Advances in Bioscience and Biotechnology
Vol.5 No.3(2014), Article ID:42705,14 pages DOI:10.4236/abb.2014.53028
Repression of oxidative stress/reactive metabolite regulated gene expression is associated with conversion of carbamazepine into a hepatotoxicant in LPS and DSS rat models
![]()
Drug Safety Sciences, Janssen Pharmaceutical Research and Development, LLC, Raritan, USA
Email: mmcmilli@its.jnj.com
Copyright © 2014 Angelique M. Leone et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Angelique M. Leone et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 30 October 2013; revised 14 January 2014; accepted 27 January 2014
KEYWORDS: Hepatotoxicity; Gene Expression; Oxidative Stress/Reactive Metabolites (OS/RM); LPS; DSS
ABSTRACT
Idiosyncratic hepatotoxicity accounts for many drug failures in the clinic and is a leading cause for blackboxed and withdrawn drugs. This toxicity has proven difficult to predict preclinically, but correlates with oxidative stress/reactive metabolites (OS/RM). As noted previously for antiepileptic compounds, many drugs causing idiosyncratic adverse drug effects are detected by OS/RM gene expression responses in the rat. In the present study, two immune activation models, low dose lipopolysaccharide (1 mg/kg IV) and 5% dextran sulphate sodium (DSS) in drinking water, were examined to determine if either would convert the non-toxic idiosyncratic toxicant carbamazepine (225 mg/kg) into a rat hepatotoxicant at 24 hours. Using the low dose LPS model, about 1/3 of the carbamazepine-treated rats either showed robust ALT and AST elevations with histopathological evidence of hepatotoxicity, or died. Rats in this LPS/carbamazepine group were subdivided based on ALT values into non-responders, responders or robust responders. Whereas most carbamazepine-induced mRNAs were repressed by LPS across all rats in this group, the OS/ RM genes aflatoxin aldehyde reductase (Afar) and glutathione transferase Ya (Gstya) were repressed only in the robust responder subgroup; it is unclear whether repression of these genes contributes to or results from hepatotoxicity. The OS/RM gene microsomal epoxide hydrolase (mEphx) showed repression across all rats. NAD(P)H: menadione oxidoreductase (Nmor) is an OS/RM-responsive gene that is also induced by LPS, confounding interpretation of its changes. After pretreatment with 5% DSS at 24 hours or for 5 days, using a protocol that reportedly produces increased endotoxin absorption, carbamazepine was not converted to a hepatototoxicant in any rats. Instead, DSS produced a pronounced (2- to 6-fold) and selective potentiation of carbamazepine induction of OS/RM-responsive mRNAs. The lack of repressive effects of DSS on these mRNAs or in converting carbamazepine to a hepatotoxicant was not due to desensitization of endotoxin responses since LPS was at least as effective when administered to DSS-pretreated rats. OS/RM gene repression may contribute to development of hepatotoxicity of carbamazepine in immune activation models. Hepatotoxicity; Gene Expression; Oxidative Stress/Reactive Metabolites (OS/RM); LPS; DSS
1. INTRODUCTION
Idiosyncratic hepatotoxicity is an expensive problem for pharmaceutical companies, since this issue generally only presents in late clinical trials or during postmarketing of a new drug. Costs include full development and marketing of the drug, and particularly litigation in the United States, with little or no return on investments [1]. While the required regulatory preclinical toxicology studies easily detect drug-induced hepatotoxicity when most animals are affected, these studies fail to detect toxicity that occurs in only one of hundreds of animals, much less in 1:10,000 which is a high enough rate to doom a drug introduced into many patient populations [2]. Affected patients can have horrific outcomes including death. The link to the offending drug is often not clear, and proper medical care may be delayed. Adverse drug effects in susceptible individuals have been linked in many cases to distinct human leukocyte antigens (HLAs), suggesting a strong immune component in idiosyncratic drug toxicities [3].
Idiosyncratic hepatotoxicity probably results from multiple mechanisms but may be predictable for many classes of compounds. Several investigators have made the connection between high dose drugs with reactive metabolites and predilection to idiosyncratic toxicities [4,5]. Many companies screen the reactivity of their compounds and metabolites by covalent binding and/or glutathione conjugations to avoid potential problems and to optimize lead selection [6,7]. Transcriptomics and toxicogenomics approaches have also been used, which give a functional readout of the reactive metabolites [8,9]. Nrf2 appears to be a critical transcription factor in protecting against the oxidative stress caused by many of the reactive metabolites [10]. In vitro screening techniques with human hepatocytes and cell lines show promise for earlier detection of problematic drug candidates [11], (however we have had difficulty in producing in vivolike oxidative stress/reactive metabolite (OS/RM)-sensitive gene expression in cultured hepatocytes, unpublished data).
Most compounds that induce OS/RM in preclinical species are acutely non-toxic. Similarly humans rarely show any toxicity to such compounds. It is clear that these compounds are generally well handled, especially by detoxifying pathways in the liver. Thus there must be factors and conditions that account for the rare conversion of these rather innocuous OS/RM compounds into hepatotoxicants. In some cases, there may be genetic predisposition, for example, loss of an important OS/RMresponsive enzyme for a particular class of reactive metabolites [12]. The immune system appears critical in this adverse response, as noted by the link to HLA alleles [3]. Mild injuries may give rise to immunogenic (danger) signals that convert the immune-tolerant liver and Kupffer cells into a drug-dependent immune-reactive state, leading to autoimmune-like toxicities.
Roth and colleagues at Michigan State have popularized a model of idiosyncratic hepatotoxicity where a low, non-toxic dose of LPS is preadministered to rats prior to a drug of interest [13]. In addition to the requisite proinflammatory effects (obvious even at low dose by microarray analysis), LPS has pronounced effects on hepatic pharmacokinetics of many compounds via inhibiting many cytochrome P450s and transporters at both enzymatic and mRNA levels [14]. Results for many drugs chosen for testing in this model are difficult to interpret based on the broad effects of LPS, but this model may be a good starting point for evaluating mechanisms common to many idiosyncratic hepatotoxicants. While many of the compounds tested in this model may be inherently hepatotoxic (eg., diclofenac [15]), and pharmacokinetic effects may explain preferential hepatotoxic effects on trovafloxacin over levoquin [15] in conversion of the former, but not the latter to a hepatotoxin in this model, modification of pharmacokinetic properties is not a trivial contribution to toxicity by increasing exposure, and may contribute to some idiosyncratic responses in humans as well. Obviously, more interesting from our perspective is the possibility that innocuous drugs with robust OS/RM gene expression signatures may be converted to hepatotoxic compounds in this model. This might provide insights into why idiosyncratic hepatotoxicants are missed preclinically and increase our chances of detecting them.
A second model, less studied from the perspective of idiosyncratic hepatotoxicity, is the dextran sulfate sodium (DSS)-induced colonic inflammation model, where endotoxin enters the bloodstream secondarily to colon/ gut damage [16]. Advantages of this model are that endogenous LPS may be involved, and DSS can be administered via drinking water. Little research has focused on hepatic involvement in this model, although glucosamine became less hepatotoxic after DSS administration, a paradoxical effect that was attributed to desensitization of toll (LPS) receptors over time (a limitation with all LPS models [16]).
In the present study we have compared the effect of the idiosyncratic drug carbamazepine in these two immune modulation models. Carbamazepine has strong OS/RM effects on rat liver at a dose of 225 mg/kg, and is relatively non-toxic, having a very high LD50 (>4000 mg/kg, due to sedation). Despite being non-toxic preclinically, carbamazepine causes idiosyncratic hepatotoxicity in some patients, although cutaneous toxicity is much more common and may derive from the same idiosyncratic characteristics of this drug.
Preliminary accounts of this work have been presented in abstract/seminar form [17,18].
2. MATERIALS AND METHODS
2.1. Animals
Healthy male, 7-week-old Sprague-Dawley rats of about 275-gram body weight (Charles River Laboratories, Inc.) were used for these studies. The animals were individually housed, on a 12 h light/dark cycle, and fed Purina Rodent Chow ad libitum. Rats were weighed, randomized, and inspected (unhealthy or small-for-age animals were removed prior to the study) on the day prior to dosing. Rats were dosed at 3 - 5 min intervals to allow exsanguination (severing vena cava and aorta under CO2 analgesia) and necropsy at exactly 24 h for all compounds. Rats were fasted after dosing until necropsy the following morning. In all instances, the animals were humanely handled in accordance with IACUC guidelines.
2.2. Compounds
Carbamazepine, LPS and other compounds were of the highest grade obtainable from Sigma-Aldrich (St. Louis, MO). LPS derived from Escherichia coli serotype O55: B5 was used only. Dextran sulfate sodium salt MW 36,000 - 50,000 was purchased from MP Biomedicals, LLC (Solon, Ohio).
