Advances in Bioscience and Biotechnology
Vol.5 No.6(2014), Article ID:46442,12 pages DOI:10.4236/abb.2014.56067
Revisiting the X-Chromosome Inactivation and Its Impact on Female Longevity
Lilian Chuaire-Noack, Magda Carolina Sánchez-Corredor, María Martínez-Agüero
Facultad de Ciencias Naturales y Matemáticas, Universidad del Rosario, Bogotá, Colombia
Email: lilian.chuaire@urosario.edu.co
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 23 February 2014; revised 6 May 2014; accepted 22 May 2014
Abstract
X-chromosome inactivation is the genetic mechanism by which X-linked gene expression is equalized between the male and female genders of all placental mammal species. Given that the probability of mutant X-linked allele expression decreases as a result of the inactivation, it has been proposed that females have biological advantages relative to males. These advantages have grabbed the attention of the scientific community in recent years and have focused it on this topic and its clinical implications. To shed some new light on this intriguing phenomenon, this article reviews the most relevant molecular events involved in this process. These events include the role of Xist, the selection mechanism for future X-chromosome inactivation, the age-related inactivation skewing, and the relationship between inactivation and the emergence of X-linked diseases, possible treatments, and longevity.
Keywords
Sex Chromosomes, Epigenetic Inheritance, Heterochromatin, DNA Methylation, Gene Therapy

1. Introduction
Ageing is one of the most complex phenotypes in metazoans, given that it results from an interaction between a diversity of genetic, environmental, and stochastic factors. For that reason, the characteristics of the senescence process, such as rate, quality, and expectancy of life, exhibit great heterogeneity, even within the same population.
It has been shown that, in approximately 25% of cases, the variation in lifespan is conditioned by genetic factors. The effect of the latter only manifests itself in a significant way during old age, especially above 60 years [1] . Thus, factors such as telomere shortening and mutations in genes involved in different metabolic, endocrine signaling, and stress response pathways act as genetic determinants of both lifespan and quality of ageing, as observed in animal models [2] [3] . In particular, gender, which results from the asymmetric inheritance of sex chromosomes, plays a momentous role in the differences in life expectancy, therefore in longevity in all human populations, making life expectancy of women invariably exceed that of men, by 4.5 years for 2010- 2015 period. This gap further widens in the more developed regions such as Northern America or Northern Europe (6.7 years) and decreases in the less developed ones, such Africa (2.4 - 3.7 years) [4] .
In humans, like in all other eutherian or metatherian mammals, the presence of X and Y heteromorphic sex chromosomes results in gene disequilibrium between the male and female genders.
The mechanism that prevents transcription of one (or more, in the case of extra X chromosomes) of the X chromosomes in the homogametic gender (XX) equalizes the dose of X-linked gene products, making it equivalent to the heterogametic gender (XY) [5] . Considering that inactivation of one X chromosome (XCI) decreases the probability of expressing recessive X-linked mutant alleles, women could have biological advantages over men starting at conception, which could impact life viability and expectancy [6] -[8] .
Approximately 50 years ago, it was discovered that, around the time of implantation, human female embryos exhibit cellular mosaicism, a consequence of the stochastic transcriptional silencing of several loci on one of the two X chromosomes. This phenomenon results in 50% of cells expressing alleles from the maternal X chromosome (Xm) and 50% of cells expressing alleles from the paternal homolog (Xp), which not only causes a compensatory effect and avoids an “overdose” of X-linked genes [9] -[11] , but also allows the expression of a diversity of cellular phenotypes that are apparently associated with a broader spectrum of responses to a variety of physiological and pathological conditions as compared with men [12] . The presence of two copies for each of the 1717 genes located on the X chromosome opens up the possibility that the phenotype corresponding to a recessive mutant allele is not expressed, as long as the other allele is wild type [7] [13] [14] . The alleles located on the Xm are always expressed in men, given that they lack an Xp. In terms of viability, this circumstance implies that deleterious alleles responsible for X-linked diseases are invariably expressed in men, which constitutes a determining factor in the appearance of the gap in mortality rates between genders (higher in men, lower in women), both in utero and during childhood [7] . In the adulthood, for example, adult mortality in men is higher than that in women in all regions of the world, except in Southern Africa, where HIV/AIDS has reduced women’s survival advantage [4] .
In contrast to the random X-inactivation, which is the only manner of X-silencing in most eutherian mammals, in certain metatherian species such as marsupials, inactivation occurs only in the paternally derived X chromosome (Xp), in what has been called imprinted X-inactivation (iXCI) [15] .
2. XCI and the Role of Xist
Human X-chromosome inactivation (XCI) is a complex process that is not yet fully understood due to, among other reasons, the limitations in human embryo manipulation to establish embryonic stem cell lines. The heterogeneity of these cell lines in terms of spontaneous differentiation and X-chromosome silencing are further limitations [16] . Regardless, the discovery of the role that the non-coding RNA Xist (X-inactive specific transcript) plays in the formation of heterochromatin on the X chromosome in cis was a milestone in this research field. The discovery of Xist was based on the observation that its deletion was lethal for female embryos. Xist is considered to be the main effector of the XCI process in placental mammals [17] .
The Xist gene encodes a polyadenylated transcript, approximately 19 kb in size, located on the long arm (q) of the X chromosome. It contains seven to eight A-repeat elements in its 5’ end, giving rise to a highly conserved motif among placental mammals. This motif is responsible for recruiting the Polycomb repressor complex 2 (PRC2) [18] , which catalyze the trimethylation of histone H3 on lysine 27 [19] . This function implies that the motif confers gene expression silencing capacity to the Xist RNA [20] .
Xist is also part of the so-called X inactivation center (Xic) (see Figure 1), whose importance was first elucidated by Rastan and Robertson through cytogenetic methods [21] . Therefore, the Rastan model posed that none of the X chromosomes in a diploid cell can be inactivated when the inactivation center is deleted in only one of them. This finding led them to consider Xic as the “minimal region that is necessary and sufficient to trigger X-chromosome inactivation” [21] .
