ryu, B.R., Ko, H.W., Jou, I., Noh, J.S. and Gwag, B.J. (1999) Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. Journal of Neurobiology, 39, 536-546. doi:10.1002/(SICI)1097-4695(19990615)39:4<536::AID-NEU7>3.0.CO;2-J
- Hebert, L.E., Scherr, P.A., Bienias, J.L., Bennett, D.A. and Evans, D.A. (2003) Alzheimer disease in the US population: Prevalence estimates using the 2000 Census. Archives of Neurology, 60, 1119-1122. doi:10.1001/archneur.60.8.1119
- Jorm, A.F. (1991) Cross-national comparisons of the occurrence of Alzheimer’s and vascular dementias. European Archives of Psychiatry and Clinical Neuroscience, 240, 218-222.
- Walsh, D.M. and Selkoe, D.J. (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron, 44, 181-193. doi:10.1016/j.neuron.2004.09.010
- Tan, J., Town, T., Paris, D., Mori, T., Suo, Z.M., Crawford, F., Mattson, M.P., Flavell, R.A. and Mullan, M. (1999) Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science, 286, 2352-2355. doi:10.1126/science.286.5448.2352
- American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders. 4th Edition, American Psychiatric Association, Washington DC.
- Kawas, C.H. (2003) Clinical practice. Early Alzheimer’s disease. New England Journal of Medicine, 349, 1056- 1063. doi:10.1056/NEJMcp022295
- McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D. and Stadlan, E.M. (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology, 34, 939-944. doi:10.1212/WNL.34.7.939
- Chui, H. and Lee, A.E. (2002) Clinical criteria for dementia subtypes. Evidence-based dementia practice, Blackwell Science, Oxford.
- Greene, J.D., Baddeley, A.D. and Hodges, J.R. (1996) Analysis of the episodic memory deficit in early Alzheimer’s disease: Evidence from the doors and people test. Neuropsychologia, 34, 537-551. doi:10.1016/0028-3932(95)00151-4
- Pillon, B., Deweer, B., Agid, Y. and Dubois, B. (1993) Explicit memory in Alzheimer’s, Huntington’s, and Parkinson’s diseases. Archives of Neurology, 50, 374-379. doi:10.1001/archneur.1993.00540040036010
- Price, B.H., Gurvit, H., Weintraub, S., Geula, C., Leimkuhler, E. and Mesulam, M. (1993) Neuropsychological patterns and language deficits in 20 consecutive cases of autopsy-confirmed Alzheimer’s disease. Archives of Neurology, 50, 931-937. doi:10.1001/archneur.1993.00540090038008
- Esteban-Santillan, C. Praditsuwan, R., Ueda, H. and Geldmacher, D.S. (1998) Clock drawing test in very mild Alzheimer’s disease. Journal of American Geriatrics Society, 46, 1266-1269.
- Kirk, A. and Kertesz, A. (1991) On drawing impairment in Alzheimer’s disease. Archives of Neurology, Vol. 48, 73-77. doi:10.1001/archneur.1991.00530130083024
- Galasko, D., Bennett, D., Sano, M., Ernesto, C., Thomas, R., Grundman, M. and Ferris, S. (1997) An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s disease Cooperative Study. Alzheimer’s disease and Associated Disorders, 11, S33-S39.
