Journal of Cancer Therapy
Vol.4 No.7(2013), Article ID:36129,11 pages DOI:10.4236/jct.2013.47138
The MMTV-PyVT Transgenic Mouse as a Multistage Model for Mammary Carcinoma and the Efficacy of Antineoplastic Treatment
![]()
Department of Diagnostic Medicine and Pathobiology, Kansas State University, Manhattan, USA.
Email: tnguyen@vet.k-state.edu
Copyright © 2013 Stephanie Shishido et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 24th, 2013; revised July 25th, 2013; accepted August 3rd, 2013
Keywords: ER; PR; p53; Ki67; MMTV-PyVT; survivin; Tamoxifen
ABSTRACT
Animal models are commonly used to analyze the mechanism of carcinogenesis as well as the development and screening of potent drugs. Although numerous animal models of breast cancer have been used in research, few display multiple stages of tumorigenesis. The transgenic strain FVB/N-Tg(MMTV-PyVT) 634 Mul/J (also known as PyVT) was established to determine the effect of mammary gland-specific expression of the polyomavirus middle T antigen. Here the PyVT model with three distinct stages of tumor development (Pre, Early, and Late stages) was used as a model system for measuring tumor growth, tumor burden, and metastasis of mammary carcinomas. Additionally the expression profile of the molecular markers, survivin and Ki-67, was determined. Three antineoplastic compounds were testes over a 14- day period to determine their efficacy at attenuating tumor growth at each stage of development. Interestingly cisplatin and paclitaxel were determined to be ineffective anticancer drugs, while tamoxifen significantly reduced tumor growth in the Pre and Early stages of tumor formation.
1. Introduction
In vivo models are important for translational research and may be used to explore the mechanism of tumorigenesis, or the pharmacodynamics and the development of therapeutics for cancer. Animal models to study cancer formation should include features such as molecular targets, drug metabolism, pharmacokinetics/dynamics, drug distribution, anatomic similarities, microenvironment, angiogenesis, and metastasis. Change in tumor size is the most widely used end point for drug efficacy, but tumor frequency, survival, and tumor burden are other important factors. Thus, specific model systems have advantages for measuring tumor burden, drug sensitivity, and metastatic potential. The antitumor activity of chemotherapeutics is commonly determined in immune deficient mice transplanted with human tumors, but these xenograft tumors have the potential loss of tumorigenicity, limited metastatic potential of many cell lines, and loss of linearity with increasing tumor volume [1].
Animal models have evolved from the transplantable syngeneic mouse tumor models to chemically induced, transgenic, and spontaneous animal tumor models. Numerous chemotherapeutic agents have shown promising results in preclinical models and yet had minimal activity in clinical settings. This has led to skepticism about xenograft and syngeneic tumor models. Newer techniques, including transgenic mouse models, have the potential to be more predictive. Tumorigenesis is a multistep process and all stages of development need to be considered in the design of more effective therapies. Since several transgenic mammary models of human breast cancer progress through well-defined cancer stages, they are useful preclinical systems to test the efficacy of chemopreventive and chemotherapeutic agents. The use of transgenic mammary carcinoma models allows for detailed study of stage-specific responses to antineoplastic agents, defining the appropriate timing for intervention with specific compounds.
The transgenic strain FVB/N-Tg(MMTV-PyVT) 634 Mul/J (also known as PyVT) is a novel in vivo model for the study of mammary carcinoma formation and metastasis with important clinical utility. The PyVT model carries the Polyoma Virus middle T antigen with the mouse mammary tumor virus (MMTV) promoter that drives mammary tissue-specific expression [2] . The PyVT oncogene activates multiple oncogenic pathways, such as src and phosphatidylinositol-3-kinase [3] , leading to an aggressive tumor phenotype. Previous studies indicate that virgin females carrying the transgene develop multi-focal, poorly differentiated, highly invasive ductal carcinoma by 10 - 12 weeks of age, with a high incidence of lung metastases stemming from the primary mammary tumor [4] . At 5 weeks of age females develop noninvasive focal lesions which are classified into four groups: simple, solid, cystic, and mixed (solid and cystic) [5] . Solid lesions consist of large foci with a dense mass of atypical cells in nodular sheets. Cystic lesions vary in size and complexity, and are lined by multilayered epithelium with significant amounts of clear fluid [5] . The premalignant tumors are morphologically heterogeneous and highly proliferative neoplastic cells with atypical nuclei that contain abnormal microvasculature, and stay within the basement membrane ADDIN EN.CITE ADDIN EN.CITE.DATA [5] .
The MMTV-PyVT expression is variable in tumors [5] , which suggests that the transgene is not necessary for the maintenance of the malignancy. The PyVT model represents a multistep process due to lesions progressing from hyperplasia to an adenoma/mammary intraepithelial neoplasia mixed phenotype, followed by early and late carcinoma with pulmonary metastasis [4,5] . Metastasis of the primary tumor to distant sites remains a significant cause of death in many cancer types, highlighting the importance for a metastatic model. This model of spontaneous mammary carcinogenesis is a powerful tool for studying the mechanism associated with tumor progression and development of novel chemotherapeutics.
This model has not been used to test the efficacy of many antineoplastic compounds despite its clinical relevance to human breast cancer. The compounds utilized here are cisplatin, paclitaxel, and tamoxifen. The mechanism of action is briefly explained. Cisplatin induces damage to tumors via formation of DNA adducts followed by cell growth inhibition and induction of apoptosis. Paclitaxel prevents cellular proliferation by binding to tubulin and inhibiting disassembly of the microtubules in the cell. Tamoxifen is a selective estrogen receptor modulator (SERM) that competitively inhibits the binding of estradiol to the estrogen receptors. The effects of these compounds on the MMTV-PyVT transgene-induced mouse have not yet been reported.
The present study reviews the characteristics of tumor development and determines the effects of antineoplastic treatment on tumorigenesis and metastasis using the MMTV-PyVT transgene-induced mammary tumor model. Interestingly with the progression of malignancy there is an increase in expression levels of Ki-67 and survivin, with a decrease in estrogen receptor (ER) and progesterone receptor (PR) expression. This supports previous work done on this model. We showed for the first time the anticancer activity of tamoxifen, not cisplatin or paclitaxel, against a multistage mammary tumor model.