Methocel (hydroxypropyl methylcellulose; F4M grade; Dow Chemicals) was used at 0.5% (weight/volume) in water for vehicle and carbamazepine dosing. Methocel is a preferred vehicle rather than water for dosing in-house proprietary compounds, as it is inert but aids in solubility for many compounds. No methocel effects on rat liver gene expression relative to non-dosed rats were observed at 24 hours.
Previously we used carbamazepine at 20, 120 and 225 mg/kg to study OS/RM gene expression [8]; the Merck Index gives an LD50 of 4025 mg/kg for rats, so 225 mg/kg was chosen to produce a pronounced OS/RM gene expression response in the absence of toxicity. For all carbamazepine studies, rats were necropsied 24 hours after a single oral dose.
2.3. Clinical Chemistry
Blood was collected at necropsy, serum was collected after centrifugation, and either stored at −80 or assayed fresh, and ALT and AST were measured using an Advia 1800 automated chemistry system and Siemens reagents (Siemens Corporation, Washington, District of Columbia).
2.4. Histopathology
Medial lobe samples were taken from some studies from each animal, were fixed in 10% neutral-buffered formalin for approximately 48 hours and subsequently processed, embedded in paraffin, sectioned at 5 microns, mounted on glass slides, deparaffinized, and stained with hematoxylin and eosin (H&E).
2.5. RNA Isolation
The livers were removed and an approximately 200 mg strip from the medial lobe was snap-frozen in liquid nitrogen. We routinely use sections from the medial lobe of the liver to assess histopathology in 5-day toxicology studies, hence, the same region was chosen for toxicogenomics studies. Liver samples were stored at −70˚C until RNA extraction. Total RNA was extracted using Qiagen RNEasy Midi kits (Qiagen, Inc. Valencia, CA) as per kit instructions. The amount of RNA in the samples was determined spectrophotometrically by absorbance ratio at 260 and 280 nm. Quality of RNA in the samples was assessed using rRNA peaks determined by an Agilent 2100 Bioanalyzer.
2.6. RT-PCR
Probe and primer sets for mRNA markers of macrophage activation, OS/RM, cytokines, or drug metabolism endpoints (Table 1) were obtained as kits from Applied Biosystems, Life Technologies (Foster City, CA), and TaqMan One Step assays were performed as per instructions. RNA samples (quantified spectrophotometrically as above) were equally diluted to 20 ng/uL, PCR reactions performed, and probes of interest quantified by cycle number (Ct) detected. Each unit increase in cycle number was taken to be a 2-fold decrease. Results for each sample were compared to their respective controls. Ribosomal 18S RNA was used for further fine correction of RNA levels in samples.
2.7. Low Dose LPS Model
All animals received IV tail vein injections with a maximum total volume of 0.3 ml of either sterile saline or LPS 1 mg/kg in a sterile saline solution. A 23G miniset vein infusion set was used to catheterize the tail vein, and the rat was restrained in a holding device briefly during the procedure. Two hours post IV injection the animals received oral carbamazepine or methocel vehicle and at 24 hours the animals were necropsied.
2.8. DSS Model
5% DSS was dissolved in the drinking water. Bottles were used to easily monitor the consumption amount of each animal. Dose and duration of treatment was derived from the literature [19,20]. Each day the animals were weighed and clinical signs of gastrointestinal dysfunction (such as blood in the feces) were assessed. 5% DSS was provided in the drinking water for 24 hours (water bottles removed the night before) or 5 days prior to oral treatment with methocel vehicle or carbamazepine and the animals were divided into separate treatment groups by the amount of weight loss while on DSS.
To assess possible desensitization of receptors for LPS, in one experiment rats were given water or 5% DSS as above then treated with saline or LPS IV for two hours, followed by methocel vehicle or carbamazepine, then necropsied at 24 hours.
3. RESULTS
Previously we reported that compounds producing oxid-
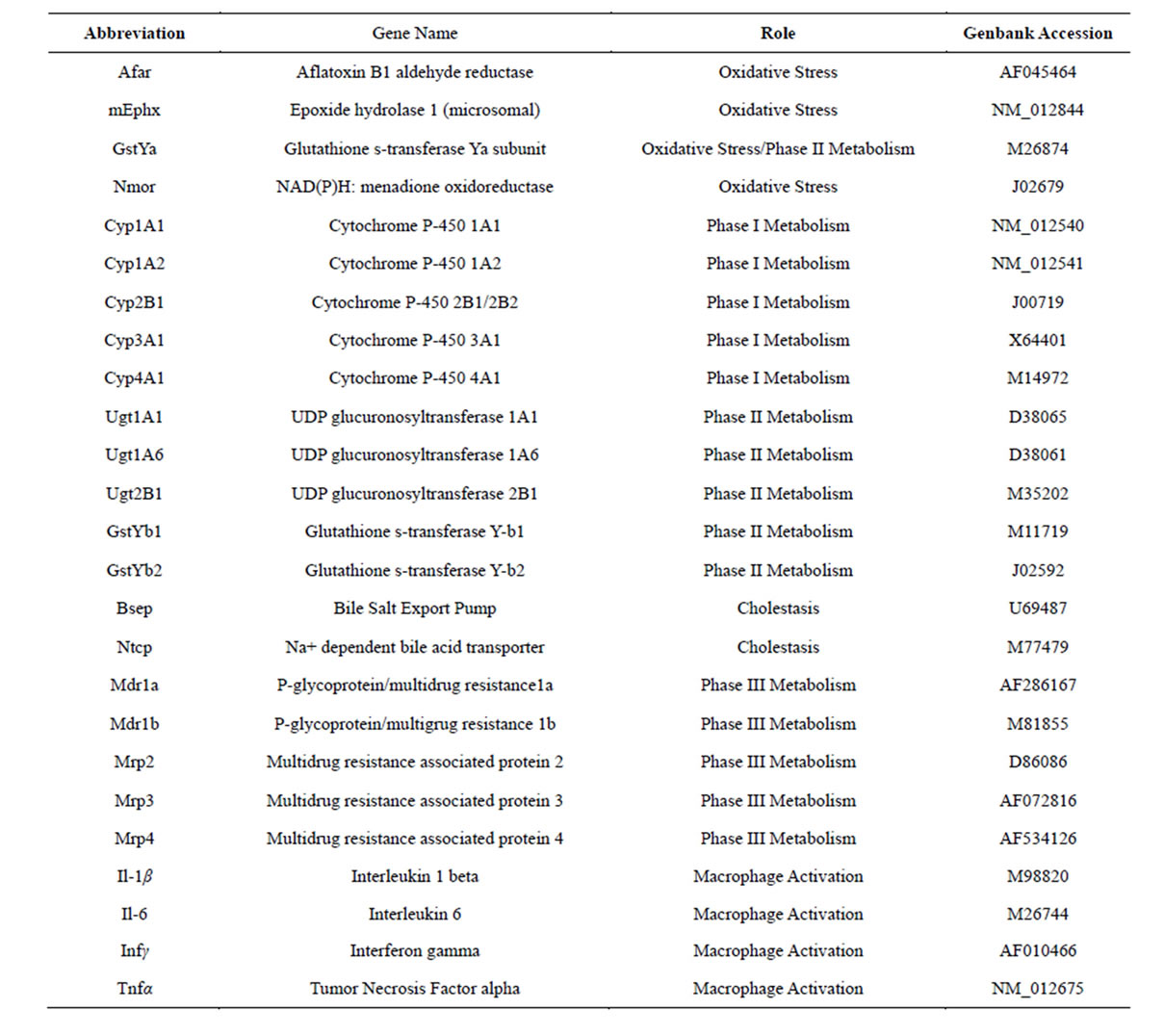
Table 1. Genes and their functions that were analyzed with RT-PCR in the present study.
ative stress due to reactive metabolites (OS/RM) produce a rat hepatic gene expression signature distinct from oxidative stress associated with macrophage activation (and damage) and with peroxisome proliferators (PPARα agonists) [21,22]. This OS/RM signature detects many non-toxic compounds that cause idiosyncratic hepatotoxicity in patients [8] (Leone et al., submitted). Hypersensitivity and idiosyncratic adverse drug effects in people appear dependent on immune cell activation and/or loss of immune-tolerance. Thus immune activation models were tested in rats to determine if a non-toxic OS/ RM-producing drug (carbamazepine) could be reproducibly converted to a hepatotoxicant. Two models were examined: the low dose LPS model popularized by Roth’s group and a DSS experimental colitis model associated with endotoxin uptake [16].