The loci of other genes regulating Xist expression are also contained in the Xic. These loci include the non-coding genes Jpx (just proximal to Xist) and FTX (five-prime to Xist) [21] , along with the protein-coding Chic1 (cysteine-rich hydrophobic 1), CDX4 (caudal X-linked gene 4), NAPIl2 (nucleosome assembly protein 1-like 2), CNBP2 (cellular nucleic acid-binding protein 2), and XPCT (X-linked PEST-containing transporter)
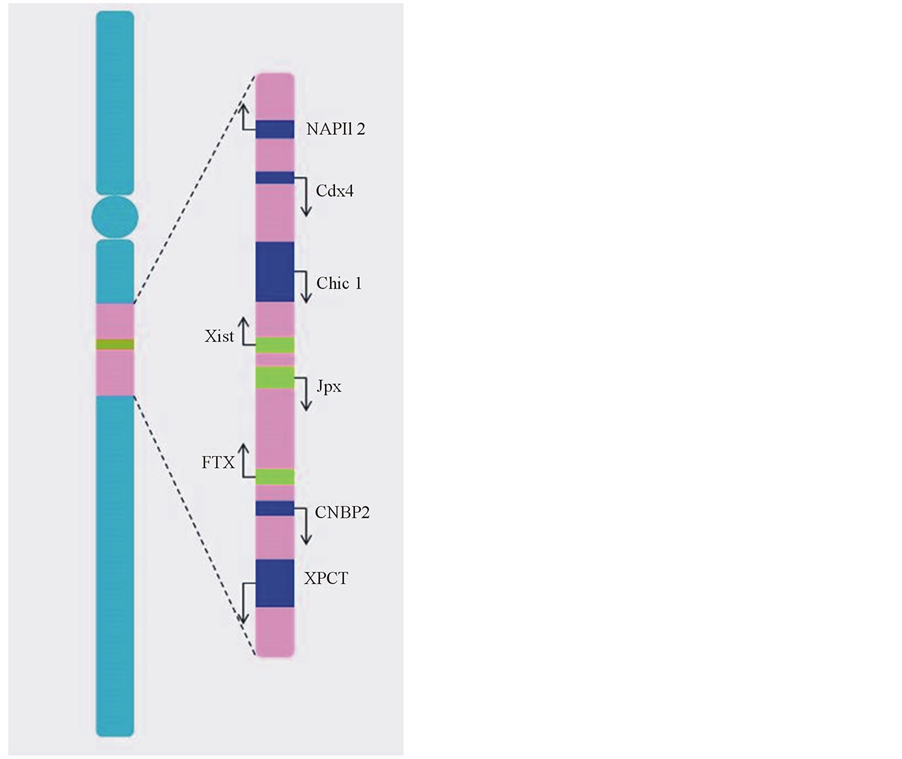
Figure 1. X inactivation center. The human XIC locus localizes on the long arm of the X chromosome and spans approximately 2300 kb. It consists of genes transcribing into long non-coding RNAs (in green) and protein-coding genes (in blue). The arrows indicate transcription direction.
genes [22] -[24] and possible binding sites for regulatory proteins.
In mice, where XCI has been most widely studied, the silencing phenomenon occurs during embryogenesis and consists of two phases. In the first phase, an unrandom imprinted X-inactivation takes place so the Xp is preferentially inactivated during the fourto eight-cell blastomere stage.
In the second phase, at the beginning of gastrulation and after Xp reactivation in epiblastic cells of the internal cell mass, either chromosome (Xm or Xp) is randomly inactivated [25] -[28] .
In addition, mice have a negative regulator of Xist, the lncRNA Tsix, which is not active in humans. This transcript is antisense oriented to Xist, thus repressing Xist transcription in cis and preventing inactivation on the future Xa. It has been demonstrated that Xist induction can be suppressed by inhibiting the short RNA RepA to recruit Polycomb repressive complex 2 (PCR2) onto the 5’ end of Xist [18] . Presumably Xist can be also silenced when DNA-methyltransferase3A (DNMT3a) is activated by Tsix on Xist promoter region or, alternatively, when RNAi machinery is recruited by Tsix, taking into account that small RNAs have been found, as a result of processing the duplex RNA Xist-Tsix by Dicer [19] [29] .
Although it is likely that the kinetics of XCI in human embryos occurs in a similar manner to the kinetics observed in mouse embryos, it has not been shown that there is preferential Xp inactivation in humans during the early stages prior to implantation. There is also no consensus regarding the onset of both Xist transcription and XCI, mostly due to the difficulty in discriminating between epiblastic cells of the internal cell mass and cells of the trophectoderm. The differences in embryo culture conditions further complicate this process. Okamoto et al. [30] reported that, in both male and female human embryos conceived in vitro that have not yet been implanted, transcription begins in the fourto eight-cell stage. Furthermore, female embryos start accumulating Xist RNA on both X chromosomes at this stage, regardless of origin. However, van der Berg et al. [31] reported a notable and random up-regulation of Xist transcription during the eight-cell stage in only one of the two X chromosomes. This event implies that XCI has already begun and that the Xist allele is methylated in the homologous chromosome [32] . In the absence of evidence for heterochromatin formation on Xist-coated X chromosomes in early or late female human blastocysts cultured in the presence of physiological oxygen levels, Okamoto et al. [30] proposed that, during this stage and under their experimental conditions, embryonic stem cells retain their pre-XCI state, which is in agreement with the results of Lengner et al. [16] , who determined that these cells only irreversibly leave the pre-XCI state when they are chronically subjected to atmospheric oxygen concentrations, suggesting that XCI could occur in response to cellular stress, including oxidative stress. However, regarding Xist expression in male human blastocysts, Okamoto et al. [30] proposed that Xist expression decreases around day 7, together with the emergence of a heterochromatic Y body. These observations coincide with the identification of very low or nonexistent levels of Xist RNA, which could be considered evidence that early expression of Xist, in either gender, does not have a functional relevance.
Regardless of the moment at which Xist transcription starts in female human embryos, selection of the future X-inactive (Xi) chromosome seems to happen after up-regulation of Xist RNA in cis.
It has been shown that at this moment, the transcript binds its own locus, mediated by the Yin Yang 1 transcription factor [33] , allowing the Xist molecules to adjust and form a cloud-like structure over that particular X chromosome [17] and giving rise to an inert compartment where DNA consists only of non-coding repeats.
At the same time, the transcriptional machinery—which includes RNA polymerase II and the nascent transcripts, TAF10, and the TBP proteins [34] —is lost.
This loss results in the silencing of X-linked alleles and their re-location within the compartment formed by Xist, from which they were initially distant [34] [35] .
Afterwards, it has also been described that the Xi chromatin experiences several epigenetic modifications that contribute to the formation of a heterochromatic mass, known as a Barr body in somatic cells.
These changes include dimethylation of histone H3 on lysine 9 (H3K9me2) [36] , trimethylation of histone H3 on lysine 27 (H3K27me3), monomethylation of histone H4 on lysine 20 (H4K20me1) [37] , hypermethylation of CpG dinucleotides in promoter regions and also of 5’ untranslated regions of Xi-linked genes, monoubiquitination of histone H2A on lysine 119, and insertion of the repressive macroH2A histone [37] -[40] (see Figure 2). According to Ryba et al. [41] , these modifications could explain not only the origin of gene repression stability associated with XCI but also the origin of other events, such as the late replication of Xi during S-phase and the dissociation of the Xist RNA-Xi complex during mitosis followed by its subsequent reconstitution during the G1 phase of the next cycle. All of these events assume that, as Ryba et al. [41] speculated, late replication is associated with the difficulty encountered by initiation factors in accessing the heterochromatin. However, other opinions suggest that gene silencing associated with XCI cannot be exclusively attributed to the aforementioned epigenetic changes [38] . In this respect, it has been shown that expression of mutant Xist RNA lacking the sequences in its 5’ region causes chromatin modifications but not gene silencing. This difference can be explained by the fact that the motifs formed by the A-repeat elements in the 5’ region have been linked to the activation of other pathways associated with gene repression [42] . In addition, results obtained by Panova et al. [43] using female human pluripotent stem cells indicated that neither the X inactivation state, nor its late replication during S-phase, are necessarily related to the degree of condensation of the chromosome territory that is observed during interphase and that furthermore, the Xi chromosome can only be reactivated when its territory is in a de-condensed state and can only be replicated when it is synchronized with its homologue.