2. Materials and Methods
2.1. Animals
A colony of FVB-TgN(MMTV-PyVT) transgenic mice (The Jackson Laboratory; Bar Harbor, ME) was established. To identify transgenic progeny, genomic DNA was extracted from a 1.5-cm tail clipping. All mice carrying the PyVT transgene developed mammary tumors. Tumor development of positive female mice was closely monitored every 2 - 3 days. Tumor onset was recorded as the age of the animal at which palpable abnormal masses were detected. Tumor size was measured in two dimensions with calipers every 2 days. Mice at each stage of tumor development were randomly divided into four experimental groups: 1) control animals given the vehicle solvent (DMSO); 2) animals treated with 3.5 mg/kg cisplatin; 3) animals treated with 10 mg/kg paclitaxel; and 4) animals treated with 20 mg/kg tamoxifen. All treatments were administered as an intraperitoneal (IP) injection every other day for 14 days. Animal care and use protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Kansas State University, Manhattan, Kansas following NIH guidelines.
2.2. Compounds
cis-Diammineplatinum(II) dichloride (P4394), paclitaxel (T1912), and tamoxifen (T5648) were purchased from Sigma Aldrich (St. Louis, MO, USA).
2.3. Antibodies
Primary antibodies: Anti-ERα (sc-8002, mouse monoclonal), anti-ERβ (sc-8974, rabbit polyclonal), anti-PR (sc-166170, mouse monoclonal), anti-survivin (sc-374616, mouse monoclonal), and anti-Ki67 (sc-23900, mouse monoclonal), from Santa Cruz Biotechnology (Santa Cruz, CA); antiGAPDH (2118, rabbit monoclonal) from Cell Signaling (Boston, MA); anti-pan epithelial (MAB1631) and antipigment epithelium (MAB1059) from Chemicon; and anti-Her2 (AP7629e, rabbit polyclonal) and anti-p53 (AP 6266b, rabbit polyclonal) from ABGENT (San Diego, CA) were used for both western blot and immunohistochemistry (IHC).
2.4. Western Blot Analysis
Mammary gland tumor tissue was homogenized in 500 mL of lysis buffer (20 mM Tris pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 0.5% Triton X-100) with protease inhibitors at 1:1000 dilution (Sigma-Aldrich, Saint Louis, MO). Tissue was homogenized via the OMNI Bead Ruptor 24 at a speed of 5.65 m/s for 45 seconds, followed by centrifugation at 13,000 rpm for 30 minutes at 4˚C. Twenty-five μg of whole-cell extract was resolved by 10% SDS polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membrane (Midwest Scientific, Saint Louis, MO). Nitrocellulose membrane was blocked in 5% milk for an hour at room temperature and then incubated with monoclonal antibodies (1:1000). Western blots were detected by enhanced chemiluminescence detection reagents (Pierce, Rockford, IL) and visualized by Fluorchem E imaging system (ProteinSimple, Santa Clara, CA).
2.5. Immunohistochemistry (IHC)
Mammary carcinomas and organs were removed and fixed in a solution of 10% formaldehyde and embedded into paraffin prior to sectioning them onto slides at a 5 µm thickness. Paraffin sections (5 µm) were dried at 60˚C for 25 minutes. Deparaffinization was performed with 100% xylene and 100%, 90%, 75%, 50% ethanol. Antigen retrieval was performed in 1× citrate buffer solution and steam. Slides were then incubated overnight at room temperature with a polyclonal antibody (1:50 dilution). After washes in PBS, slides were successively incubated with biotinylated secondary antibodies (1:1000) for 15 minutes. Slides were washed and immunostains were amplified by incubation with Avidin Biotin Complex (ABC) for 10 minutes accordingly. Cells were visualized with 3,3-diaminobenzidine (DAB) followed by a hematoxylin counterstain. The sections were viewed and the images captured with a Nikon 80i microscope under 40× and 60× magnification.
2.6. Statistical Analysis
Significance was determined at a p-value ≤ 0.05 using a standard t-test. Data is presented as the mean ± the 95% confidence interval of a minimum of six samples per treatment group.
3. Results
FVB-TgN(MMTV-PyVT) female transgenic mice developed tumors as early as 4 weeks of age. All 10 mammary pads developed tumors with the maximum tumor burden achieved around 12 weeks of age. Tumor development was divided into 3 stages based on the extent of tumor size and the frequency of tumor formation. The Pre stage of tumor development began at 4 - 5 weeks of age, consisting of a pre-cancerous condition where no tumors were palpated and the mammary tissue appeared normal on gross observation. The Early stage of development was confined to 6 - 8 weeks of age, represented by the gross observation of 1 - 2 solid tumors within the mammary tissue. The Late stage was based on the presence of all 10 primary mammary tumors with secondary lung metastasis, which appeared after 10 weeks of age. Representative sections of the lung tissue were stained with hematoxylin and eosin (H&E) for histopathological review to determine the presence of metastases.
To confirm the PyVT tumors were of epithelial origin, immunohistochemistry was conducted at each stage of development with pan epithelium, pigment epitheliumderived factor, and E-cadherin (Figure 1). There was a strong positive staining for pan epithelium in the Pre and Early stages, while the Late stage showed weak staining. This was consistent for the pigment epithelium-derived factor (PEDF) and E-cadherin. This is indicative of a transition in cellular phenotype, demonstrating the process of epithelial-mesenchymal transition (EMT) in this model.
Breast cancers are routinely assessed for hormone receptor status [estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2)] due to their relation to different subtypes and impact on prognosis, treatment, and overall survival. PyVT tumors isolated from all stages of development were shown to be ERand HER2-positive (Figure 2). Only the Early and Late stages tumors expressed detectable levels of PR. This was confirmed with Western blot analysis (data not shown). Estrogen receptor (ER), progesterone receptor (PR), and expression of human epidermal growth factor receptor 2 (HER-2) are recognized prognostic and predictive factors. ER found in 50% - 80% of breast cancers [6]. PR is a surrogate marker of a functional ER and is expressed in 60% - 70% invasive breast carcinomas with a higher positivity in older age and postmenopausal women [7]. HER-2/neu also known as C-erb B2 (HER-2), is a protooncogene located on chromosome 17 and the protein (HER-2) is overexpressed in 15% - 25% of invasive breast carcinoma with associated poor prognosis [8].