3.1. Low Dose LPS Model
In initial studies, low dose LPS (1 mg/kg IV) was compared to high dose LPS (5 mg/kg IV) and gene expression results are shown in Table 2. Low dose LPS had no effect on AST or ALT in these pilot studies and was selected for further study; although a small percentage of rats did show toxic responses in later studies, see below, no liver lesions were detected in histopathological sections from low dose LPS treated rats. Pronounced effects were observed in response to low dose LPS with many Macrophage Activator (MA) Gene Expression Signature [22] mRNAs (Table 2). High dose LPS killed about half
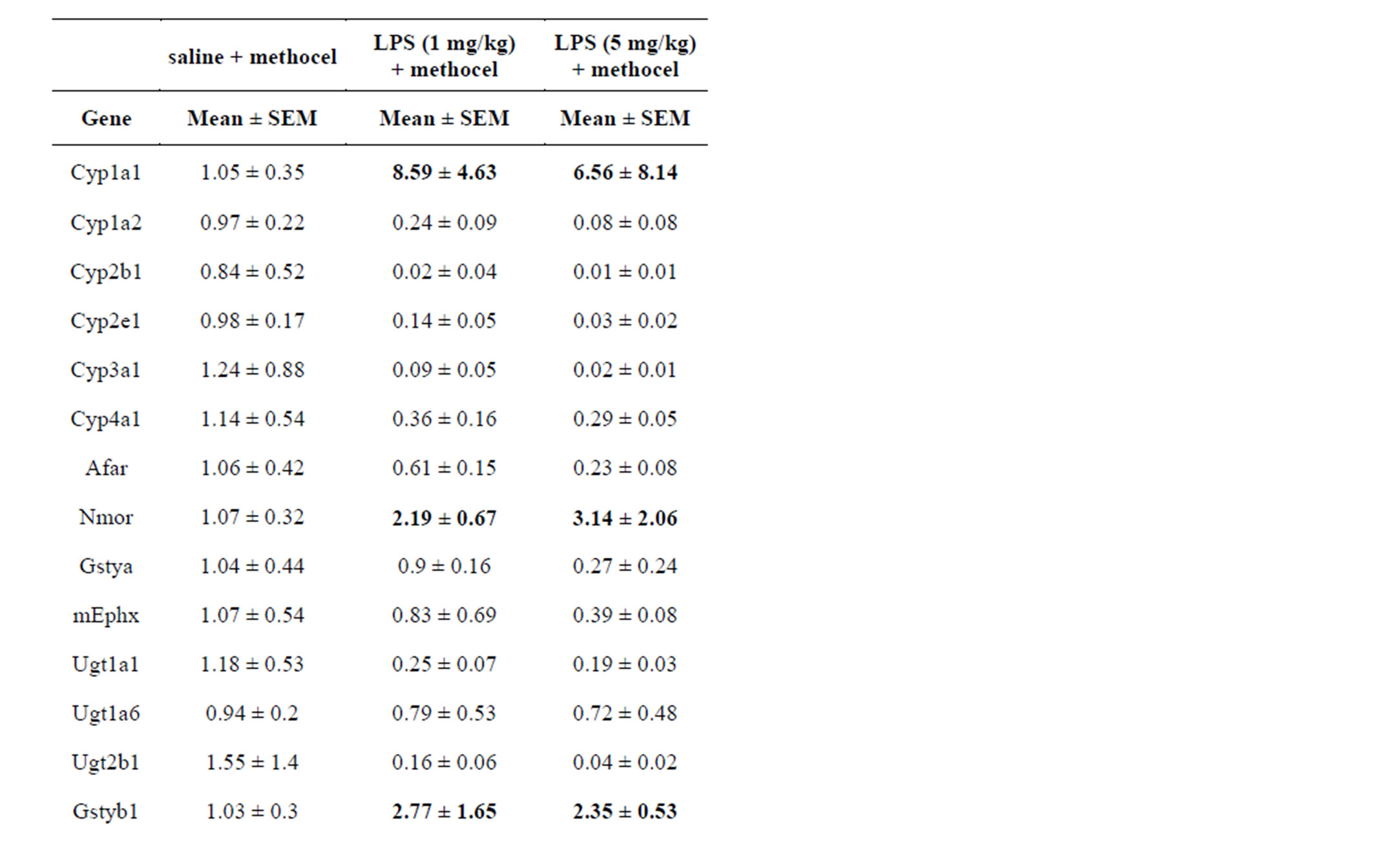
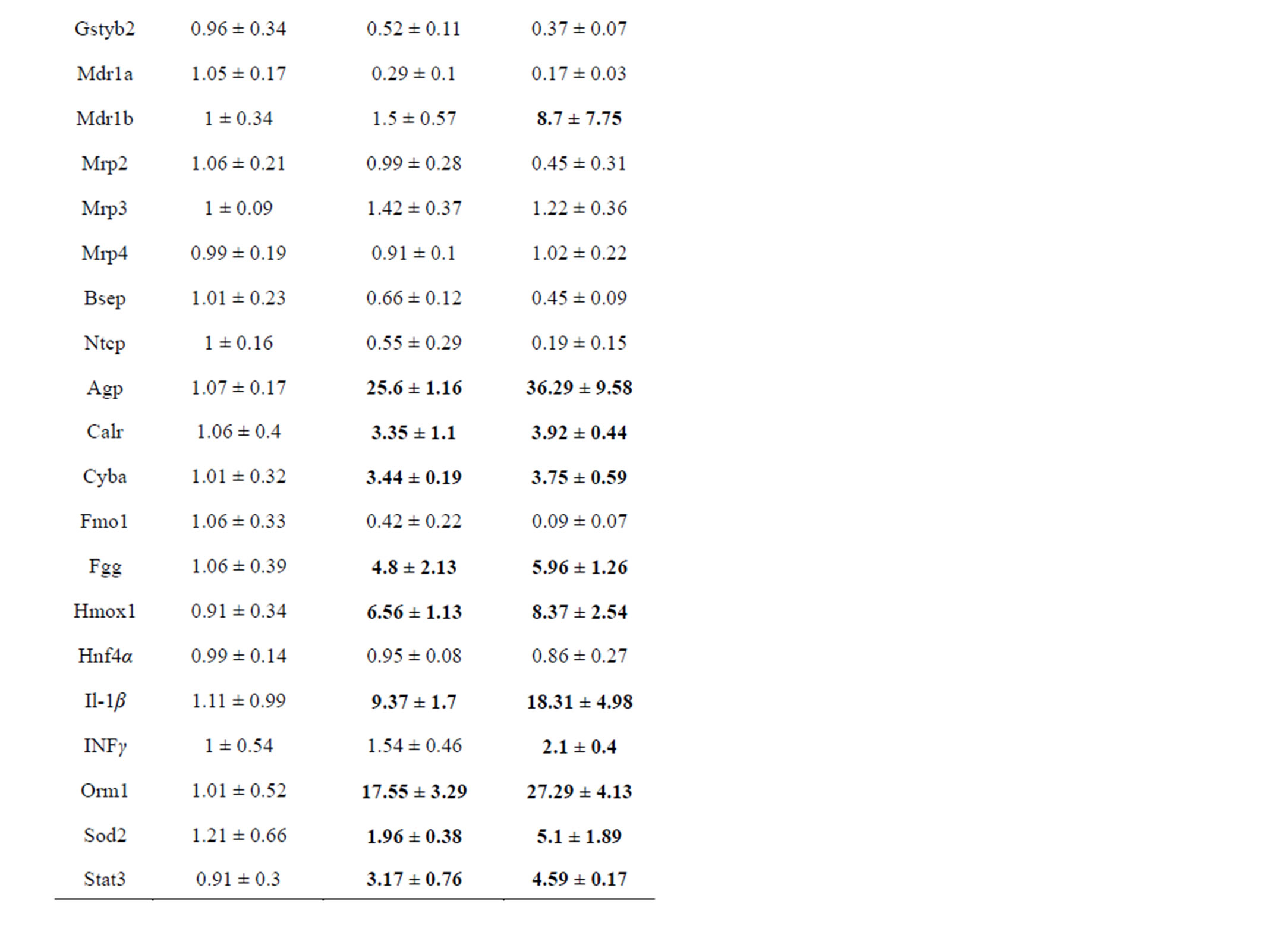

Table 2. Comparison of rat hepatic gene responses at 24 hr to low dose (1 mg/kg) and high dose (5 mg/kg) LPS.
of the injected rats, and elevated ALT (mean 168 U/L) and AST (mean 350 U/L) about 3-fold over ULN in the surviving rats. High dose LPS was generally more effective than low dose LPS at repression of genes, but was only slightly more effective at inducing MA-responsive genes (Table 2); due to scatter in the responses these differences were not significant. Several proinflammatory cytokines (interleukin 1 beta, interleukin 6, interferon gamma, and tumor necrosis factor alpha) were selected for further study, as reportedly involved in hepatotoxicity, whereas most acute phase and hepatic MA response genes (alpha 1-acid glycoprotein, calreticulin, orosomucoid, cytochrome b-245 alpha polypeptide, fibrinogen, heme oxygenase 1, superoxide dismutase 2, signal transducer and activator of transcription 3, tissue inhibitor of metalloproteases, and inducible nitric oxide synthase 2) were not further studied despite robust responses (Table 2). Despite much variability in responses, Cyp1a1, Nmor, Gstyb1 and Mdr1b mRNAs were shown to have small inductions in response to LPS in some rats (Table 2), which confounds interpretation when combined with test drugs.
As a further check of the low dose LPS, pilot experiments were run replicating the conversion of diclofenac (100 mg/kg) to a hepatotoxicant in the rat (data not shown; [15]).