3. What Determines Which X Chromosome Will Be Inactivated?
In human beings, the stochastic inactivation of the X chromosome follows a similar pattern as in other placental mammals. The inactivation occurs in all female cells and in male cells containing more than one X chromosome. The process consists of several known stages:
• Initiation Prior to the selection of the X chromosome to become inactivated (Xi), the cell assesses the ratio of X chro-
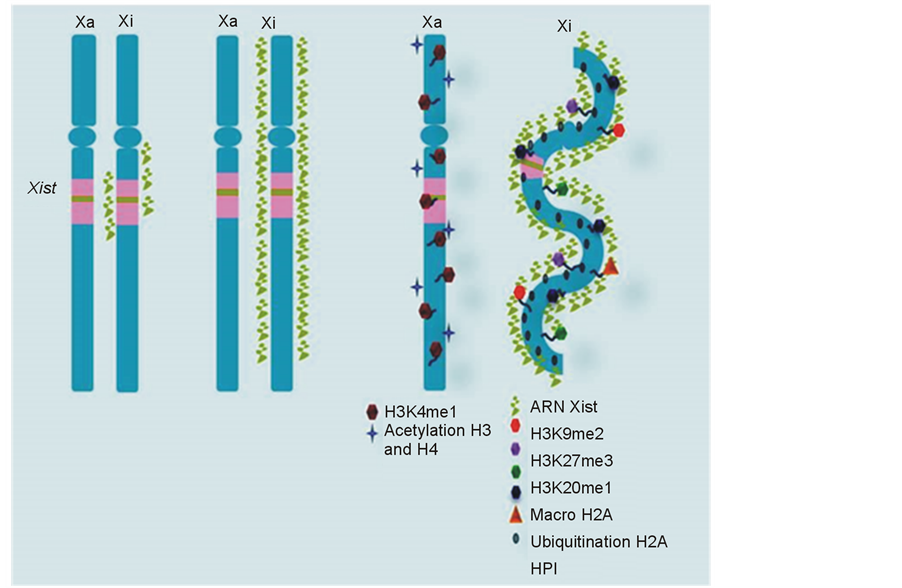
Figure 2. Heterochromatin formation on Xi. The histone code is involved in XCI through histone modifications, such as H3K9me2, H3K27me3, and H4K20me1, that occur after the Xist RNA “cloud” has formed and the Polycomb repressor complex 2 (PRC2) has been activated. These changes allow for the recruitment of epigenetic modulators, such as heterochromatin protein HPI, which binds H3K9me2 to cover the X and ensure its heterochromatinization. Hypoacetylation of histones H3 and H4 further contribute to this event, as does the insertion of the repressor histone variant macroH2A and the monoubiquitination of histone H2A.
mosomes to diploid sets of autosomal chromosomes. Afterwards, it selects one of the X chromosomes.
Different models aiming to explain the counting and selection process have been proposed, all based on evidence indicating that autosomal ploidy determines the number of X chromosomes to become inactivated. These models suggest that transactivating factors exist, which are produced by the autosomal chromosomes and can interact with the X chromosomes. In this respect, it is worth mentioning the symmetry-breaking model [44] holding that some components of the so-called “blocking factor complex”, which are present in both X and autosomal chromosomes, can bind the inactivation center of one X chromosome (future Xa). This binding causes loss of symmetry between both X chromosomes, and the other X chromosome (future Xi) becomes the target of inactivation and inhibition of Xist up-regulation. However, it should be noted that the formation over the Xa has not yet been shown.
Other recent hypotheses about the counting and selection processes are based on several aspects different than the autosomal ploidy, which include the “alternate state” model [45] and the hypothesis about the association between loci of homologous chromosomes, which emerged from observations in murine embryonic stem cells. The first model proposes that, before inactivation occurs, the X chromosomes display differences in the allelic cohesion prior to DNA replication. These differences cause the sister chromatids of the future Xi to come in closer contact than the chromatids of its homolog (future Xa), indicating that local diffusion of Xist has already occurred in cis. However, this model, proposed by Mlynarczyk-Evans et al. [45] , does not explain the existing differences in cohesion between X chromosomes that are observed in murine cells with deletions in Xist or in its negative regulator in mice Tsix (antisense Xist transcript). Thus, chromatid participation in Xi selection has not yet been proven. The second model proposes that associations between loci of homologous chromosomes cause opposing transcriptional activities in homologous alleles. Therefore, to initiate XCI, a co-localization or pairing of the Xics in both X chromosomes must first occur. This pairing would occur between the Xist loci and the so-called “X pairing region” (Xpr), which is localized −400 kb upstream of Xist. The latter appears to be crucial in evaluating the existence of more than one specific X-linked allele [46] -[49] . While it has been suggested that pairing at the Xpr loci facilitates both the subsequent co-localization of the Xist loci and the coordination of the monoallelic expression of Tsix and the reciprocal expression of Xist [47] [49] [50] , neither the nature of said relationship, nor the role of Xist in it, have been proven.
• Spreading of the inactivation state Transcriptional silencing of most genes on the Xi spreads throughout the entire chromosome through DNA methylation of promoter regions, which occurs in a progressive way and refractory to reactivation [34] . However, the process by which Xist localizes to chromatin and extends across the X-chromosome is not yet fully understood. In this respect, Riggs and Gartler [51] first proposed the presence of “way stations”, i.e., cis-elements, probably Xist RNA-binding sites, that could strengthen inactivation wave, as it propagates away from Xic. More recently, Engreitz et al. [52] demonstrated that Xist lncRNA initially moves from its transcription locus in searching for possible distal target sites in the future Xi, by a mechanism independent of its A-repeat domain. Then, it gradually extends across the active genes in a process depending on its A-repeat-domain that leads to transcriptional silencing. Engreitz et al. suggested that in order to achieve its purpose, Xist lncRNA uses their ability to alter chromatin structure and therefore chromosome architecture by the recruitment of PCR2 and the repositioning of the silenced genes in its compartment.
• Maintenance of the inactive state throughout successive cell divisions During maintenance, Xist lncRNA accesses and coats the entire Xi. It has been shown that there are different levels of Xist enrichment along the entire length of the inactive X-chromosome, in a strongly correlated manner with H3K27me3 levels [52] .