Tumors were shown to express p53 (Figure 2). P53 is an additional prognostic marker for inflammatory breast cancer, a more aggressive form than locally advanced breast cancer [9]. It is a sensor of cellular stress and master regulator of apoptotic programming [10]. The role of p53 protein is to maintain genomic stability as a multifunctional transcriptional regulator participating in the cell cycle [11]. In cells with active p53, it functions as a survival gene, and its loss sensitizes the cell to genotoxic stress [12]. Elevated levels of p53 expression have been associated with poor prognoses [13].
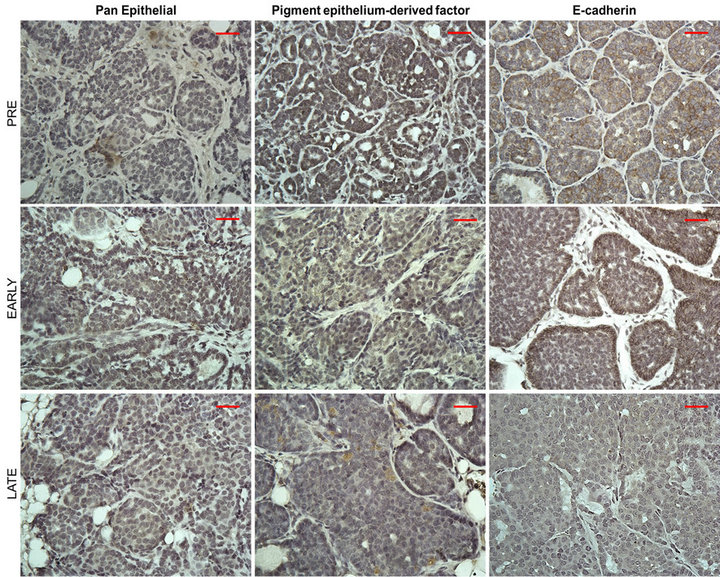
Figure 1. Immunohistochemistry of tumor phenotype from PyVT females during tumor development. Paraffin-embedded sections stained with antibodies against pan epithelial, pigment epithelium-derived factor, and E-cadherin at Pre, Early, and Late stages. Proteins staining: brown, counterstaining: blue (hematoxylin). Images represent only 1 of n = 3 per group at a 40× magnification. Scale bar = 50 µm.
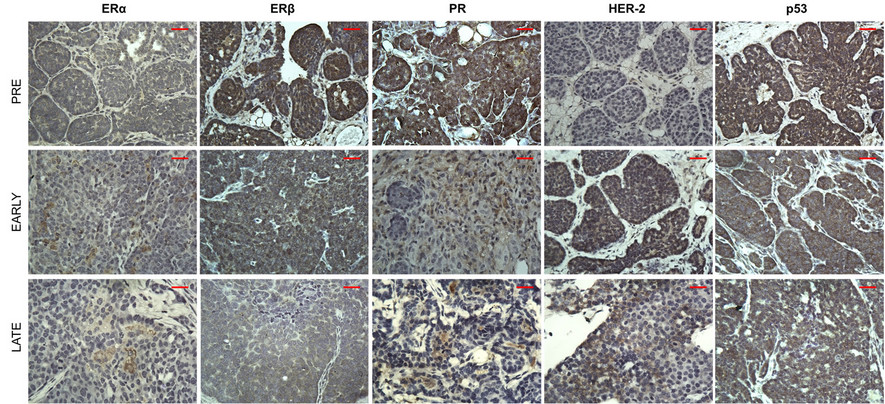
Figure 2. Immunohistochemistry of tumor phenotype from PyVT females during tumor development. Paraffin-embedded sections stained with antibodies against estrogen receptor (ERα and ERβ), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER-2), and tumor protein 53 (p53) at Pre, Early, and Late stages. Proteins staining: brown, counterstaining: blue (hematoxylin). Images represent only 1 of n = 6 per group at a 40× magnification. Scale bar = 50 µm.
Neoplastic cells have multiple survival techniques against death signals, such as the use of inhibitors of apoptosis. The anti-apoptotic protein survivin is a member of the inhibitors of apoptosis protein family expressed in a range of tumor types that regulates mitosis [14-16]. Survivin expression correlates with chemotherapeutic resistance and accelerated relapse [17]. Immunoblot analysis of tumor homogenate indicated that the expression of survivin increased as the tumor developed (Figure 3(a)). This was confirmed by immunohistochemistry (Figure 3(c)).
Cellular proliferation was determined by the expression of Ki-67, which is detectable during all activate phases of the cell cycle (G1, S, G2, and mitosis), but absent in the resting cell (G0) [18]. Elevated Ki-67 expression is associated with increased breast cancer recurrence and poor patient survival [19]. Ki-67 is one of only five genes for proliferation, out of 16 cancer-associated genes, that contribute significantly to the Oncotype score [20]. Immunoblot and immunohistochemistry of Ki-67 on PyVT tumors showed an increase in expression levels as the tumor progressed to a malignant phenotype (Figure 3(b)). This was confirmed by immunohistochemistry (Figure 3(c)).
Histopathological examination of the mammary tumors was conducted for each treatment group in the three stages of tumor development. When present, tumors were categorized as adenoma/mammary intraepithelial neoplasia (MIN), early carcinoma, or late carcinoma (Figure 4). Adenoma/MIN involved expansion of acini and ducts by a proliferation of polygonal neoplastic epithelial cells with multifocal coalescence of the affected ducts and acini. Neoplastic cells exhibited minimal cellular atypia and a low mitotic index (0-2/400x field). The neoplastic proliferation was confined by the basement membrane and there was a lack of fibrous connective tissue within the neoplasm. Early carcinomas were unencapsulated and moderately well-demarcated, with closely packed nests and acini of neoplastic cells with mild to moderate cellular atypia and 1 - 3 mitotic figures per high powered field. Neoplastic cells breached the basement membrane and were multifocally separated by a small to moderate amount of fibrovascular stroma. Late carcinomas were unencapsulated, poorly demarcated and invasive, composed of sheets of tightly packed nest and acini of neoplastic cells separated by moderate amounts of fibrovascular stroma. Anisocytosis and anisokaryosis were moderate and mitoses average 1-3/400x field. The Pre tumors were either adenoma/MIN or early carcinomas, while the Early tumors were all early carcinomas. The Late tumors were both late carcinomas.