Carbamazepine is a drug causing idiosyncratic reactions in patients that produces a strong OS/RM response yet is non-toxic at high dose (225 mg/kg) in rats [8,9]. Although some rats showed obvious hepatotoxicity (Figure 1) in response to combined low dose LPS + carbamazepine as expected, the response was quite variable, so the rats were divided into responders and non-responders (Table 3) to assess differences in gene expression. Of 22 rats treated with LPS + carbamazepine, 3 (14%) died (no clinical chemistries or gene expression could be determined due to autolysis), 5 (23%) showed robust ALT and AST elevations, 7 (31%) showed elevations of ALT and AST above ULN, and 7 (31%) showed
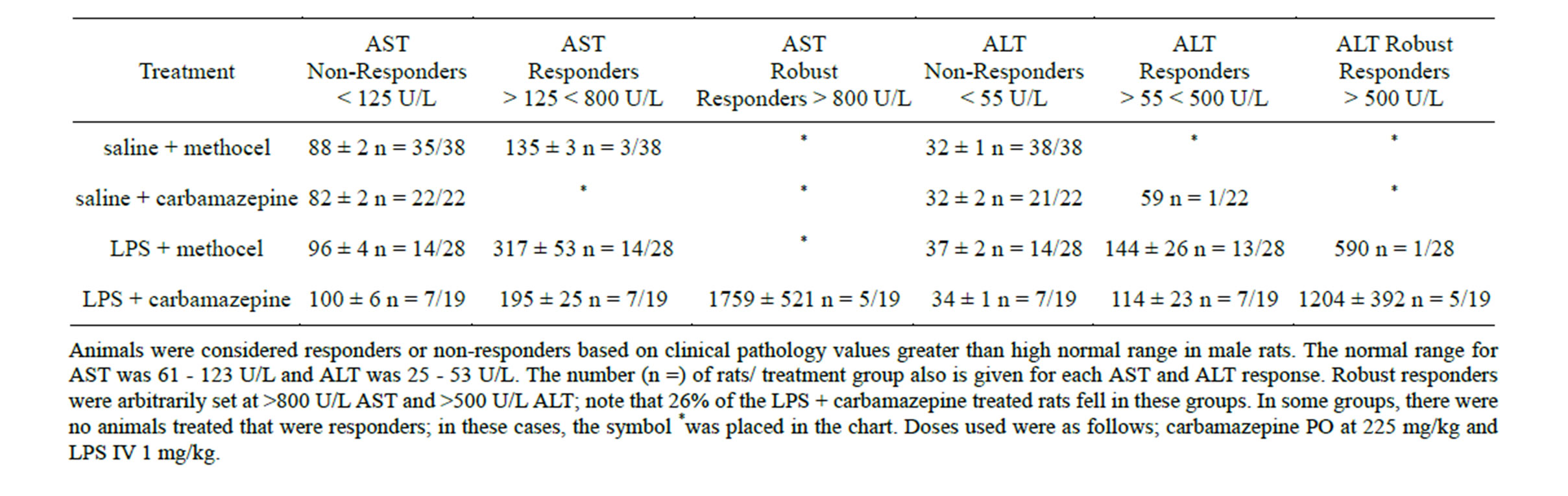
Table 3. ALT and AST values in Non-Responder, Responder, and Robust Responder Rats.
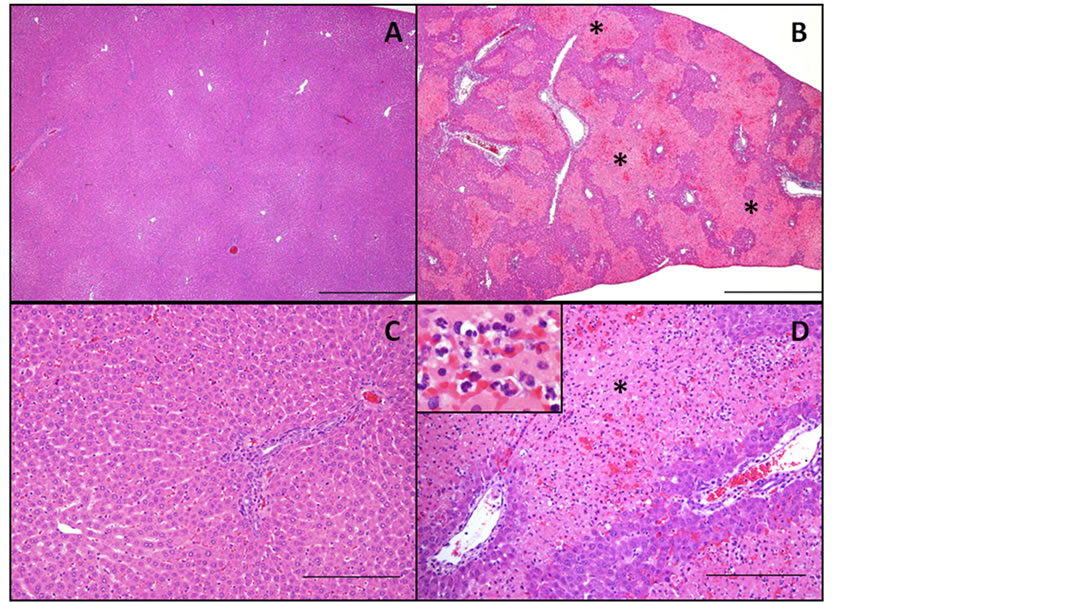
Figure 1. Histopathology in rat liver after co-treatment with low dose LPS and Carbamazepine. A & C: Liver. Rat No. 1001 treated with saline IV + methocel PO has no significant hepatocellular changes; A is 4X H&E with bar = 1 mm and C is 20X H&E with bar = 200 µm. B & D: Liver. Rat No. 6004 treated with LPS 1 mg/kg + carbamazepine 255 mg/kg PO has moderate multifocal hepatocellular necrosis (denoted with asterisks) that bridges adjacent lobules and is most prominent within the intermediate and periportal regions. The inset within D highlights the numerous neutrophils admixed with mild hemorrhage within areas of necrosis; B is 4X H&E with bar = 1 mm and D is 20X H&E with bar = 200 µm.
no change in these enzymes (Table 3). There was a positive correlation between serum increases in ALT and AST and the degree of necrosis observed in the intermediate zone of the liver (Figure 1) Despite no effects in pilot studies, low dose LPS elevated ALT and AST elevations above ULN in 14/28 (50%) rats, and robustly elevated ALT in one rat (4%; Table 3). The carbamazepine group had one (4%) of 22 rats with ALT barely above ULN, and the control vehicle group had none (Table 3).
When gene expression was examined across LPS + carbamazepine non-responder and responder groups and compared to the saline + carbamazepine group, it was clear that the robust responders showed the most severe repressions by LPS (Table 4). For OS/RM-regulated mRNAs Afar and Gstya, only the robust responder groups showed inhibition relative to the saline + carbamazepine group (Table 4). Glutathione transferase Yb2 mRNAs also showed repression only in the robust responder group. Several mRNAs showed a “response-de-
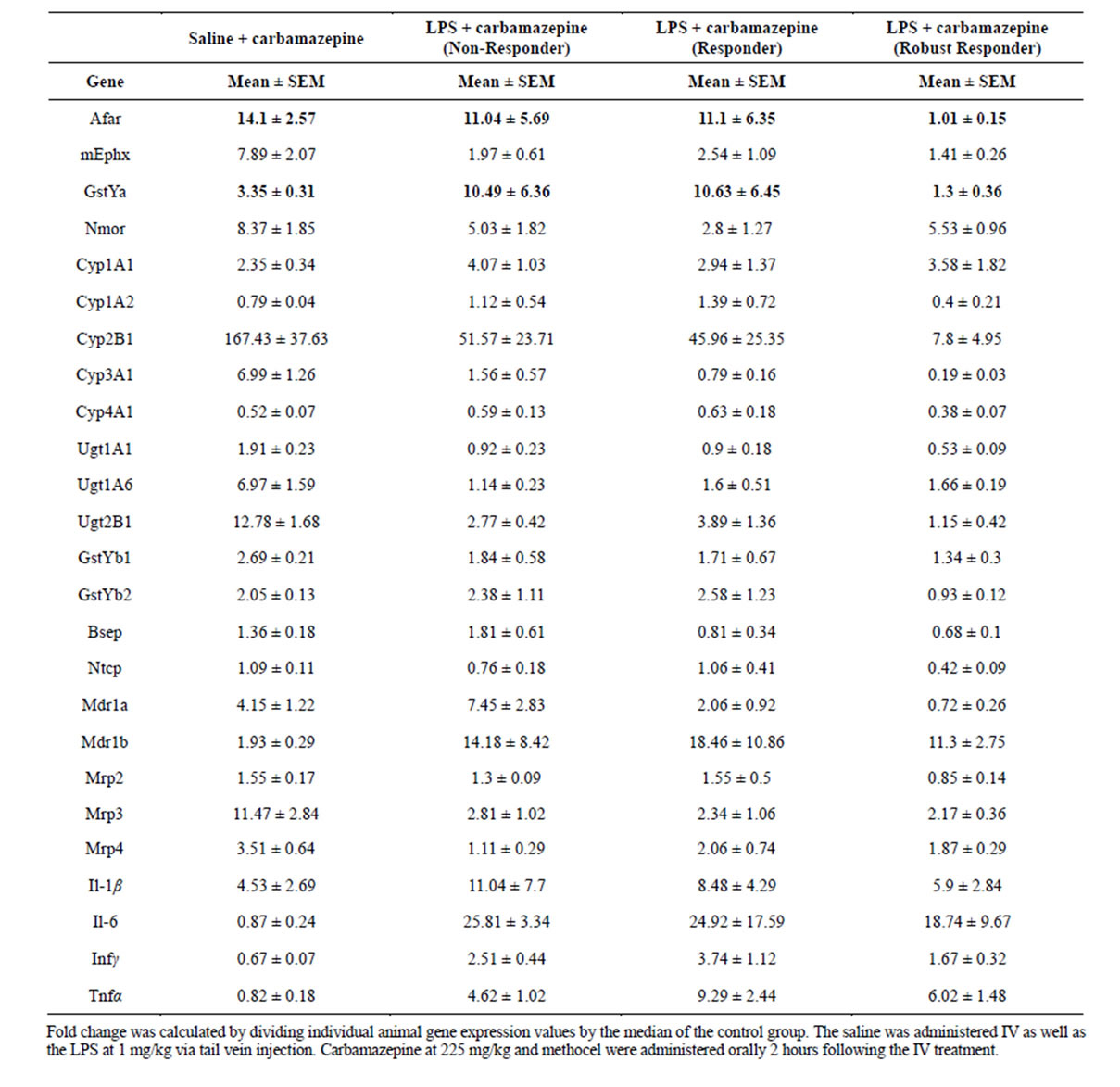
Table 4. Gene Responses to Carbamazepine and Low Dose LPS + Carbamazepine Separated by Non-Responders, Responders and Robust responders (as in Table 3).