The previously described DNA methylation events on the X chromosome occur lately during XCI. In addition, genes such as Orc2 (origin recognition complex 2) and HP1 have been linked to maintaining the inactive state of the X chromosome. These associations are based on the observation that certain small interfering RNAs cause de-repression of Xi gene expression [53] . Another epigenetic modifier, the structural protein SMCHD1 (structural maintenance of chromosomes hinge domain containing 1), also contributes to the maintenance of the inactive Xi. The observation that its absence causes embryonic lethality exclusively in women during the mid-gestation period [54] [55] led to the proposal that SMCHD1 plays a critical role towards the end of the epigenetic cascade of events leading to XCI, determining that, in case there is a mutation in the SMCHD1 gene, XCI maintenance, but not its initiation, fails. Concomitantly, other genes linked to the X chromosome selected for inactivation are not hypermethylated in the CpG islands of their promoters. If they already have been hypermethylated, they can escape the inactivation state shortly after silencing is initiated [56] .
It is worth noting that primordial germ cells constitute the only cell lineage in which epigenetic changes that occurred during inactivation are not maintained [34] .
4. Age-Dependent Skewing in XCI
In women, along with females from other eutherian species, XCI loses its stochastic nature during early childhood [57] due to genetic, environmental, or random causes. Consequently, inactivation takes on a gradual skewing, either towards the Xm or the Xp, to establish a pattern that will be maintained in a clonal fashion [58] -[60] . It has been proposed that X-linked loci participate in the selection process, as demonstrated by Christensen et al. [58] . They observed that monozygotic twins show a strong skewing towards the same hematopoietic cell line. Nonetheless, close to 15% of genes on the inactivated chromosome seem to randomly escape silencing, favoring compensation mechanisms [61] . This fact potentially increases X-linked cellular protein contents and adds diversity to cellular responses in women as compared with men [12] . Either way, there is still a biological advantage for the female gender [61] . It has been shown that most genes involved in this “escape” localize to the recent X-added region (XAR) and have homologues on the Y chromosome, unlike genes on the X-conserved region (XCR), which, with very few exceptions, are completely silenced [62] . The importance of incomplete silencing of the inactivated X chromosome becomes evident when individuals with aneuploidies, such as Turner syndrome (monosomy X0), are taken into account. Theoretically, if there was a complete X inactivation in these individuals, the absence of an X chromosome should not cause the physical anomalies associated with this type of monosomy.
In metatherian species, although many X-linked genes can escape paternal XCI, the reasons for that are different than in eutherians. First of all, marsupials lack of both Xist gene and neighboring non-coding RNAs within the Xic. Instead, the untranslated nuclear RNA Rsx can spread across the future Xi for repressing gene transcription [63] .
Unlike eutherian Xi, the marsupial Xi is not capable to recruit the inactive marks, e.g. H3K27me3 and probably H3K9me2, in accordance with the absence of Xist [64] . This fact could explain the relative unstability of the Xp silencing state in methaterian as well as the frequent reactivation of X-linked genes like glucose-6-phosphate dehydrogenase (G6pd) and phosphoglycerate kinase A (Pgk1). In addition, the reactivation of these X-linked genes has shown to be tissue and specie-specific [15] .
5. XCI, Longevity, and Gender
Although the current hypothesis that X-chromosome inactivation is the cause of greater longevity in the homogametic gender throughout most of the animal kingdom is widely accepted, it has not been shown to be the only determining factor in gender-specific longevity differences in certain kinds of mammals. These species include members of the Eutheria (placental) infraclass, such as some small bat species of the Myotis gender, guinea pigs, the golden hamster, and some mouse strains, in which males live longer than females, along with members of the Metatheria (marsupials) infraclass, such as some small dasyurids of the Antechinus genus [6] . In the particular case of the Antechinus agilis marsupial, the differences in longevity between both genders are not dramatic [65] . On the other hand, males are semelparous, dying shortly after the mating season due to the elevated testosterone and cortisol levels associated with the competition demands of mating with several females [66] . These facts further support the hypothesis that, in species with semelparous males, females present with higher mortality rates compared to species with iteroparous males [67] . Therefore, it is reasonable to note that, as previously stated by Austad [6] , while the homogametic hypothesis of longevity may be valid in humans, it does not necessarily fully apply to all animal species. It must be taken into account that there are other concurrent mechanisms affecting the differences in longevity between genders. These mechanisms could operate simultaneously and include the insulin/IGF-1, TOR, and NF-κβ regulatory signaling pathways, along with sex hormones [68] , differential telomere shortening [69] , and dimorphic mechanisms to combat oxidative stress [8] , among others.
On the other hand, the role of epigenetics in aging and longevity has not been completely elucidated. In this respect, the age-dependent changes in genomic DNA methylation in several human tissues are now increasingly recognized as involved in aging and age-associated diseases [70] -[72] . Therefore, epigenome may suffer stochastic changes as a result of its inability to maintain established patterns of DNA methylation or histone modification. Such epimutations could alter the balance of transcription factors in the cell and therefore interfere with its transcriptional activity, provoking cell dysfunction as well as abnormal responses to diverse environmental circumstances. In the case of inactivated X, hypomethylation is evident that occurs at some point in life, thus explaining the relationship between derepression of the inactive X-linked alleles and the aforementioned age-dependent skewing of XCI [70] [71] .
Adding strength to the beneficial effect of X inactivation on women longevity, it is worth highlighting the results of studies conducted in centenarians and their offspring. These studies indicate that the smaller the deviation or inactivation skewing, or the older the age of skewing onset, the higher the probability of a longer life. Both events, the increased longevity and the so-called “healthy ageing”, which result in a smaller skewing or a greater delay in X inactivation, could be explained by the fact that, as long as the deviation process has not yet begun and the two cellular populations (Xp and Xm) remain thus in a 50:50 ratio, the compensatory effects that arise effectively counteract the expression of recessive mutant alleles [73] . On the other side of the coin, age-dependent XCI skewing can be also involved in favoring the expression of deleterious alleles, as evidenced by the late-onset of certain X-linked disorders such as Graves’ disease and Hashimoto thyroiditis, scleroderma and X-linked sideroblastic anaemia, and cancer, among others [74] -[77] .
Therefore, according to Gentilini et al. [70] , the older age of skewing onset must be correlated with both a better maintenance of DNA methylation patterns during life and a better control in signal transmission through epigenetic mechanisms and consequently, with an increased female longevity.
It has been postulated that an accelerated cellular skewing may also occur in response to conditions such as acute inflammation, cell necrosis and apoptosis, albeit its maintenance or its possible reversibility after recovery depends on how stem cells are affected [12] .
In spite of women’s longevity advantage at all ages, interesting and curious differences by gender have been shown regarding the health status of centenarian individuals, as first reported by Franceschi et al. [78] in its Italian Multicenter Study on Centenarians. Thus, centenarian men were healthier than centenarian women, presumably due to factors that does not affect them equally, i.e. genetic and immunological. In this respect, it is also possible to think that at such advanced ages, the likelihood that women express deleterious alleles increases in a remarkable manner, thanks to the age-related skewed X-inactivation.