- Mega, M.S., Cummings, J.L., Fiorello, T. and Gornbein, J. (1996) The spectrum of behavioral changes in Alzheimer’s disease. Neurology, 46, 130-135. doi:10.1212/WNL.46.1.130
- Knopman, D.S., DeKosky, S.T., Cummings, J.L., Chui, H., Corey-Bloom, J., Relkin, N., Small, G.W., Miller, B. and Stevens, J.C. (2001) Practice parameter: Diagnosis of dementia (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology, 56, 1143-1153. doi:10.1212/WNL.56.9.1143
- Silverman, D.H., Small, G.W., Chang, C.Y., Lu, C.S., Kung de Aburto, M.A., Chen, W., Czernin, J., Rapoport, S.I., Pietrini, P., Alexander, G.E., Schapiro, M.B., Jagust, W.J., Hoffman, J.M., Welsh-Bohmer, K.A., Alavi, A., Clark, C.M., Salmon, E., de Leon, M.J., Mielke, R., Cummings, J.L., Kowell, A.P., Gambhir, S.S., Hoh, C.K. and Phelps, M.E. (2001) Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. Journal of American Medical Association, 286, 2120-2127. doi:10.1001/jama.286.17.2120
- Cummings, J.L. (2004) Alzheimer’s disease. New England Journal of Medicine, 351, 56-67. doi:10.1056/NEJMra040223
- Hardy, J. and Selkoe, D.J. (2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science, 297, 353-356. doi:10.1126/science.1072994
- Butterfield, D.A., Castegna, A., Lauderback, C.M. and Drake, J. (2002) Evidence that amyloid beta peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiology of Aging, 23, 655-664. doi:10.1016/S0197-4580(01)00340-2
- Carter, D.B. Dunn, E., McKinley, D.D., Stratman, N.C., Boyle, T.P. Kuiper, S.L. Oostveen, J.A. Weaver, R.J. Boller, J.A. and Gurney, M.E. (2001) Human apolipoprotein E4 accelerates beta amyloid deposition in APPsw transgenic mouse brain. Annals of Neurology, 50, 468- 475. doi:10.1002/ana.1134
- Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., Yang, F. and Cole, G., (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science, 274, 99-102. doi:10.1126/science.274.5284.99
- Mesulam, M.M. (1999) Neuroplasticity failure in Alzheimer’s disease: Bridging the gap between plaques and tangles. Neuron, 24, 521-529. doi:10.1016/S0896-6273(00)81109-5
- Hock, C., Konietzko, U., Streffer, J.R., Tracy, J., Signorell, A., Müller-Tillmanns, B., Lemke, U., Henke, K., Moritz, E., Garcia, E., Wollmer, M.A., Umbricht, D., de Quervain, D.J., Hofmann, M., Maddalena, A., Papassotiropoulos, A. and Nitsch, R.M. (2003) Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron, 38, 547-554. doi:10.1016/S0896-6273(03)00294-0
- Pappas, B.A., Bayley, P.J., Bui, B.K., Hansen, L.A. and Thal, L.J. (2000) Choline acetyltransferase activity and cognitive domain scores of Alzheimer’s patients. Neurobiology of Aging, 21, 11-17. doi:10.1016/S0197-4580(00)00090-7
- Palmer, A.M., Stratmann, G.C., Procter, A.W. and Bowen, D.M. (1988) Possible neurotransmitter basis of behavioral changes in Alzheimer’s disease. Annals of Neurology, 23, 616-620. doi:10.1002/ana.410230616
- Aisen, P.S. (1997) Inflammation and Alzheimer’s disease: Mechanisms and therapeutic strategies. Gerontology, 43, 143-149.
- Perlmutter, L.S. Barron, E. and Chui, H.C. (1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neuroscience Letters, 119, 32-36. doi:10.1016/0304-3940(90)90748-X
- McGeer, P.L., Kawamata, T., Walker, D.G., Akiyama, H., Tooyama, I. and McGeer, E.G. (1993) Microglia in degenerative neurological disease. Glia, 7, 84-92. doi:10.1002/glia.440070114
- Fetler, L. and Amigorena, S. (2005) Neuroscience. Brain under surveillance: The microglia patrol. Science, 309, 392-393. doi:10.1126/science.1114852
- Liu, B. and Hong, J.S. (2003) Role of Microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. Journal of Pharmacology and Experimental Therapeutics, 304, 1-7. doi:10.1124/jpet.102.035048
- Kreutzberg, G.W. (1996) Microglia: A sensor for pathological events in the CNS. Trends in Neurosciences, 19, 312-318. doi:10.1016/0166-2236(96)10049-7
- Griffin, W.S., Sheng, J.G., Royston, M.C., Gentleman, S.M., McKenzie, J.E., Graham, D.I., Roberts, G.W. and Mrak, R.E. (1998) Glial-neuronal interactions in Alzheimer’s disease: The potential role of a “cytokine cycle” in disease progression. Brain Pathology, 8, 65-72. doi:10.1111/j.1750-3639.1998.tb00136.x
- D’Andrea, M.R., Cole, G.M. and Ard, M.D. (2004) The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiology of Aging, 25, 675-683. doi:10.1016/j.neurobiolaging.2003.12.026
- Dickson, D.W., Lee, S.C., Mattiace, L.A., Yen, S.H. and Brosnan, C. (1993) Microglia and cytokines in neurological disease, with special reference to AIDs and Alzheimer’s disease. Glia, 7, 75-83. doi:10.1002/glia.440070113
- Barger, S.W. and Harmon, A.D. (1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature, 388, 878-881. doi:10.1038/42257
- DeGiorgio, L.A., Shimizu, Y., Chun, H.S., Kim, Y.S., Sugama, S., Son, J.H., Joh, T.H. and Volpe, B.T. (2002) Amyloid precursor protein gene disruption attenuates degeneration of substantia nigra compacta neurons following axotomy. Brain Research, 938, 38-44.