The mammary carcinoma developed by the PyVT transgenic model is characterized by a metastatic patterninvolving the lung tissue. No lung metastasis was observed for the Pre stage of tumor development. Metastatic foci were not commonly found in the Early stage mice, but a few did develop small lesions by approximately 8 weeks of age (Figure 5(a)). Mice in the Late stage of tumor development formed large secondary tumors in the lung epithelium, as determined by gross observation and histopathology (Figures 5(b)-(d)). Lung tumors exhibited similar histopathologic features as those in the mammary tissue.
Tumor growth was monitored for two weeks during each stage of development. The initial tumor volume for Pre stage mice was 7.57 ± 10.77 mm3 at 5 weeks of age. Two weeks after the initial measurement of the Pre stage mice, the tumors grew to an averaged total of 309.04 ± 16.63 mm3 (Figure 6(a)). During the Early stage of development the initial tumor volume was measured at 6 weeks of age to be 133.33 ± 76.59 mm3. By 8 weeks of age, 14 days post initial volume measurement, the Early stage mice developed an average total tumor volume of 450.71 ± 39.56 mm3 (Figure 6(b)). The Late stage of tumor development began at 10 weeks of age, when mice had an initial tumor volume of 588.3 ± 78.87 mm3. After 14 days post initial volume measurement, total tumor volume was 1727.21 ± 50.82 mm3 (Figure 6(c)).
The effect of three antineoplastic compounds was tested on the development of tumors in the PyVT mouse model. Treatment with cisplatin or paclitaxel did not significantly attenuate tumor growth during the Pre, Early, or Late stage of development (Data not shown). However treatment with tamoxifen did significantly attenuated tumor growth during the Pre and Early stages of tumor development (Figures 6(a) and (b)). There was a significant difference in tumor volumes between tamoxifen and DMSO treated mice during the Pre stage of tumor development from day 10 to day 14 (Figure 6(a)). The change in tumor volume over this 14 day period showed a significant reduction of 124 mm3 with tamoxifen treatment compared to control (p-values = 0.018). During the Early stage of development there was a significant difference in tumor volumes between tamoxifen and control groups at day 14 (Figure 6(b)). Tamoxifen significantly decreased the initial tumor volume of 148 mm3 to a final volume of 306 mm3 after 14 days of treatment (p-value = 0.013). The average size of tumors after 14 days of tamoxifen treatment was 144 mm3 smaller than the final control volume, which is a reduction of 31%. Tamoxifen treatment did not significantly reduced tumor growth during the Late stage of tumor development (Figure 6(c)). There was no observable change in the formation of metastatic lung lesions.
4. Discussion
Throughout history, scientists have utilized a multitude
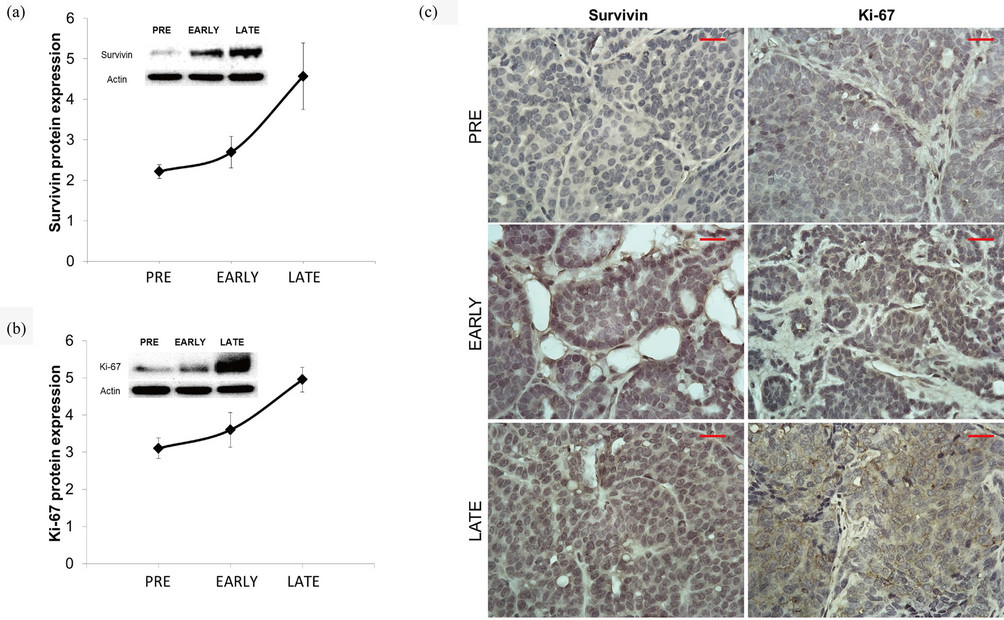
Figure 3. Expression of molecular markers in tumors from each stage of development. (a) Raw and graphical representation of protein expression in tumors from Western blot analysis. Data presented as fold-pixel intensity of survivin in PyVT female tumors in each of the three stages of tumor development. n = 4. (b) Image and graphical representation of protein expression in tumors from Western blot analysis. Data presented as fold-pixel intensity of Ki-67 in PyVT female tumors in each of the three stages of tumor development. Actin is the loading control. n = 4. (c) Immunohistochemisty of tumors from PyVT females. Paraffin-embedded sections stained with antibodies against survivin and Ki-67 from PyVT females at Pre, Early, or Late stage of tumor development. Proteins staining: brown, counterstaining: blue (hematoxylin). Images represent only 1 of n = 6 per group at a 60× magnification. Scale bar = 20 µm.

Figure 4. Pathological evaluation of hematoxylin and eosin (H&E) stained female PyVT mammary tumors. (a) Adenoma/ MIN: expansion of acini and ducts by a proliferation of polygonal neoplastic epithelial cells which exhibited minimal cellular atypia and a low mitotic index (0-2/400x field). The neoplastic proliferation was confined by the basement membrane and there was a lack of fibrous connective tissue within the neoplasm. (b) Early Carcinoma: unencapsulated and moderately well-demarcated, with closely packed nests and acini of neoplastic cells with mild to moderate cellular atypia and 1 - 3 mitotic figures per high powered field. Neoplastic cells breached the basement membrane and were multifocally separated by a small to moderate amount of fibrovascular stroma. (c) Late Carcinoma: unencapsulated, poorly demarcated and invasive, composed of sheets of tightly packed nest and acini of neoplastic cells separated by moderate amounts of fibrovascular stroma. Anisocytosis and anisokaryosis were moderate and mitoses average 1-3/40x field. Images represent only 1 of n = 3 per group at 40× magnification. Scale bar = 50 µm.