pendent” repression with even the non-responding group showing repression and the robust responders showing the most repression: Cyp2b1, Cyp3a1, UDP glucuronyltransferase 2b1, p glycoprotein Mdr1a, and the OS/RM regulated microsomal epoxide hydrolase (mEphx, Table 4). mRNAs for UDP glucuronyltransferase 1a6 and transporters MRP3 and MRP4 were similarly repressed in all LPS + carbamazepine groups. There was no further induction of proinflammatory cytokine mRNAs in the robust responder group (generally slightly repressed relative to other LPS + carbamazepine groups). The LPSsensitive mRNAs—Cyp1a1, Nmor, Gstyb1 and Mdr1b— also showed similar responses across LPS + carbamazepine groups (Table 4).
3.2. DSS (Dextran Sulphate Sodium) Model
The hope was that 5% DSS in drinking water would provide a much more reproducible model than LPS tail vein injections. After 5 days (which due to weight loss and blood in feces was as long as feasible [16,19,20]) there was no elevation in serum ALT or AST and no conversion of carbamazepine to a hepatotoxicant by clinical chemistry measurements. As for the non-responding LPS + carbamazepine-treated animals, no histopathological lesions were observed in H&E stained liver sections from DSS + carbamazepine-treated rats (data not shown).
The second immune activation model gave gene expression results which were strikingly different from those observed with the low dose LPS model. Rats dosed with carbamazepine 225 mg/kg (24 hr) after being pretreated with 5% DSS for 5 days had a surprising pronounced increased induction of OS/RM-regulated genes (Table 5). There were mild effects of DSS alone, but DSS potentiation of carbamazepine induction of OS/RM genes ranged from 2-fold for mEphx and Nmor (and glutathione transferase Yb2), 3-fold aflatoxin aldehyde reductase and 6-fold for Gstya (Table 5). Much milder increases in carbamazepine induction were observed with Cyp, Phase II conjugation enzymes and transporter
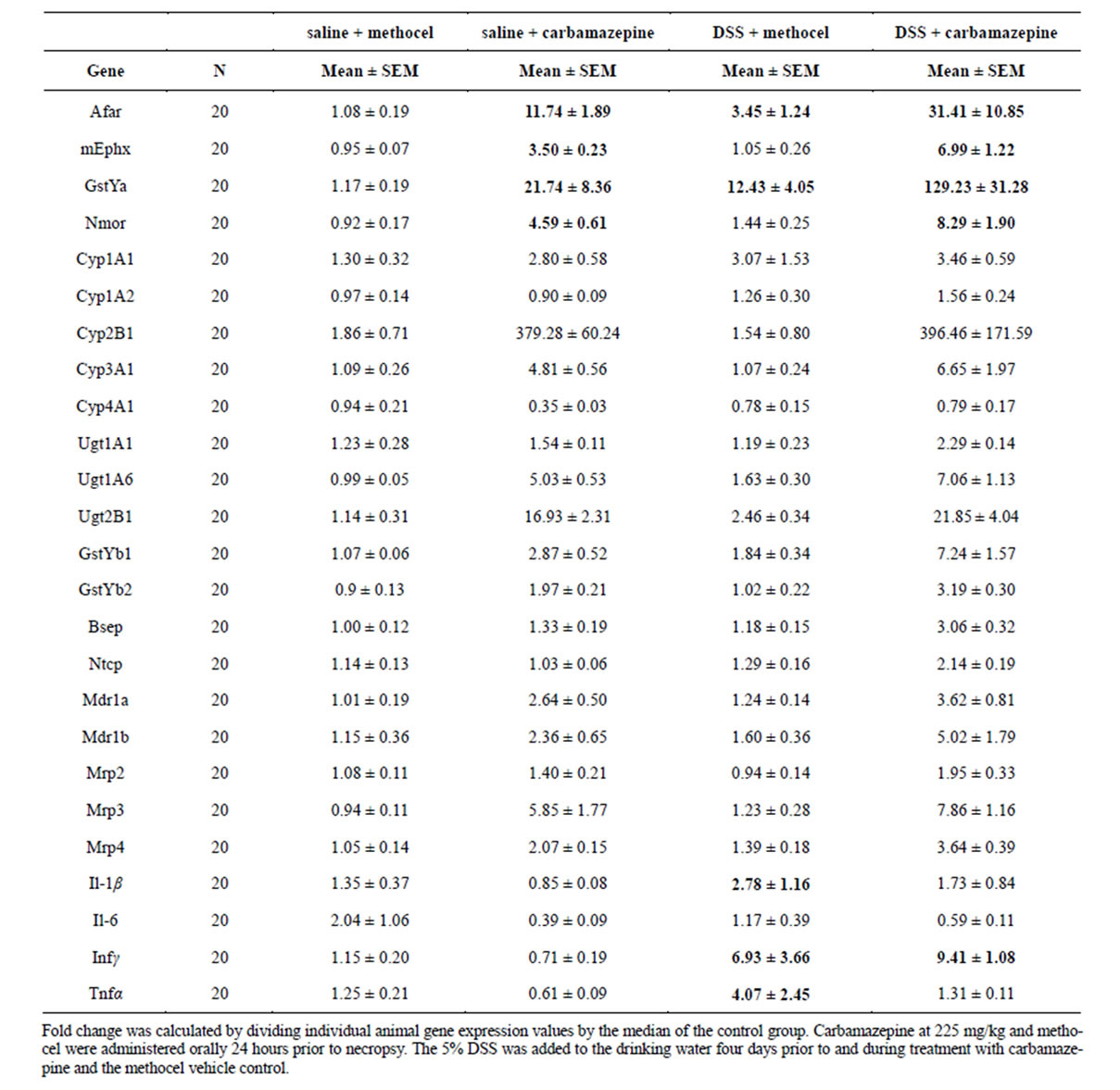
Table 5. Liver Gene Expression Responses (PCR) At 24 hours after Dosing with Carbamazepine, DSS (5 days) or the Combination of DSS (5 days) + Carbamazepine.
mRNAs. There was evidence of pro-inflammatory effects of DSS; cytokine mRNAs were induced 2.5 to 7- fold after DSS treatment (Table 5).
The groups given 5% DSS plus carbamazepine 225 mg/kg were subdivided prior to oral carbamazepine dosing by the amount of weight lost during the DSS administration. A larger gene expression response was seen in the animals that lost the most weight, but again no increases in the ALT or AST were observed in any of these groups (data not shown).