- Permanne, B., Adessi, C., Saborio, G.P., Fraga, S., Frossard, M.J., Van Dorpe, J., Dewachter, I., Banks, W.A., Van Leuven, F. and Soto, C. (2002) Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer’s disease by treatment with a β-sheet breaker peptide. Federation of American Societies for Experimental Biology, 16, 860-862.
- Combs, C.K., Karlo, J.C., Kao, S.C. and Landreth, G.E. (2001) β-amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. Journal of Neuroscience, 21, 1179-1188.
- Ho, G.J., Drego, R., Hakimian, E. and Masliah, E. (2005) Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Current Drug Targets-Inflammation and Allergy, 4, 247-256. doi:10.2174/1568010053586237
- Bach, J.H., Chae, H.S., Rah, J.C., Lee, M.W., Park, C.H., Choi, S.H., Choi, J.K., Lee, S.H., Kim, Y.S., Kim, K.Y., Lee, W.B., Suh, Y.H. and Kim, S.S. (2001) C-terminal fragment of amyloid precursor protein induces astrocytosis. Journal of Neurochemistry, 78, 109-120. doi:10.1046/j.1471-4159.2001.00370.x
- Beglopoulos, V., Sun, X., Saura, C.A., Lemere, C.A., Kim, R.D. and Shen, J. (2004) Reduced β-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. Journal of Biological Chemistry, 279, 46907-46914. doi:10.1074/jbc.M409544200
- Brunkan, A.L. and Goate, A.M. (2005) Presenilin function and g-secretase activity. Journal of Neurochemistry, 93, 769-792. doi:10.1111/j.1471-4159.2005.03099.x
- Frautschy, S.A., Yang, F., Irrizarry, M., Hyman, B., Saido, T.C., Hsiao, K. and Cole, G.M. (1998) Microglial response to amyloid plaques in APPsw transgenic mice. American Journal of Pathology, 152, 307-317.
- Wyss-Coray, T., Loike, J.D., Brionne, T.C., Lu, E., Anankov, R., Yan, F., Silverstein, S.C. and Husemann, J. (2003) Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nature Medicine, 9, 453-457. doi:10.1038/nm838
- Rossner, S., Lange-Dohna, C., Zeitschel, U. and PerezPolo, J.R. (2005) Alzheimer’s disease β-secretase BACE1 is not a neuron-specific enzyme. Journal of Neurochemistry, 92, 226-234. doi:10.1111/j.1471-4159.2004.02857.x
- Nagele, R.G., D’Andrea, M.R., Lee, H., Venkataraman, V. and Wang, H.Y. (2003) Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Research, 971, 197-209. doi:10.1016/S0006-8993(03)02361-8
- Heneka, M.T., Wiesinger, H., Dumitrescu-Ozimek, L., Riederer, P., Feinstein, D.L. and Klockgether, T. (2001) Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 60, 906-916.
- Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E.K., Jones, P.K., Ghanbari, H., Wataya, T., Shimohama, S., Chiba, S., Atwood, C.S., Petersen, R.B. and Smith, M.A. (2001) Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 60, 759-767.