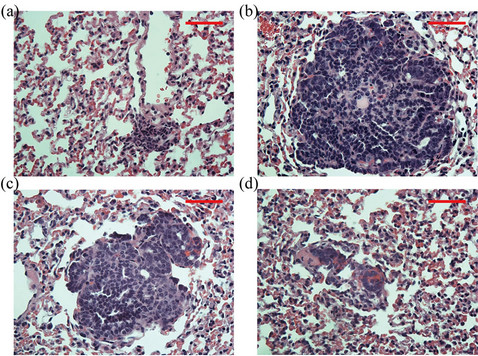
Figure 5. Representative images of hematoxylin and eosin (H&E) stained mammary tumors identified in the lung epithelium. Lung tissue collected from female PyVT mice at the (a) Early and (b)-(d) Late stage of tumor development. The lung contains large metastatic foci of neoplastic cells. Additional morphologically similar foci were noted throughout the lungs. Images are representative of n = 3 at a magnification of 40×. Scale bar = 50 µm.
of animal models to solve medical problems, develop new techniques and treatments, and cure disease. Genetically engineered mice are pursued in the cancer field, allowing researchers to investigate multiple aspects of cancer. Many types of transgenic animal have been developed, most of which have been used in small numbers as tools for investigating gene function in vivo. Several characteristics of the PyVT model make it ideal for research, such as its colony stability, predictable tumor growth behavior, metastatic phenotype, and its clinical similarity to human neoplasms.
The process of EMT involves epithelial carcinoma cells acquisitioning mesenchymal markers, such as vimentin, for increased metastatic potential [20,21] , as well as loss of epithelial cell adhesion molecules [21,22] . Alteration in E-cadherin expression is the typical epithelial cell marker of EMT [23] ; additionally loss of E-cadherin functionality promotes EMT [24]. The observed changes in epithelial marker expression in the isolated PyVT tumors are consistent with the EMT process. In addition to the decrease in PEDF, a multifunctional secreted protein with anti-angiogenic and anti-tumorigenic functions, is associated with progression towards malignancy and poor patient outcome [25] .
The PyVT mammary tumors were shown to be ER+, PR+, P53+, and HER-2+ via immunohistochemistry. This contradicts the results from Maglione et al. ADDIN EN.CITE ADDIN EN.CITE.DATA [5] , which indicated that tumors identified as mammary intraepithelial neoplasia (MIN) had no detectable levels of PR antigen. Maglione et al. used 9-week-old virgin females with the PyVT transgene; this is in the age range between the Early and Late stages of development defined here. The difference between results may be due to the dilution ratio of the primary antibody used for immunohistochemistry. Maglione et al. used a ratio of 1:1800, resulting in no antigen presentation. Here we used a ratio of 1:50 according to the manufacturer’s recommendation, providing more antibodies for binding to a small amount of antigen. Additionally expression of hormone receptors shows a cyclic change during the estrous cycle in mice [26,27]. This variation implies that the phase of the cycle, and thus the time of tissue harvest, may determine the expression of PR in the mammary carcinomas collected.
Other studies on the PyVT tumor phenotype indicate a loss of ERα and PR during tumor progression towards malignancy [4]. The results presented here support these conclusions. In addition ERβ is shown to also decrease with increased malignancy (Figure 2). The ER genes share a large proportion of homology, but differ in their distribution and functions in many tissue types. While ERα is a known prognostic marker in breast cancer, the role of ERβ is less clear. A decrease in ERβ protein expression has been shown in the transition of the normal mammary gland to a pre-invasive tumor [28]. The loss of ERβ protein in invasive carcinomas correlates with reported mRNA levels in breast tumors [29]. Overall, these results demonstrate for the first time a downregulation of ERβ during carcinogenesis in the PyVT model.
Interestingly the expression of ER and Ki-67 is mutually exclusive in normal premenopausal breast tissue, but coexpression occurs in ER positive breast cancer. With age the ER+ cell population increases, while the number of Ki-67+ cells decreases in the healthy mammary tissue [30] . The transformation of normal tissue to a neoplasia may reverse this phenotype. The results shown here indicate that ER+ cell decrease and Ki-67+ cells increase with tumor progression towards a malignant phenotype. The observed increase in Ki-67 expression corresponds to previous finding that cyclin D1, a regulator of the cell cycle also used as a molecular marker for proliferation, increases during PyVT tumor progression ADDIN EN.CITE ADDIN EN.CITE.DATA [4] .
Strong expression of survivin is observed in the majority of cancer types, and is associated with tumor progression and chemoresistance [31]. Elevated survivin expression is associated with aggressive disease and has a strong correlation with poor patient outcome. The PyVT model has an increased expression of survivin with progression of the tumor phenotype towards malignancy. Survivin can regulate the cell cycle, cytokinesis, and apoptosis through multiple interactions, such as with heat shock protein 90 [32], Smac/Diablo [33], Cdk4 [34], and p53 [35]. The observable increase in survivin expression suggests it plays a role in tumor formation and develop-

Figure 6. Tumor growth (mm3) in PyVT female mice. Tumors measured in two dimensions with calipers every 2 days for Pre (a), Early (b), and Late (c) stages of tumor development. The tumor size is expressed over a 14 day period for treatment with tamoxifen (20 mg/kg) of the vehicle control (DMSO) via 7 IPs. Days 0 - 12 represent the days of the 7 IP injections, day 14 represents the end of the study with measurements prior to tissue harvest. *p-value < 0.05 compared to controls.
ment in the PyVT model.
The p53 protein is a tumor suppressor that prevents progression of the cell cycle and may induce apoptosis [36,37]. The PyVT tumors were shown to be p53 positive at each stage of development. Interestingly, the survivin promoter sequence has two p53-binding sites [38]. Overexpression of the survivin protein saves neoplastic cells from p53-induced apoptosis [35]. The increase in survivin expression with tumor progression combined with a strong expression of p53 suggests that the cell is avoiding p53-dependent apoptosis through the upregulation of survivin.