6. Effect of XCI on Women’s Longevity in X-Linked Diseases
Considering that X-chromosome inactivation is a random process in approximately 90% of normal women and gives rise to two mosaic cell populations, the interactions between both cell types determine the “quality” of the phenotype that is expressed. This fact is especially valid in certain X-linked diseases, such as Hunter syndrome or Fabry disease. Both of these diseases are lysosomal disorders in which the normal cells of heterozygote women carriers secrete enzymes that make up for the deficient abnormal cells carrying the mutant allele. Thus, carriers usually do not manifest any symptoms. This phenomenon is called “metabolic cooperation” and is impossible to achieve in men, considering that their only X chromosome will always express the mutant allele [7] . Given that metabolic cooperation is a dose-compensation mechanism, both at the cellular and tissue levels, it can better explain women vs. men survival. This cooperation not only applies to the aforementioned instances but also to many other X-linked diseases, such as the Lesch-Nyhan neurogenetic syndrome, and some immunodeficiency diseases, such as Wiskott-Aldrich syndrome, in which it is feasible, in this vein, that the coexistence of two different cell lines in all organs promotes survival when X inactivation occurs [58] . Furthermore, some of these diseases, such as autoimmune disorders, certain kinds of anemia, primary biliary cirrhosis, and scleroderma, are late-onset, which is further proof that, according to Gentilini et al. [73] , the skewing in X-inactivation most likely acts at a genomic level to favor expression of multiple deleterious alleles.
7. X Reactivation and Possible Clinical Applications
It has been proposed that reactivation of the wild-type allele on the Xi could represent an effective therapy for women affected by X-linked disorders. This concept has become a hot research subject, given the great clinical applications.
In addition to the possibility of expressing the wild-type allele in the presence of recessive mutant alleles without any type of intervention, another biological advantage conferred to women by XCI lies in promising evidence that reactivation of the wild-type allele on the Xi is possible in some X-linked diseases. In this respect, there are a few studies conducting female induced human pluripotent stem cells (ihPSCs) undergoing reprogramming, in which both X chromosomes are active [79] [80] . When obtained from patients themselves, post-XCI ihPSCs are also epigenetically dynamic and, therefore, reprogrammable. Due to the randomness of XCI, their progeny will consist of both affected (carrying the mutant allele) and normal cells (carrying the wild-type allele), conferring great potential as a source for cell therapy [81] . It has been postulated that treatment with normal cells derived from ihPSCs could effectively restore the function of mutant genes, thus improving the symptoms associated with X-linked disorders. This strategy has been proposed as an alternative for treating women suffering from the neurodevelopmental disorder known as Rett syndrome. These patients are heterozygous for mutations in the X-linked MECP2 (methyl-CpG binding protein 2) gene, thus presenting with cellular mosaicism: some of their cells express the wild-type allele, while others express the mutant MECP2 allele [82] . This strategy could also be applied to other X-linked diseases, such as fragile X, Wiskott-Aldrich, Lesch-Nyhan, and Coffin-Lowry syndromes, along with Duchenne muscular dystrophy and α-thalassemia.
Thus, in the context of regenerative medicine, the reactivation of Xi-linked genes constitutes a plausible treatment strategy for diseases arising due to mutations in X-linked genes in women. For men, it is evident that X-linked mutant alleles will always be expressed, therefore requiring the development of different silencing strategies.
References
- vB Hjelmborg, J., Iachine, I., Skytthe, A., Vaupel, J.W., McGue, M., Koskenvuo, M., et al. (2006) Genetic Influence on Human Lifespan and Longevity. Human Genetics, 119, 312-321.
- Kenyon, C. (2005) The Plasticity of Aging: Insights from Long-Lived Mutants. Cell, 120, 449-460. http://dx.doi.org/10.1016/j.cell.2005.02.002
- Christensen, K., Johnson, T.E. and Vaupel, J.W. (2006) The Quest for Genetic Determinants of Human Longevity: Challenges and Insights. Nature Reviews Genetics, 7, 436-448. http://dx.doi.org/10.1038/nrg1871
- United Nations, Department of Economic and Social Affairs/Population Division (2012) World Mortality Report 2011. http://www.un.org/en/development/desa/population/publications/pdf/mortality/worldMortalityReport2011.pdf
- Lyon, M.F. (1961) Gene Action in the X-Chromosome of the Mouse (Musmusculus L.). Nature, 190, 372-373. http://dx.doi.org/10.1038/190372a0
- Austad, S.N. (2006) Why Women Live Longer than Men: Sex Differences in Longevity. Gender Medicine, 3, 79-92. http://dx.doi.org/10.1016/S1550-8579(06)80198-1
- Migeon, B.R. (2007) Why Females Are Mosaics, X-Chromosome Inactivation, and Sex Differences in Disease. Gender Medicine, 4, 97-105. http://dx.doi.org/10.1016/S1550-8579(07)80024-6
- Seifarth, J.E., McGowan, C.L. and Milne, K.J. (2012) Sex and Life Expectancy. Gender Medicine, 9, 390-401. http://dx.doi.org/10.1016/j.genm.2012.10.001
- Graves, J.A. and Gartler, S.M. (1986) Mammalian X Chromosome Inactivation Testing the Hypothesis of Transcriptional Control. Somatic Cell and Molecular Genetics, 12, 275-280. http://dx.doi.org/10.1007/BF01570786
- Christensen, K., Ørstavik, K.H. and Vaupel, J.W. (2001) The X Chromosome and the Female Survival Advantage. Annals of the New York Academy of Sciences, 954, 175-183. http://dx.doi.org/10.1111/j.1749-6632.2001.tb02752.x
- van den Berg, I.M., Laven, J.S., Stevens, M., Jonkers, I., Galjaard, R.J., Gribnau, J., et al. (2009) X Chromosome Inactivation Is Initiated in Human Preimplantation Embryos. The American Journal of Human Genetics, 84, 771-779. http://dx.doi.org/10.1016/j.ajhg.2009.05.003
- Spolarics, Z. (2007) The X-Files of Inflammation: Cellular Mosaicism of X-Linked Polymorphic Genes and the Female Advantage in the Host Response to Injury and Infection. Shock, 27, 597-604. http://dx.doi.org/10.1097/SHK.0b013e31802e40bd
- Tower, J. and Arbeitman, M. (2009) The Genetics of Gender and Life Span. Journal of Biology, 8, 38. http://dx.doi.org/10.1186/jbiol141
- MapViewer Annotation Release 104. http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=9606&build=104.0
- Shevchenko, A.I., Zakharova, I.S. and Zakian, S.M. (2013) The Evolutionary Pathway of X Chromosome Inactivation in Mammals. ActaNaturae, 5, 40-53.