- Cagnin, A., Brooks, D.J., Kennedy, A.M., Gunn, R.N., Myers, R., Turkheimer, F.E., Jones, T. and Banati, R.B. (2001) In-vivo measurement of activated microglia in dementia. Lancet, 358, 461-567. doi:10.1016/S0140-6736(01)05625-2
- Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G.M., Cooper, N.R., Eikelenboom, P., Emmerling, M., Fiebich, B.L., Finch, C.E., Frautschy, S., Griffin, W.S., Hampel, H., Hull, M., Landreth, G., Lue, L., Mrak, R., Mackenzie, I.R., McGeer, P.L., O’Banion, M.K., Pachter, J., Pasinetti, G., Plata-Salaman, C., Rogers, J., Rydel, R., Shen, Y., Streit, W., Strohmeyer, R., Tooyoma, I., Van Muiswinkel, F.L., Veerhuis, R., Walker, D., Webster, S., Wegrzyniak, B., Wenk, G. and Wyss-Coray, A. (2000) Inflammation and Alzheimer’s disease. Neurobiology of Aging, 21, 383-421. doi:10.1016/S0197-4580(00)00124-X
- Nicoll, J.A., Mrak, R.E., Graham, D.I., Steward, J., Wilcock, G., MacGowan, S., Esiri, M.M., Murray, L.S., Dewar, D., Love, S., Moss, T. and Griffin, W.S. (2000) Association of interleukin-1 gene polymorphisms with Alzheimer’ disease. Annals of Neurology, 47, 365-368. doi:10.1002/1531-8249(200003)47:3<365::AID-ANA13>3.0.CO;2-G
- Papassotiropoulos, A., Bagli, M., Jessen, F., Bayer, T.A., Maier, W., Rao, M.L. and Heun, R. (1999) Genetic variation of the inflammatory cytokine IL-6 delays the initial onset, and reduces the risk for sporadic Alzheimer’s disease. Annals of Neurology, 45, 666-668. doi:10.1002/1531-8249(199905)45:5<666::AID-ANA18>3.0.CO;2-3
- McCusker, S.M., Curran, M.D., Dynan, K.B., McCullagh, C.D., Urquhart, D.D., Middleton, D., Patterson, C.C., Mcllroy, S.P. and Passmore, A.P. (2001) Association between polymorphism in regulatory region of gene encoding TNF-α and risk of Alzheimer disease and vascular dementia: A case-control study. Lancet, 357, 436-439. doi:10.1016/S0140-6736(00)04008-3
- Perry, R.T., Collins, J.S., Wiener, H., Acton, R. and Go, R.C. (2001) The role of TNF and its receptors in Alzheimer’s disease. Neurobiology of Aging, 22, 873-883. doi:10.1016/S0197-4580(01)00291-3
- Kamboh, M.I., Sanghera, D.K., Ferrell, R.E. and DeKosky, S.T. (1995) APOE*4-associated Alzheimer’s disease risk is modified by alpha 1-antichymotrypsin polymorphism. Nature Genetics, 10, 486-488. doi:10.1038/ng0895-486
- Benzing, W.C., Wujek, J.R., Ward, E.K., Shaffer, D., Ashe, K.H., Younkin, S.G. and Brunden, K.R. (1999) Evidence for glial-mediated inflammation in aged APPSW transgenic mice. Neurobiology of Aging, 20, 581-589. doi:10.1016/S0197-4580(99)00065-2
- Sly, L.M., Krzesicki, R.F., Brashler, J.R., Buhl, A.E., McKinley, D.D., Carter, D.B. and Chin, J.E. (2001) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Research Bulletin, 56, 581-588. doi:10.1016/S0361-9230(01)00730-4
- Apelt J. and Schliebs, R. (2001) β-amyloid-induced glial expression of both proand anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Research, 894, 21-30. doi:10.1016/S0006-8993(00)03176-0
- Matsuoka, Y., Picciano, M., Malester, B., LaFrancois, J., Zehr, C., Daeschner, J.M., Olschowka, J.A., Fonseca, M.I., O’Banion, M.K., Tenner, A.J., Lemer, C.A. and Duff, K. (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. American Journal of Pathology, 158, 1345-1354. doi:10.1016/S0002-9440(10)64085-0
- Bellucci, A., Westwood, A.J., Ingram, E., Casamenti, F. Goedert, M. and Spillantini, M.G. (2004) Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. American Journal of Pathology, 165, 1643-1652.
- Sastre, M., Klockgether, T. and Heneka, M.T. (2006) Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. International Journal of Developmental Neuroscience, 24, 167-176. doi:10.1016/S0002-9440(10)63421-9
NOTES
*Corresponding author.