Previous investigations on the PyVT transgenic model have been limited to studies focusing on gene function or histological characterization to prove clinical similarity to human breast cancer. This model has not been utilized to test the efficacy of antineoplastic compounds despite its multistage tumor development and similarities to human breast cancer. The compounds cisplatin, paclitaxel, and tamoxifen were chosen due their popularity in cancer treatment. Interestingly cisplatin and paclitaxel were not effective anticancer treatments, while tamoxifen successfully attenuated tumor formation in the PyVT model. The lack of response to cisplatin and paclitaxel suggests potential mechanisms of resistance, such as a reduction in the accumulation of compound insides the neoplastic cells due to membrane barriers or increased efflux, faster repair mechanism, or modulation of the apoptotic pathways.
There is a pressing need for effective and low-toxicity chemotherapeutics and chemopreventive agents against mammary carcinomas. Here the inhibitory effect of tamoxifen is demonstrated on neoplasm development in the MMTV-PyVT model. Tamoxifen has been extensively studied, but this is the first time, it is shown to be effective in a multistage mammary carcinoma model with lung metastasis. Exposure to tamoxifen prior to appearance of palpable mammary tumors significantly reduced tumor burden and attenuated tumor growth. However when tamoxifen exposure began after the gross appearance of mammary tumors, it was less effective at attenuating tumor growth and metastasis, without changing primary tumor burden. The means by which tamoxifen suppressed tumor development may be associated with the MMTV promoter sequence in this transgene-induced model. The MMTV promoter sequence contains a hormone-responsive element (HRE) that can bind progestin, glucocorticoid and androgen receptors. Hormone that has bound to its receptor can enter the nucleus where the complex can bind the HRE and stimulate the MMTV sequence. MMTV is activated by high levels of estrogen and progestin, upregulating expression of the target genes and inducing cancer formation [39]. Exogenous estradiol and progesterone treatment were shown to increase MMTV mRNA expression [40]. Therefore by treating with tamoxifen, hormone is unable to bind the ER, translocate to the nucleus, and bind the HRE in the MMTV promoter sequence. This prevents promoter activation and reduces the expression of the PyVT antigen.
Tamoxifen was shown to be effective only during the Pre and Early stages of tumor development. Previous research showed that the tumors had variable expression of the PyVT antigen, indicating an importance of the transgene expression in initiation of tumor formation but not in the perpetuation of a malignant phenotype [5]. This suggests that during the Pre and Early stages of tumor development, when the transgene is more highly expressed, tamoxifen may prevent MMTV activation and subsequent PyVT antigen expression. Additionally the finding that tamoxifen efficiently suppressed the spontaneous mammary tumor development at these stages provides evidence that this compound may be beneficially utilized as a chemopreventive and chemotherapeutic in early intervention to reduce morbidity and mortality associated with this neoplastic disease.
The long terminal repeat (LTR) region of the MMTV promoter has previously been shown not to be downmodulated by the antiestrogenic effects of tamoxifen [41], but this study was conducted in the T47D human breast cancer cell line stably transfected with the LTR of the MMTV. There is no data indicating that systemic estrogen concentrations or tamoxifen treatments in the transgenic mouse model do not alter promoter activation. Multiple studies conducted on transgenic mice using the MMTV promoter sequence do show anticancer effects with the use of tamoxifen [42-45], though they do not indicate if the promoter sequence plays a role in the compound’s efficacy.
This study looks at the transgenic PyVT mouse as a model for breast cancer research and drug development. Tumor growth was monitored from a precancerous stage to a metastatic stage. The tumor phenotype was determined for three distinct periods of development emphasizing a change in cellular phenotype (i.e. EMT) and hormone receptor expression, proliferative shift, survival techniques, and a detailed histopathological analysis of mammary carcinoma lesions. For the first time this model was used to determine the efficacy of three common antineoplastic compounds in the development of spontaneous mammary carcinomas. Tamoxifen can attenuate the formation of breast carcinoma if given early enough to inhibit transformation of the normal cell. The mechanism of tamoxifen chemoprevention may involve a reduction of the number of total cells transformed by the MMTV-PyVT transgene. These data suggest that tamoxifen, not cisplatin or paclitaxel, may improve the clinical outcome in patients prior to diagnosis of metastatic disease, and could potentially reduce morbidity and mortality associated with breast cancer.
5. Acknowledgements
The authors would like to thank the financial support of Merial Animal Health and National Institute of Health (NIH-1R15CA152922). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Merial Company and NIH.
REFERENCES
- T. Vargo-Gogola and J. M. Rosen, “Modelling Breast Cancer: One Size Does Not Fit All,” Nature Reviews. Cancer, Vol. 7, No. 9, 2007, pp. 659-672. doi:10.1038/nrc2193
- C. T. Guy, R. D. Cardiff and W. J. Muller, “Induction of Mammary Tumors by Expression of Polyomavirus Middle T Oncogene: A Transgenic Mouse Model for Metastatic Disease,” Molecular and Cellular Biology, Vol. 12, No. 3, 1992, pp. 954-961.
- M. A. Webster, J. N. Hutchinson, M. J. Rauh, S. K. Muthuswamy, M. Anton, C. G. Tortorice, et al., “Requirement for Both Shc and Phosphatidylinositol 3 Kinase Signaling Pathways in Polyomavirus Middle T-Mediated Mammary Tumorigenesis,” Molecular and Cellular Biology, Vol. 18, 1998, pp. 2344-2359.
- E. Y. Lin, J. G. Jones, P. Li, L. Zhu, K. D. Whitney, W. J. Muller, et al., “Progression to Malignancy in the Polyoma Middle T Oncoprotein Mouse Breast Cancer Model Provides a Reliable Model for Human Diseases,” The American Journal of Pathology, Vol. 163, 2003, pp. 2113- 2126. doi:10.1016/S0002-9440(10)63568-7
- J. E. Maglione, D. Moghanaki, L. J. Young, C. K. Manner, L. G. Ellies, S. O. Joseph, et al., “Transgenic Polyoma Middle-T Mice Model Premalignant Mammary Disease,” Cancer Research, Vol. 61, No. 22, 2001, pp. 8298- 8305.