- Lengner, C.J., Gimelbrant, A.A., Erwin, J.A., Cheng, A.W., Guenther, M.G., Welstead, G.G., et al. (2010) Derivation of Pre-X Inactivation Human Embryonic Stem Cells under Physiological Oxygen Concentrations. Cell, 141, 872-883. http://dx.doi.org/10.1016/j.cell.2010.04.010
- Penny, G.D., Kay, G.F., Sheardown, S.A., Rastan, S. and Brockdorff, N. (1996) Requirement for Xist in X Chromosome Inactivation. Nature, 379, 131-137. http://dx.doi.org/10.1038/379131a0
- Zhao, J., Sun, B.K., Erwin, J.A., Song, J.J. and Lee, J.T. (2008) Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science, 322, 750-756. http://dx.doi.org/10.1126/science.1163045
- Jeon, Y., Sarma, K. and Lee, J.T. (2012) New and Xisting Regulatory Mechanisms of X Chromosome Inactivation. Current Opinion in Genetics & Development, 22, 62-71. http://dx.doi.org/10.1016/j.gde.2012.02.007
- Wutz, A., Rasmussen, T.P. and Jaenisch, R. (2002) Chromosomal Silencing and Localization Are Mediated by Different Domains of Xist RNA. Nature Genetics, 30, 167-174. http://dx.doi.org/10.1038/ng820
- Rastan, S. and Robertson, E.J. (1985) X-Chromosome Deletions in Embryo-Derived (EK) Cell Lines Associated with Lack of X-Chromosome Inactivation. Journal of Embryology and Experimental Morphology, 90, 379-388.
- Lee, J.T. and Lu, N. (1999) Targeted Mutagenesis of Tsix Leads to Nonrandom X Inactivation. Cell, 99, 47-57. http://dx.doi.org/10.1016/S0092-8674(00)80061-6
- Johnston, C.M., Newall, A.E., Brockdorff, N. and Nesterova, T.B. (2002) Enox, a Novel Gene that Maps 10 kb Upstream of Xist and Partially Escapes X Inactivation. Genomics, 80, 236-244. http://dx.doi.org/10.1006/geno.2002.6819
- Chureau, C., Prissette, M., Bourdet, A., Barbe, V., Cattolico, L., Jones, L., Eggen, A., Avner, P. and Duret, L. (2002) Comparative Sequence Analysis of the X-Inactivation Center Region in Mouse, Human, and Bovine. Genome Research, 12, 894-908.
- Huynh, K.D. and Lee, J.T. (2003) Inheritance of a Pre-Inactivated Paternal X Chromosome in Early Mouse Embryos. Nature, 426, 857-862. http://dx.doi.org/10.1038/nature02222
- Okamoto, I., Otte, A.P., Allis, C.D., Reinberg, D. and Heard, E. (2004) Epigenetic Dynamics of Imprinted X Inactivation during Early Mouse Development. Science, 303, 644-649. http://dx.doi.org/10.1126/science.1092727
- Patrat, C., Okamoto, I., Diabangouaya, P., Vialon, V., Le Baccon, P., Chow, J. and Heard, E. (2009) Dynamic Changes in Paternal X-Chromosome Activity during Imprinted X-Chromosome Inactivation in Mice. Proceedings of the National Academy of Sciences of the United States of America, 106, 5198-5203. http://dx.doi.org/10.1073/pnas.0810683106
- Navarro, P., Oldfield, A., Legoupi, J., Festuccia, N., Dubois, A., Attia, M., Schoorlemmer, J., Rougeulle, C., Chambers, I. and Avner, P. (2010) Molecular Coupling of Tsix Regulation and Pluripotency. Nature, 468, 457-460. http://dx.doi.org/10.1038/nature09496
- Ogawa, Y., Sun, B.K. and Lee, J.T. (2008) Intersection of the RNA Interference and X-Inactivation Pathways. Science, 320, 1336-1341. http://dx.doi.org/10.1126/science.1157676
- Okamoto, I., Patrat, C., Thépot, D., Peynot, N., Fauque, P., Daniel, N., Diabangouaya, P., Wolf, J.P., Renard, J.P., Duranthon, V. and Heard, E. (2011) Eutherian Mammals Use Diverse Strategies to Initiate X-Chromosome Inactivation during Development. Nature, 472, 370-374. http://dx.doi.org/10.1038/nature09872
- van den Berg, I.M., Galjaard, R.J., Laven, J.S. and van Doorninck, J.H. (2011) XCI in Preimplantation Mouse and Human Embryos: First There Is Remodeling. Human Genetics, 130, 203-215. http://dx.doi.org/10.1007/s00439-011-1014-9
- Dvash, T., Lavon, N. and Fan, G.P. (2010) Variations of X Chromosome Inactivation Occur in Early Passages of Female Human Embryonic Stem Cells. PLoS One, 5, e11330. http://dx.doi.org/10.1371/journal.pone.0011330
- Jeon, Y. and Lee, J.T. (2011) YY1 Tethers Xist RNA to the Inactive X Nucleation Center. Cell, 146, 119-133. http://dx.doi.org/10.1016/j.cell.2011.06.026
- Escamilla-Del-Arenal, M., da Rocha, S.T. and Heard, E. (2011) Evolutionary Diversity and Developmental Regulation of X-Chromosome Inactivation. Human Genetics, 130, 307-327. http://dx.doi.org/10.1007/s00439-011-1029-2
- Chow, J.C., Ciaudo, C., Fazzari, M.J., Mise, N., Servant, N., Glass, J.L., et al. (2010) LINE-1 Activity in Facultative Heterochromatin Formation during X Chromosome Inactivation. Cell, 141, 956-969. http://dx.doi.org/10.1016/j.cell.2010.04.042
- Heard, E., Rougeulle, C., Arnaud, D., Avner, P., Allis, C.D. and Spector, D.L. (2001) Methylation of Histone H3 at Lys-9 Is an Early Mark on the X Chromosome during X Inactivation. Cell, 107, 727-738. http://dx.doi.org/10.1016/S0092-8674(01)00598-0
- Kohlmaier, A., Savarese, F., Lachner, M., Martens, J., Jenuwein, T. and Wutz, A. (2004) A Chromosomal Memory Triggered by Xist Regulates Histone Methylation in X Inactivation. PLoS Biology, 2, e171. http://dx.doi.org/10.1371/journal.pbio.0020171
- Ohhata, T. and Wutz, A. (2013) Reactivation of the Inactive X Chromosome in Development and Reprogramming. Cellular and Molecular Life Sciences, 70, 2443-2461. http://dx.doi.org/10.1007/s00018-012-1174-3
- Mermoud, J.E., Popova, B., Peters, A.H., Jenuwein, T. and Brockdorff, N. (2002) Histone H3 Lysine 9 Methylation Occurs Rapidly at the Onset of Random X Chromosome Inactivation. Current Biology, 12, 247-251. http://dx.doi.org/10.1016/S0960-9822(02)00660-7
- Plath, K., Fang, J., Mlynarczyk-Evans, S.K., Cao, R., Worringer, K.A., Wang, H.B., de la Cruz, C.C., Otte, A.P., Panning, B. and Zhang, Y. (2003) Role of Histone H3 Lysine 27 Methylation in X Inactivation. Science, 300, 131-135. http://dx.doi.org/10.1126/science.1084274
- Ryba, T., Hiratani, I., Lu, J.J., Itoh, M., Kulik, M., Zhang, J.F., Schulz, T.C., Robins, A.J., Dalton, S. and Gilbert, D.M. (2010) Evolutionarily Conserved Replication Timing Profiles Predict Long-Range Chromatin Interactions and Distinguish Closely Related Cell Types. Genome Research, 20, 761-770. http://dx.doi.org/10.1101/gr.099655.109
- Agrelo, R., Souabni, A., Novatchkova, M., Haslinger, C., Leeb, M., Komnenovic, V., et al. (2009) SATB1 Defines the Developmental Context for Gene Silencing by Xist in Lymphoma and Embryonic Cells. Developmental Cell, 16, 507- 516. http://dx.doi.org/10.1016/j.devcel.2009.03.006
- Panova, A.V., Nekrasov, E.D., Lagarkova, M.A., Kiselev, S.L. and Bogomazova, A.N. (2013) Late Replication of the Inactive X Chromosome Is Independent of the Compactness of Chromosome Territory in Human Pluripotent Stem Cells. Acta Naturae, 5, 54-61.