Masubuchi and Horie have proposed that prolonged DSS treatment desensitizes endotoxin receptors and pathways, and thus blocks hepatotoxicity of d-glucosamine [16]. However, when rats were administered 5% DSS in the drinking water for only 24 hours rather than 5 days, we observed a similar potentiation of carbamazepine induction of the OS/RM mRNAs and (similar potentiation of Cyp 2b1 and Mrp3 mRNAs (Table 6). Again there was no evidence of hepatotoxicity by histopathology or by ALT and AST evaluation (all DSS 24 hour data shown in Figure 2). Proinflammatory cytokine mRNA induction responses after 24 hour DSS and DSS + carbamazepine responses were variable (Tables 6 and 7). This would argue that any desensitization of the gut endotoxin response occurs quickly after DSS treatment, or that this treatment is behaving very differently from low dose LPS.
To address these possibilities, carbamazepine interactions with 5% DSS (24 hours in drinking water), LPS (1 mg/kg IV 2 hours prior), or combined treatment with DSS + LPS were examined (Table 7). In this experiment the 2- to 4-fold potentiation of carbamazepine induction was again noted for all OS/RM-responsive mRNAs except the LPS-sensitive NMOR (which was not potentiated in this experiment). Moreover Cyp2b1 and MRP3 (potentiated by DSS at 24 hours in Table 6) were repressed by DSS in this experiment (Table 7), and the DSS/carbamazepine combination elevated proinflammatory cytokines, (Table 7, but not observed in Table 6). The basis for variability in some responses across these two experiments is not clear, but may result from the mild proinflammatory response in the second experiment (Tables 6 and 7). Low dose LPS repressed carbamazepine-induced OS/RM-responsive mRNAs (and many other genes, as noted above, Tables 2, 4, and 7).
The point of this experiment was to address whether DSS had desensitized toll receptors. Clearly LPS effectively still repressed carbamazepine-induced OS/RM-responsive mRNAs, as well as other genes, and a proinflammatory cytokine response persisted (Table 7). Moreover, these repressed gene responses were associated with elevated ALT and AST in some carbamazepine/ DSS/LPS-treated rats (Figure 2), showing that a hepatotoxic response can occur in these rats “desensitized” to
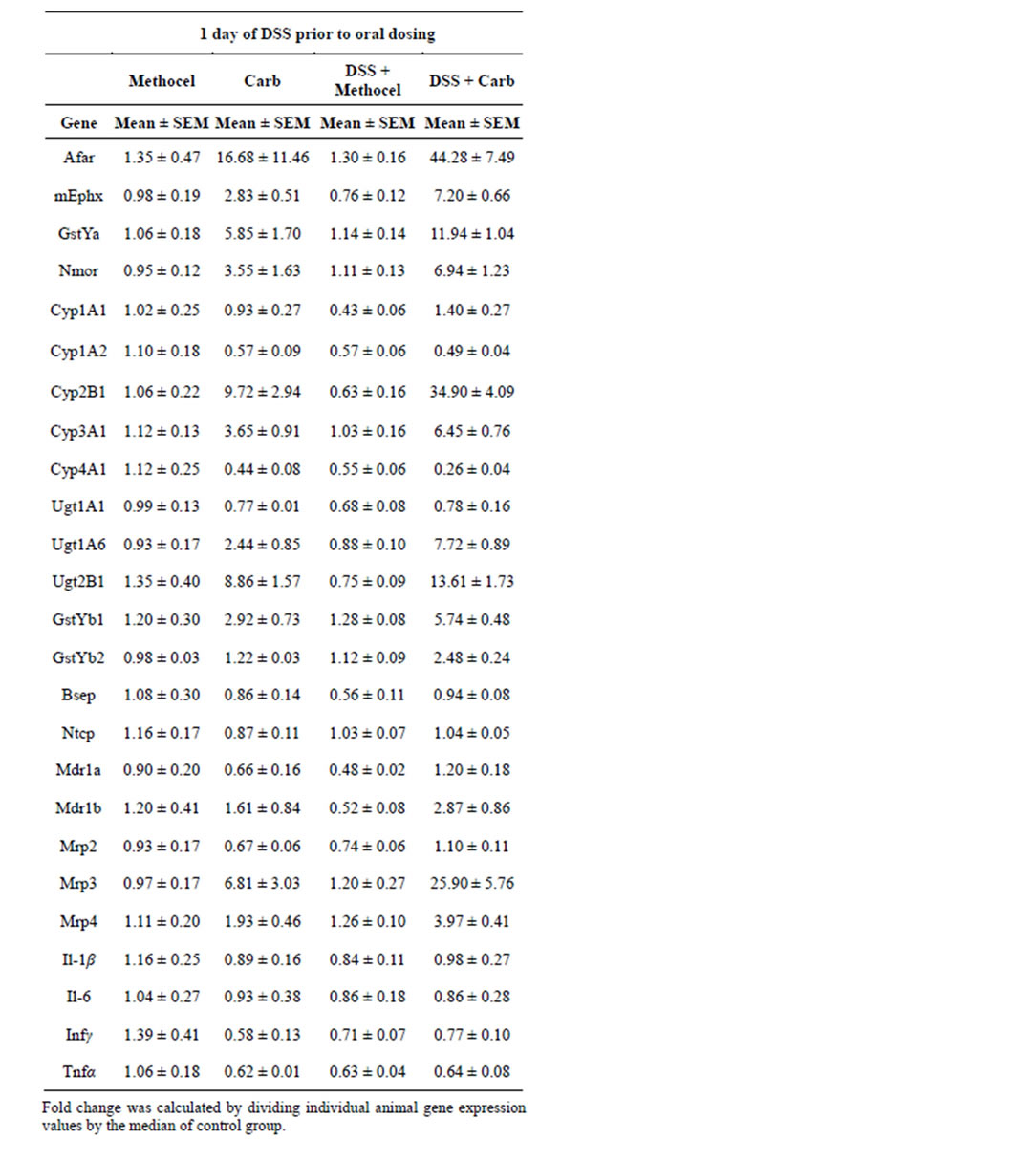
Table 6. Liver Gene Expression Responses (PCR) at 24 hours after Dosing with Carbamazepine, DSS (24 hours) or the Combination of DSS (24 hours) + Carbamazepine.
endogenous endotoxins by DSS treatment.
4. DISCUSSION
Drugs, and candidate drugs, that produce OS/RM-regulated gene expression are frequently innocuous in pre-
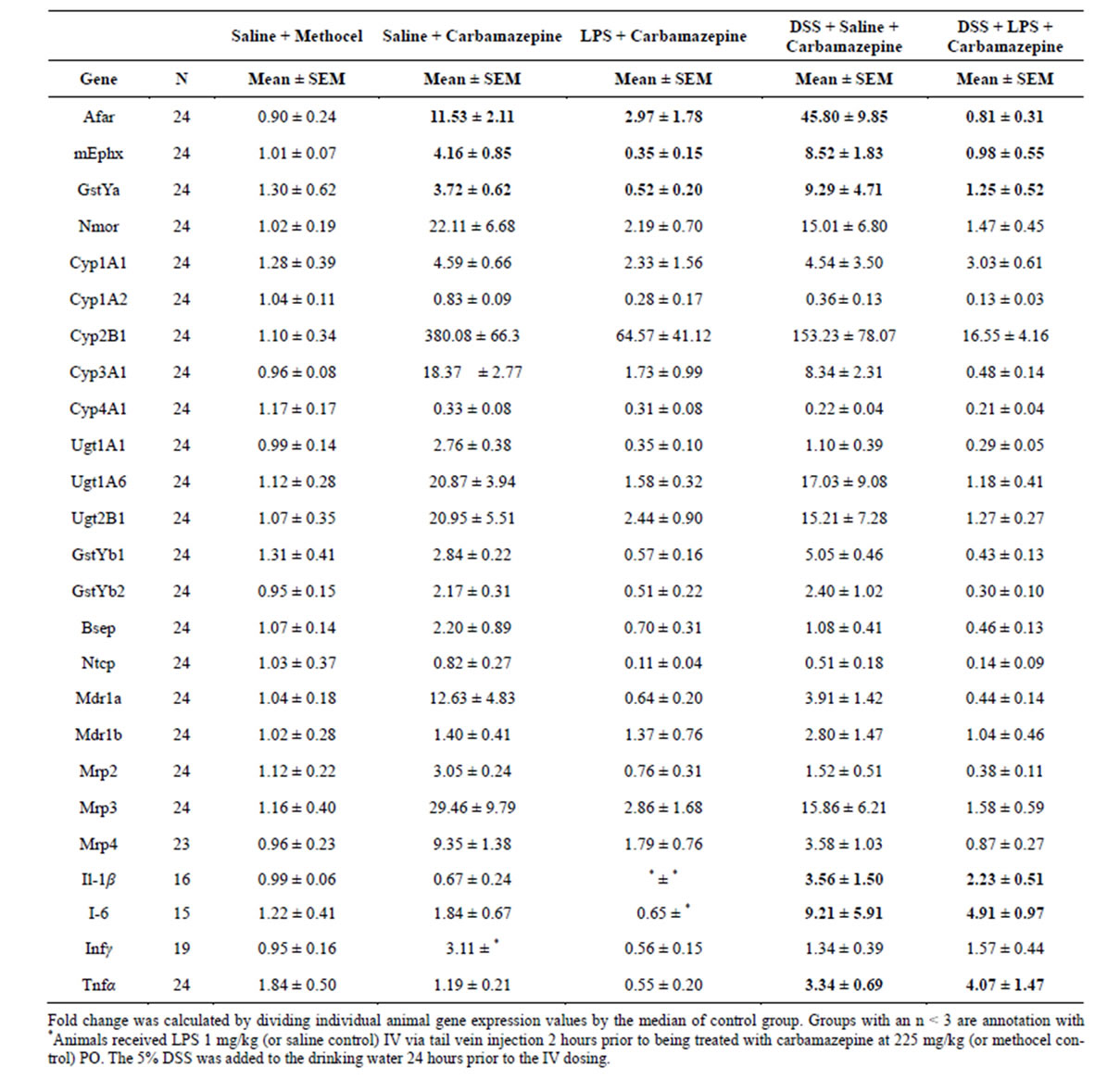
Table 7. Liver Gene Expression Responses (PCR) at 24 hours after Dosing with Carbamazepine, or the Combinations of LPS (2 hours prior) + Carbamazepine, DSS (24 hours) + Carbamazepine, or DSS + LPS + Carbamazepine.