- G. M. Clark, C. K. Osborne and W. L. McGuire, “Correlations between Estrogen Receptor, Progesterone Receptor, and Patient Characteristics in Human Breast Cancer,” Journal of Clinical Oncology, Vol. 2, 1984, pp. 1102- 1109.
- X. Sastre-Garau, M. Jouve, B. Asselain, A. Vincent-Salomon, P. Beuzeboc, T. Dorval, et al., “Infiltrating Lobular Carcinoma of the Breast. Clinicopathologic Analysis of 975 Cases with Reference to Data on Conservative Therapy and Metastatic Patterns,” Cancer, Vol. 77, No. 1, 1996, pp. 113-120. doi:10.1002/(SICI)1097-0142(19960101)77:1<113::AID-CNCR19>3.0.CO;2-8
- D. J. Slamon, G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich and W. L. McGuire, “Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the Her-2/Neu Oncogene,” Science, Vol. 235, No. 4785, 1987, pp. 177-182. doi:10.1126/science.3798106
- S. A. Aziz, S. Pervez, S. Khan, N. Kayani, S. I. Azam and M. H. Rahbar, “Case Control Study of Prognostic Markers and Disease Outcome in Inflammatory Carcinoma Breast: A Unique Clinical Experience,” The Breast Journal, Vol. 7, No. 6, 2001, pp. 398-404. doi:10.1046/j.1524-4741.2001.07604.x
- S. Haupt, M. Berger, Z. Goldberg and Y. Haupt, “Apoptosis—The P53 Network,” Journal of Cell Science, Vol. 116, No. 20, 2003, pp. 4077-4085. doi:10.1242/jcs.00739
- S. Jin and A. J. Levine, “The P53 Functional Circuit,” Journal of Cell Science, Vol. 114, No. 23, 2001, pp. 4139-4140.
- C. G. Ferreira, C. Tolis and G. Giaccone, “P53 and Chemosensitivity,” Annals of Oncology: Official Journal of the European Society for Medical Oncology, Vol. 10, 1999, pp. 1011-1021.
- D. Kandioler-Eckersberger, C. Ludwig, M. Rudas, S. Kappel, E. Janschek, C. Wenzel, et al., “Tp53 Mutation and P53 Overexpression for Prediction of Response to Neoadjuvant Treatment in Breast Cancer Patients,” Clinical Cancer Research, Vol. 6, No. 1, 2000, pp. 50-56.
- C. Adida, P. L. Crotty, J. McGrath, D. Berrebi, J. Diebold and D. C. Altieri, “Developmentally Regulated Expression of the Novel Cancer Anti-Apoptosis Gene Survivin in Human and Mouse Differentiation,” The American Journal of Pathology, Vol. 152, No. 1, 1998, pp. 43-49.
- D. C. Altieri, “Validating Survivin as a Cancer Therapeutic Target,” Nature Reviews. Cancer, Vol. 3, No. 1, 2003, pp. 46-54. doi:10.1038/nrc968
- G. Ambrosini, C. Adida and D. C. Altieri, “A Novel AntiApoptosis Gene, Survivin, Expressed in Cancer and Lymphoma,” Nature Medicine, Vol. 3, No. 8, 1997, pp. 917- 921. doi:10.1038/nm0897-917
- T. Dohi, E. Beltrami, N. R. Wall, J. Plescia and D. C. Altieri, “Mitochondrial Survivin Inhibits Apoptosis and Promotes Tumorigenesis,” Journal of Clinical Investigation, Vol. 114, No. 8, 2004, pp. 1117-1127. doi:10.1172/JCI22222
- F. Lopez, F. Belloc, F. Lacombe, P. Dumain, J. Reiffers, P. Bernard, et al., “Modalities of Synthesis of Ki67 Antigen during the Stimulation of Lymphocytes,” Cytometry, Vol. 12, 1991, pp. 42-49. doi:10.1002/cyto.990120107
- P. Kronqvist, T. Kuopio, M. Nykanen, H. Helenius, J. Anttinen and P. Klemi, “Predicting Aggressive Outcome in T1n0m0 Breast Cancer,” British Journal of Cancer, Vol. 91, No. 2, 2004, pp. 277-281. doi:10.1038/sj.bjc.6601948
- C. Oakman, S. Bessi, E. Zafarana, F. Galardi, L. Biganzoli and A. Di Leo, “Recent Advances in Systemic Therapy: New Diagnostics and Biological Predictors of Outcome in Early Breast Cancer,” Breast Cancer Research, Vol. 11, No. 2, 2009, p. 205. doi:10.1186/bcr2238
- E. W. Thompson, J. Torri, M. Sabol, C. L. Sommers, S. Byers, E. M. Valverius, et al., “Oncogene-Induced Basement Membrane Invasiveness in Human Mammary Epithelial Cells,” Clinical & Experimental Metastasis, Vol. 12, No. 3, 1994, pp. 181-194. doi:10.1007/BF01753886
- C. Xue, D. Plieth, C. Venkov, C. Xu and E. G. Neilson, “The Gatekeeper Effect of Epithelial-Mesenchymal Transition Regulates the Frequency of Breast Cancer Metastasis,” Cancer Research, Vol. 63, 2003, pp. 3386-3394.
- E. D. Hay and A. Zuk, “Transformations between Epithelium and Mesenchyme—Normal, Pathological, and Experimentally-Induced,” American Journal of Kidney Diseases, Vol. 26, 1995, pp. 678-690. doi:10.1016/0272-6386(95)90610-X
- M. A. Huber, N. Kraut and H. Beug, “Molecular Requirements for Epithelial-Mesenchymal Transition during Tumor Progression,” Current Opinion in Cell Biology, Vol. 17, No. 5, 2005, pp. 548-558. doi:10.1016/j.ceb.2005.08.001
- H. Uehara, M. Miyamoto, K. Kato, Y. Ebihara, H. Kaneko, H. Hashimoto, et al., “Expression of Pigment Epithelium-Derived Factor Decreases Liver Metastasis and Correlates with Favorable Prognosis for Patients with Ductal Pancreatic Adenocarcinoma,” Cancer Research, Vol. 64, 2004, pp. 3533-3537. doi:10.1158/0008-5472.CAN-03-3725
- N. Gava, C. L. Clarke, K. Byth, R. L. Arnett-Mansfield and A. deFazio, “Expression of Progesterone Receptors a and B in the Mouse Ovary during the Estrous Cycle,” Endocrinology, Vol. 145, No. 7, 2004, pp. 3487-3494. doi:10.1210/en.2004-0212
- M. Vasei, N. Azarpira and A. Talei, “Status of Estrogen and Progesterone Receptors in Various Phases of the Menstrual Cycle in Breast Cancer,” Archives of Iranian medicine, Vol. 9, No. 3, 2006, pp. 250-253.