- Nicodemi, M. and Prisco, A. (2007) Symmetry-Breaking Model for X-Chromosome Inactivation. Physical Review Letters, 98, 108104. http://dx.doi.org/10.1103/PhysRevLett.98.108104
- Mlynarczyk-Evans, S., Royce-Tolland, M., Alexander, M.K., Andersen, A.A., Kalantry, S., Gribnau, J. and Panning, B. (2006) X Chromosomes Alternate between Two States Prior to Random X-Inactivation. PLoS Biology, 4, e159. http://dx.doi.org/10.1371/journal.pbio.0040159
- Bacher, C.P., Guggiari, M., Brors, B., Augui, S., Clerc, P., Avner, P., Eils, R. and Heard, E. (2006) Transient Colocalization of X-Inactivation Centres Accompanies the Initiation of X Inactivation. Nature Cell Biology, 8, 293-299. http://dx.doi.org/10.1038/ncb1365
- Augui, S., Filion, G.J., Huart, S., Nora, E., Guggiari, M., Maresca, M., Stewart, A.F. and Heard, E. (2007) Sensing X Chromosome Pairs before X Inactivation via a Novel X-Pairing Region of the Xic. Science, 318, 1632-1636. http://dx.doi.org/10.1126/science.1149420
- Barakat, T.S., Jonkers, I., Monkhorst, K. and Gribnau, J. (2010) X-Changing Information on X Inactivation. Experimental Cell Research, 316, 679-687. http://dx.doi.org/10.1016/j.yexcr.2010.01.015
- Masui, O., Bonnet, I., Le Baccon, P., Brito, I., Pollex, T., Murphy, N., Hupé, P., Barillot, E. and Belmont, A.S. (2011) Live-Cell Chromosome Dynamics and Outcome of X Chromosome Pairing Events during ES Cell Differentiation. Cell, 145, 447-458. http://dx.doi.org/10.1016/j.cell.2011.03.032
- Scialdone, A. and Nicodemi, M. (2008) Mechanics and Dynamics of X-Chromosome Pairing at X Inactivation. PLoS Computational Biology, 4, e1000244. http://dx.doi.org/10.1371/journal.pcbi.1000244
- Gartler, S.M. and Riggs, A.D. (1983) Mammalian X-Chromosome Inactivation. Annual Review of Genetics, 17, 155- 190. http://dx.doi.org/10.1146/annurev.ge.17.120183.001103
- Engreitz, J.M., Pandya-Jones, A., McDonel, P., Shishkin, A., Sirokman, K., Surka, C., Kadri, S., Xing, J., Goren, A., Lander, E.S., Plath, K. and Guttman, M. (2013) The Xist lncRNA Exploits Three-Dimensional Genome Architecture to Spread across the X Chromosome. Science, 341, 1237973. http://dx.doi.org/10.1126/science.1237973
- Chan, K.M., Zhang, H., Malureanu, L., van Deursen, J. and Zhang, Z. (2011) Diverse Factors Are Involved in Maintaining X Chromosome Inactivation. Proceedings of the National Academy of Sciences of the United States of America, 108, 16699-16704. http://dx.doi.org/10.1073/pnas.1107616108
- Blewitt, M.E., Gendrel, A.V., Pang, Z., Sparrow, D.B., Whitelaw, N., Craig, J.M., Apedaile, A., Hilton, D.J., Dunwoodie, S.L., Brockdorff, N., Kay, G.F. and Whitelaw, E. (2008) SmcHD1, Containing a Structural-Maintenance-ofChromosomes Hinge Domain, Has a Critical Role in X Inactivation. Nature Genetics, 40, 663-669. http://dx.doi.org/10.1038/ng.142
- Mould, A.W., Pang, Z., Pakusch, M., Tonks, I.D., Stark, M., Carrie, D., Mukhopadhyay, P., Seidel, A., Ellis, J.J., Deakin, J., Wakefield, M.J., Krause, L., Blewitt, M.E. and Kay, G.F. (2013) Smchd1 Regulates a Subset of Autosomal Genes Subject to Monoallelic Expression in Addition to Being Critical for X Inactivation. Epigenetics & Chromatin, 6, 19. http://dx.doi.org/10.1186/1756-8935-6-19
- Gendrel, A.V., Apedaile, A., Coker, H., Termanis, A., Zvetkova, I., Godwin, J., et al. (2012) Smchd1-Dependent and -Independent Pathways Determine Developmental Dynamics of CpG Island Methylation on the Inactive X Chromosome. Developmental Cell, 23, 265-279. http://dx.doi.org/10.1016/j.devcel.2012.06.011
- Wong, C.C.Y., Caspi, A., Williams, B., Houts, R., Craig, I.W. and Mill, J. (2011) A Longitudinal Twin Study of Skewed X Chromosome-Inactivation. PLoS One, 6, e17873. http://dx.doi.org/10.1371/journal.pone.0017873
- Christensen, K., Kristiansen, M., Hagen-Larsen, H., Skytthe, A., Bathum, L., Jeune, B., Andersen-Ranberg, K., Vaupel, J.W. and Orstavik, K.H. (2000) X-Linked Genetic Factors Regulate Hematopoietic Stem-Cell Kinetics in Females. Blood, 95, 2449-2451.