clinical testing, but produce idiosyncratic toxicities in humans, with hepatotoxicity being a particular concern. In the present study, two models of immune activation— low dose LPS and 5% DSS in the drinking water—were evaluated to see if either or both would convert the nontoxic paradigm OS/RM drug carbamazepine [8] into a rat hepatotoxicant. Low dose LPS converted carbamazepine (225 mg/kg) into a robust toxicant but only in a third of the treated rats (and three died). However, in a pilot study with low dose LPS and diclofenac, there was much less variability and most rats showed hepatotoxicity in agreement with published results [15], so some of the variability resulted from the choice of carbamazepine. There was also considerable variability in gene expression responses in both models.
Of four OS/RM-responsive genes previously studied [8], Afar and Gstya were robustly repressed only in “robust responder” rats that showed ALT and AST elevations, and damage by histopathology. It is unknown if these repressions of protective enzyme mRNAs contri-

Figure 2. Clinical Chemistries of serum samples 24 hours after oral dosing of water or 5% DSS in drinking water, 0.5% Methocel or Carbamazepine, and IV Normal Saline or IV LPS (1 mg/kg). The normal range for AST was 61 - 123 U/L and ALT was 25 - 53 U/L; Upper Limit of Normal (ULN) is marked for AST and ALT. Data are graphed from individual rats. Doses used were as follows; carbamazepine PO at 225 mg/kg and LPS IV 1 mg/kg. Note that only the DSS/LPS/carbamazepine group shows responders consistent with hepatotoxicity.
bute to hepatotoxicity in these robust responders, or if the repressions are due to the damage in this group. Ephx mRNA was repressed in all LPS-treated groups including “non-responders”; as the most previously characterized Cyps, Phase II conjugation enzymes and transporter mRNAs [14] induced by carbamazepine; generally these repressions were more robust in the rats showing hepatotoxicity. Nmor, like Cyp1a1, Gstyb1 and Mdr1b mRNAs showed variable inductions by LPS as did the proinflammatory cytokines postulated by others to be involved in LPS-potentiated toxicities [23,24]. In multiple experiments with DSS treated rats, after both 24 hours and 5 days, there was no conversion of carbamazepine to a hepatotoxicant, but instead a robust potentiation of carbamazepine induction of the protective OS/ RM genes Afar, Gstya, Ephx and (usually) Nmor. DSS potentiation of OS/RM genes did not reflect desensitization of LPS receptors, since adding on low dose LPS produced repression and hepatotoxicity in DSS/carbamazepine-treated rats. Only OS/RM genes showed a reproducible potentiation of carbamazepine induction by DSS.
The present results generally confirm the Michigan State low dose LPS model, although our results are too variable for routine use for screening idiosyncratic potential of drug candidates. This model has been championed for paired comparisons of idiosyncratic hepatotoxicants and less toxic, but structurally similar, drugs such as ranitidine and famotidine [25] and trovafloxacin and levofloxacin [23]. Our results are also in full agreement with those from Morgan’s lab, who additionally has shown rapid downregulation of protein and enzyme responses along with mRNAs for hepatic Cyps and transporters after immune activation [14]. Repression of OS/ RM-regulated mRNAs by low dose LPS is of obvious interest when studying drug candidates that produce OS/ RM, and considerable further work is required to determine if protein and activities of these protective enzymes rapidly follow changes in mRNA as is the case for Cyps and transporters. In humans, some pathological conditions, such as psoriasis produce enough systemic cytokines to repress Cyp activities, and effective treatments can normalize these Cyp activities, (which can be problematic when people are on multiple medications [26,27]).
Although carbamazepine has been extensively studied, its toxicity remains poorly understood. Induction of the OS/RM-regulated enzyme microsomal epoxide hydroxylase by carbamazepine may protect the rat from toxicity, as a relatively long-lived epoxide of carbamazepine has been postulated to contribute to toxicity. In human in vitro models, decreased microsomal mEphx activity is associated with more carbamazepine epoxide formation and more cytotoxicity, as expected [12,28,29]. However, most adverse drug responses to carbamazepine appear to be further downstream from carbamazepine epoxide formation and covalent binding. While loss of mEphx activity by inhibition, repression of gene expression or loss of a functional allele might contribute in rare cases, differences in HLA alleles [30] and their effects in immune responses to covalent carbamazepine epoxide binding appear to explain adverse drug responses to this compound.
The basis for the conversion of non-toxic carbamazepine to hepatotoxicant by low-dose LPS also remains unclear. We found no evidence of increased proinflammatory cytokine production in the LPS/carbamazepine groups. Preliminary work suggests that low dose LPS delays carbamazepine absorption (which conceivably could contribute to LPS gene expression repression); still, LPS repressions of carbamazepine metabolism should lead to less reactive metabolite formation (although one could argue that shunting to an LPS-insensitive pathway might produce the important reactive metabolite toxicant; there is no ecvidence for such a hypothesis). Such speculation also extends to DSS; more or less bioavailability of carbamazepine cannot explain the selective DSS potentiation of carbamazepine induction of OS/RM genes that was observed.
Unfortunately the convenient treatment of 5% DSS in water did not convert carbamazepine into a hepatotoxicant in the present studies. This lack of a toxic effect is speculated to result from the non-pathological GI flora of commercially available lab rats. Others have suggested that GI flora can be relatively easily manipulated since rats are coprophagic [31,32]. Desensitization of endotoxin responses by DSS has been invoked to explain the lack of potentiation of galactosamine-induced hepatotoxicity in rats [16]. However, this likely reflects a similar non-hepatotoxic response to DSS as observed in the present study. DSS treatment thus provides a more complex model than low dose LPS, but eventually with modifications may prove amenable to testing of idiosyncratic hepatotoxicants.
REFERENCES
- Bond, C.A. and Raehl, C.L. (2006) Clinical pharmacy services, pharmacy staffing, and adverse drug reactions in United States hospitals, pharmacotherapy. The Journal of Human Pharmacology and Drug Therapy, 26, 735-747. http://dx.doi.org/10.1592/phco.26.6.735
- Olsen, K.M., Rebuck, J.A., Weidenbach, T. and Fish, D.N. (2000) Pharmacokinetics of intravenous trovafloxacin in critically Ill adults, pharmacotherapy. The Journal of Human Pharmacology and Drug Therapy, 20, 400- 404. http://dx.doi.org/10.1592/phco.20.5.400.35056
- Bharadwaj, M., Illing, P., Theodossis, A., Purcell, A.W. Rossjohn, J. and McCluskey, J. (2012) Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annual Review of Pharmacology and Toxicology, 52, 401-431. http://dx.doi.org/10.1146/annurev-pharmtox-010611-134701
- Uetrecht, J. (2007) Idiosyncratic drug reactions: Current understanding. Annual Review of Pharmacology and Toxicology, 47, 513-539. http://dx.doi.org/10.1146/annurev.pharmtox.47.120505.105150
- Srivastava, A., Maggs, J.L., Antoine, D.J., Williams, D.P., Smith, D.A. and Park, B.K. (2008) Role of reactive metabolites in drug-induced hepatotoxicity. Handbook Exp Pharmacology, 196,165-194.