- P. Roger, M. E. Sahla, S. Makela, J. A. Gustafsson, P. Baldet and H. Rochefort, “Decreased Expression of Estrogen Receptor Beta Protein in Proliferative Preinvasive Mammary Tumors,” Cancer Research, Vol. 61, No. 6, 2001, pp. 2537-2541.
- E. Leygue, H. Dotzlaw, P. H. Watson and L. C. Murphy, “Altered Estrogen Receptor Alpha and Beta Messenger Rna Expression During Human Breast Tumorigenesis,” Cancer Research, Vol. 58, No. 15, 1998, pp. 3197-3201.
- B. S. Shoker, C. Jarvis, R. B. Clarke, E. Anderson, J. Hewlett, M. P. A. Davies, et al., “Estrogen Receptor-Positive Proliferating Cells in the Normal and Precancerous Breast,” American Journal of Pathology, Vol. 155, No. 6, 1999, pp. 1811-1815.
- D. C. Altieri, “Survivin, Versatile Modulation of Cell Division and Apoptosis in Cancer,” Oncogene, Vol. 22, No. 53, 2003, pp. 8581-8589. doi:10.1038/sj.onc.1207113
- P. Fortugno, E. Beltrami, J. Plescia, J. Fontana, D. Pradhan, P. C. Marchisio, et al., “Regulation of Survivin Function by Hsp90,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 24, 2003, pp. 13791-13796. doi:10.1073/pnas.2434345100
- Z. Y. Song, X. B. Yao and M. Wu, “Direct Interaction between Survivin and Smac/Diablo Is Essential for the Anti-Apoptotic Activity of Survivin during Taxol-Induced Apoptosis,” Journal of Biological Chemistry, Vol. 278, No. 25, 2003, pp. 23130-23140. doi:10.1074/jbc.M300957200
- A. Suzuki and K. Shiraki, “Tumor Cell “Dead or Alive: Caspase and Survivin Regulate Cell Death, Cell Cycle and Cell Survival,” Histology and Histopathology, Vol. 16, No. 2, 2001, pp. 583-593.
- A. Mirza, M. McGuirk, T. N. Hockenberry, Q. Wu, H. Ashar, S. Black, et al., “Human Survivin Is Negatively Regulated by Wild-Type P53 and Participates in P53- Dependent Apoptotic Pathway,” Oncogene, Vol. 21, No. 17, 2002, pp. 2613-2622. doi:10.1038/sj.onc.1205353
- R. V. Sionov and Y. Haupt, “The Cellular Response to P53: The Decision between Life and Death,” Oncogene, Vol. 18, No. 45, 1999, pp. 6145-6157. doi:10.1038/sj.onc.1203130
- B. Vogelstein, D. Lane and A. J. Levine, “Surfing the P53 Network,” Nature, Vol. 408, No. 6810, 2000, pp. 307- 310.
- L. Wang, Q. Wu, P. Qiu, A. Mirza, M. McGuirk, P. Kirschmeier, et al., “Analyses of P53 Target Genes in the Human Genome by Bioinformatic and Microarray Approaches,” The Journal of Biological Chemistry, Vol. 276, No. 47, 2001, pp. 43604-43610. doi:10.1074/jbc.M106570200
- V. Theodorou, M. A. Kimm, M. Boer, L. Wessels, W. Theelen, J. Jonkers, et al., “Mmtv Insertional Mutagenesis Identifies Genes, Gene Families and Pathways Involved in Mammary Cancer,” Nature Genetics, Vol. 39, No. 6, 2007, pp. 759-769. doi:10.1038/ng2034
- Y. Yin, Z. Yang and S. Zhang, “Combined Treatment with Exogenous Estradiol and Progesterone Increases the Incidence of Breast Cancer in Ta2 Mice without Ovaries,” Cancer Letters, Vol. 311, No. 2, 2011, pp. 171-176. doi:10.1016/j.canlet.2011.07.011
- J. F. Glover and P. D. Darbre, “Multihormone Regulation of Mmtv-Ltr in Transfected T-47-D Human Breast Cancer Cells,” Journal of Steroid Biochemistry, Vol. 32, 1989, pp. 357-363. doi:10.1016/0022-4731(89)90207-0
- R. Fuchs-Young, S. H. Shirley, I. Lambertz, J. K. Colby, J. Tian, D. Johnston, et al., “P53 Genotype as a Determinant of Er Expression and Tamoxifen Response in the Mmtv-Wnt-1 Model of Mammary Carcinogenesis,” Breast Cancer Research and Treatment, Vol. 130, No. 2, 2011, pp. 399-408. doi:10.1007/s10549-010-1308-y
- S. Menard, P. Aiello, E. Tagliabue, C. Rumio, P. L. Lollini, M. I. Colnaghi, et al., “Tamoxifen Chemoprevention of a Hormone-Independent Tumor in the Proto-Neu Transgenic Mice Model,” Cancer Research, Vol. 60, No. 2, 2000, pp. 273-275.
- P. Nanni, G. Nicoletti, C. De Giovanni, L. Landuzzi, E. Di Carlo, M. Iezzi, et al., “Prevention of Her-2/Neu Transgenic Mammary Carcinoma by Tamoxifen Plus Interleukin 12,” International Journal of Cancer, Vol. 105, No. 3, 2003, pp. 384-389. doi:10.1002/ijc.11092
- N. Sidell, N. Kirma, E. T. Morgan, H. Nair and R. R. Tekmal, “Inhibition of Estrogen-Induced Mammary Tumor Formation in Mmtv-Aromatase Transgenic Mice by 4-Chlorophenylacetate,” Cancer Letters, Vol. 251, 2007, pp. 302-310. doi:10.1016/j.canlet.2006.11.031