- Sharp, A., Robinson, D. and Jacobs, P. (2000) Ageand Tissue-Specific Variation of X Chromosome Inactivation Ratios in Normal Women. Human Genetics, 107, 343-349. http://dx.doi.org/10.1007/s004390000382
- Avner, P. and Heard, E. (2001) X-Chromosome Inactivation: Counting, Choice and Initiation. Nature Reviews Genetics, 2, 59-67. http://dx.doi.org/10.1038/35047580
- Carrel, L. and Willard, H.F. (2005) X-Inactivation Profile Reveals Extensive Variability in X-Linked Gene Expression in Females. Nature, 434, 400-404. http://dx.doi.org/10.1038/nature03479
- Al Nadaf, S., Deakin, J.E., Gilbert, C., Robinson, T.J., Graves, J.A. and Waters, P.D. (2012) A Cross-Species Comparison of Escape from X Inactivation in Eutheria: Implications for Evolution of X Chromosome Inactivation. Chromosoma, 121, 71-78. http://dx.doi.org/10.1007/s00412-011-0343-8
- Grant, J., Mahadevaiah, S.K., Khil, P., Sangrithi, M.N., Royo, H., Duckworth, J., McCarrey, J.R., VandeBerg, J.L., Renfree, M.B., Taylor, W., Elgar, G., Camerini-Otero, R.D., Gilchrist, M.J. and Turner, J.M.A. (2012) Rsx Is a Metatherian RNA with Xist-Like Properties in X-Chromosome Inactivation. Nature, 487, 254-258.
- Deakin, J.E., Chaumeil, J., Hore, T.A. and Marshall Graves, J.A. (2009) Unravelling the Evolutionary Origins of X Chromosome Inactivation in Mammals: Insights from Marsupials and Monotremes. Chromosome Research, 17, 671- 685. http://dx.doi.org/10.1007/s10577-009-9058-6
- Braithwaite, R.W. and Lee, A.K. (1979) A Mammalian Example of Semelparity. American Naturalist, 113, 151-156. http://dx.doi.org/10.1086/283372
- Naylor, R., Richardson, S.J. and McAllan, B.M. (2008) Boom and Bust: A Review of the Physiology of the Marsupial Genus Antechinus. Journal of Comparative Physiology B, 178, 545-562. http://dx.doi.org/10.1007/s00360-007-0250-8
- Kraaijeveld, K., Kraaijeveld-Smit, F.J.L. and Adcock, G.J. (2003) Does Female Mortality Drive Male Semelparity in Dasyurid Marsupials? Proceedings of the Royal Society of London. Series B, 270, S251-S253.
- Pan, Z. and Chang, C. (2012) Gender and the Regulation of Longevity: Implications for Autoimmunity. Autoimmunity Reviews, 11, A393-A403. http://dx.doi.org/10.1016/j.autrev.2011.12.004
- Barrett, E.L. and Richardson, D.S. (2011) Sex Differences in Telomeres and Lifespan. Aging Cell, 10, 913-921. http://dx.doi.org/10.1111/j.1474-9726.2011.00741.x
- Gentilini, D., Mari, D., Castaldi, D., Remondini, D., Ogliari, G., Ostan, R., et al. (2013) Role of Epigenetics in Human Aging and Longevity: Genome-Wide DNA Methylation Profile in Centenarians and Centenarians’ Offspring. Age, 35, 1961-1973. http://dx.doi.org/10.1007/s11357-012-9463-1
- Gravina, S. and Vijg, J. (2010) Epigenetic Factors in Aging and Longevity. Pflügers Archiv, 459, 247-258. http://dx.doi.org/10.1007/s00424-009-0730-7
- West, J., Widschwendter, M. and Teschendorff, A.E. (2013) Distinctive Topology of Age-Associated Epigenetic Drift in the Human Interactome. Proceedings of the National Academy of Sciences of the United States of America, 110, 14138-14143. http://dx.doi.org/10.1073/pnas.1307242110
- Gentilini, D., Castaldi, D., Mari, D., Monti, D., Franceschi, C., Di Blasio, A.M. and Vitale, G. (2012) Age-Dependent Skewing of X Chromosome Inactivation Appears Delayed in Centenarians’ Offspring. Is There a Role for Allelic Imbalance in Healthy Aging and Longevity? Aging Cell, 11, 277-283. http://dx.doi.org/10.1111/j.1474-9726.2012.00790.x
- Simmonds, M.J., Kavvoura, F.K., Brand, O.J., Newby, P.R., Jackson, L.E., Hargreaves, C.E., et al. (2014) Skewed X Chromosome Inactivation and Female Preponderance in Autoimmune Thyroid Disease: An Association Study and Meta-Analysis. Journal of Clinical Endocrinology & Metabolism, 99, E127-E131. http://dx.doi.org/10.1210/jc.2013-2667
- Uz, E., Loubiere, L.S., Gadi, V.K., Ozbalkan, Z., Stewart, J., Nelson, J.L. and Ozcelik, T. (2008) Skewed X-Chromosome Inactivation in Scleroderma. Clinical Reviews in Allergy & Immunology, 34, 352-355. Http://dx.doi.org/10.1007/S12016-007-8044-z
- Cazzola, M., May, A., Bergamaschi, G., Cerani, P., Rosti, V. and Bishop, D.F. (2000) Familial-Skewed X-Chromosome Inactivation as a Predisposing Factor for Late-Onset X-Linked Sideroblastic Anemia in Carrier Females. Blood, 96, 4363-4365.
- Chen, H.T., Wu, Y.C., Chen, S.T., Tsai, H.C. and Chien, Y.C. (2014) Androgen Receptor CAG Repeats, Non-Random X Chromosome Inactivation, and Loss of Heterozygosity at Xq25 in Relation to Breast Cancer Risk. BMC Cancer, 14, 144.
- Franceschi, C., Motta, L., Valensin, S., Rapisarda, R., Franzone, A., Berardelli, M., et al. (2000) Do Men and Women Follow Different Trajectories to Reach Extreme Longevity? Italian Multicenter Study on Centenarians (IMUSCE). Aging (Milano), 12, 77-84.
- Hanna, J., Cheng, A.W., Saha, K., Kim, J., Lengner, C.J., Soldner, F., Cassady, J.P., Muffat, J., Carey, B.W. and Jaenisch, R. (2010) Human Embryonic Stem Cells with Biological and Epigenetic Characteristics Similar to Those of Mouse ESCs. Proceedings of the National Academy of Sciences of the United States of America, 107, 9222-9227. http://dx.doi.org/10.1073/pnas.1004584107
- Wang, W., Yang, J., Liu, H., Lu, D., Chen, X., Zenonos, Z., Campos, L.S., Rad, R., Guo, G., Zhang, S.J., Bradley, A. and Liu, P.T. (2011) Rapid and Efficient Reprogramming of Somatic Cells to Induced Pluripotent Stem Cells by Retinoic Acid Receptor Gamma and Liver Receptor Homolog 1. Proceedings of the National Academy of Sciences of the United States of America, 108, 18283-18288. http://dx.doi.org/10.1073/pnas.1100893108
- Tchieu, J., Kuoy, E., Chin, M.H., Trinh, H., Patterson, M., Sherman, S.P., et al. (2010) Female Human iPSCs Retain an Inactive X Chromosome. Cell Stem Cell, 7, 329-342. http://dx.doi.org/10.1016/j.stem.2010.06.024
- Cheung, A.Y., Horvath, L.M., Carrel, L. and Ellis, J. (2012) X-Chromosome Inactivation in Rett Syndrome Human Induced Pluripotent Stem Cells. Frontiers in Psychiatry, 3, 24. http://dx.doi.org/10.3389/fpsyt.2012.00024