- Evans, D.C., Watt, A.P., Nicoll-Griffith, D.A. and Baillie, T.A. (2003) Drug-protein adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chemical Research in Toxicology, 17, 3-16. http://dx.doi.org/10.1021/tx034170b
- Obach, R.S., Kalgutkar, A.S., Soglia, J.R. and Zhao, S.X. (2008) Can in vitro metabolism-dependent covalent binding data in liver microsomes distinguish hepatotoxic from nonhepatotoxic drugs? An analysis of 18 drugs with consideration of intrinsic clearance and daily dose. Chemical Research in Toxicology, 21, 1814-1822. http://dx.doi.org/10.1021/tx034170b
- Leone, A.M., Kao, L.M., McMillian, M.K., Nie, A.Y., Parker, J.B., Kelley, M.F., Usuki, E., Parkinson, A., Lord, P.G. and Johnson, M.D. (2007) Evaluation of felbamate and other antiepileptic drug toxicity potential based on hepatic protein covalent binding and gene expression. Chemical Research in Toxicology, 20, 600-608. http://dx.doi.org/10.1021/tx600351g
- Lu, W., Li, X. and Uetrecht, J.P. (2008) Changes in gene expression induced by carbamazepine and phenytoin: Testing the danger hypothesis. Journal of Immunotoxicology, 5, 107-113. http://dx.doi.org/10.1080/15476910802085723
- Morio, L.A., Leone, A., Sawant, S.P., Nie, A.Y., Brandon Parker, J., Taggart, P., Barron, A.M., McMillian, M.K. and Lord, P. (2006) Hepatic expression of heme oxygenase-1 and antioxidant response element-mediated genes following administration of ethinyl estradiol to rats. Toxicology and Applied Pharmacology, 216, 416-425. http://dx.doi.org/10.1016/j.taap.2006.06.016
- Xu, J.J., Henstock, P.V., Dunn, M.C., Smith, A.R., Chabot, J.R. and de Graaf, D. (2008) Cellular imaging predictions of clinical drug-induced liver injury. Toxicological Sciences, 105, 97-105. http://dx.doi.org/10.1093/toxsci/kfn109
- Pirmohamed, M., Kitteringham, N.R., Breckenridge, A.M. and Park, B.K. (1992) Detection of an autoantibody directed against human liver microsomal protein in a patient with carbamazepine hypersensitivity. British Journal of Clinical Pharmacology, 33,183-186. http://dx.doi.org/10.1111/j.1365-2125.1992.tb04022.x
- Zou, W., Roth, R.A. and Ganey, P.E. (2012) Animal models of idiosyncratic, drug-induced liver injury: Emphasis on the inflammatory stress hypothesis, Encyclopedia of drug metabolism and interactions. John Wiley & Sons, Inc., Hoboken.
- Morgan, E.T., Goralski, K.B., Piquette-Miller, M., Renton, K.W., Robertson, G.R., Chaluvadi, M.R., Charles, K.A., Clarke, S.J., Kacevska, M., Liddle, C., Richardson, T.A., Sharma, R. and Sinal, C.J. (2008) Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metabolism and Disposition, 36, 205-216. http://dx.doi.org/10.1124/dmd.107.018747
- Deng, X., Stachlewitz, R.F., Liguori, M.J., Blomme, E.A.G., Waring, J.F., Luyendyk, J.P., Maddox, J.F., Ganey, P.E. and Roth, R.A. (2006) Modest inflammation enhances diclofenac hepatotoxicity in rats: Role of neutrophils and bacterial translocation. Journal of Pharmacology and Experimental Therapeutics, 319, 1191-1199. http://dx.doi.org/10.1124/jpet.106.110247
- Masubuchi, Y. and Horie, T. (2004) Endotoxin-mediated disturbance of hepatic cytochrome P450 function and development of endotoxin tolerance in the rat model of dextran sulfate sodium-induced experimental colitis. Drug Metabolism and Disposition, 32, 437-441. http://dx.doi.org/10.1124/dmd.32.4.437
- Leone, A., McMillian, M., Proctor, J., Kao, M., Troisi, J., Fermier, A., Varacallo, L., Taggart, P., McCarty, J., Sawant, S., Collins, A., Nie, A., Taylor, E., Lord, P. and Kelley, M. (2008) Idiosyncratic hepatotoxicity: A rat Model of Converting Oxidative Stress/Reactive Metabolites to Hepatotoxicants. Society of Toxicologic Pathology Annual Meeting (47th), Seattle, Washington, 16-20 March.
- McMillian, M.K., Leone, A.M., Kelley, M.F., Proctor, S.J.E., Kao, L.M., Troisi, J., Fermier, A., Varacallo, L., Taggart, P. and McCarty, J. (2008) Toxicogenomics, oxidative stress and hepatotoxicity. Proceedings of 17th International Symposium on Microsomes and Drug Oxidations, 17, 25-30.
- Vicario, M., Crespí, M., Franch, À., Amat, C., Pelegrí, C. and Moretó, M. (2005) Induction of colitis in young rats by dextran sulfate sodium. Digestive Diseases and Sciences, 50, 143-150. http://dx.doi.org/10.1007/s10620-005-1292-y
- Vicario, M., Amat, C., Rivero, M., Moretó, M. and Pelegrí, C. (2007) Dietary glutamine affects mucosal functions in rats with mild dss-induced colitis. The Journal of Nutrition, 137, 1931-1937.
- McMillian, M., Nie, A., Parker, J.B., Leone, A., Kemmerer, M., Bryant, S., Herlich, J., Yieh, L., Bittner, A., Liu, X., Wan, J., Johnson, M.D. and Lord, P. (2005) Drug-induced oxidative stress in rat liver from a toxicogenomics perspective. Toxicology and Applied Pharmacology, 207, 171-178. http://dx.doi.org/10.1016/j.taap.2005.02.031
- McMillian, M., Nie, A.Y., Parker, J.B., Leone, A., Bryant, S., Kemmerer, M., Herlich, J., Liu, Y., Yieh, L., Bittner, A., Liu, X., Wan, J. and Johnson, M.D. (2004) A gene expression signature for oxidant stress/reactive metabolites in rat liver. Biochemical Pharmacology, 68, 2249- 2261. http://dx.doi.org/10.1016/j.bcp.2004.08.003
- Waring, J.F., Liguori, M.J., Luyendyk, J.P., Maddox, J.F., Ganey, P.E., Stachlewitz, R.F., North, C., Blomme, E.A.G. and Roth, R.A. (2006) Microarray analysis of lipopolysaccharide potentiation of trovafloxacin-induced liver injury in rats suggests a role for proinflammatory chemokines and neutrophils. Journal of Pharmacology and Experimental Therapeutics, 316, 1080-1087. http://dx.doi.org/10.1124/jpet.105.096347
- Blazka, M.E., Wilmer, J.L., Holladay, S.D., Wilson, R.E. and Luster, M.I. (1995) Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicology and Applied Pharmacology, 133, 43-52. http://dx.doi.org/10.1006/taap.1995.1125
- Luyendyk, J.P., Maddox, J.F., Cosma, G.N., Ganey, P.E., Cockerell, G.L. and Roth, R.A. (2003) Ranitidine treatment during a modest inflammatory response precipitates idiosyncrasy-like liver injury in rats. Journal of Pharmacology and Experimental Therapeutics, 307, 9-16. http://dx.doi.org/10.1124/jpet.103.054288
- Dickmann, L.J., Patel, S.K., Rock, D.A., Wienkers, L.C. and Slatter, J.G. (2011) Effects of interleukin-6 (IL-6) and an anti-il-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metabolism and Disposition, 39, 1415-1422.
- Dallas, S., Sensenhauser, C., Batheja, A., Singer, M., Markowska M., Zakszewski, C., Mamidi, R.N.V.S., McMillia, M., Han, C., Zhou, H. and Silva, J. (2012) De-risking bio-therapeutics for possible drug interactions using cryopreserved human hepatocytes. Current Drug Metabolism, 13, 923-929. http://dx.doi.org/10.2174/138920012802138589
- Pirmohamed, M., Kitteringham, N.R., Guenthner, T.M., Breckenridge, A.M. and Park, B.K. (1992) An investigation of the formation of cytotoxic, protein-reactive and stable metabolites from carbamazepine in vitro. Biochemical Pharmacology, 43, 1675-1682. http://dx.doi.org/10.1016/0006-2952(92)90696-G
- Ketter, T.A., Frye, M.A., Corá-Locatelli, G., Kimbrell, T.A. and Post, R.M. (1999) Metabolism and excretion of mood stabilizers and new anticonvulsants. Cellular and Molecular Neurobiology, 19, 511-532. http://dx.doi.org/10.1023/A:1006990925122
- Yip, V.L., Marson, A.G., Jorgensen, A.L., Pirmohamed, M. and Alfirevic, A. (2012) HLA genotype and carbamazepine-induced cutaneous adverse drug reactions: A systematic review. Clinical Pharmacology & Therapeutics, 92, 757-765. http://dx.doi.org/10.1038/clpt.2012.189
- Rath, H.C., Schultz, M., Freitag, R., Dieleman, L.A., Li, F.L., Linde, H.-J., Schölmerich, J. and Sartor, R.B. (2001) Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infection and Immunity, 69, 2277-2285. http://dx.doi.org/10.1128/IAI.69.4.2277-2285.2001
- Damman, C.J., Miller, S.I., Surawicz, C.M. and Zisman, T.L. (2012) The microbiome and inflammatory bowel disease: Is there a therapeutic role for fecal microbiota transplantation[quest]. The American Journal of Gastroenterology, 107, 1452-1459. http://dx.doi.org/10.1038/ajg.2012.93