Advances in Bioscience and Biotechnology
Vol.4 No.6(2013), Article ID:33546,17 pages DOI:10.4236/abb.2013.46099
High yield expression of proteins in E. coli for NMR studies
![]()
1NMR Research Centre, Indian Institute of Science, Bangalore, India
2Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore, India
Email: *hsatreya@gmail.com
Copyright © 2013 Somnath Mondal et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 1 April 2013; revised 12 May 2013; accepted 25 May 2013
Keywords: E. Coli; Recombinant DNA Technology; Structural Biology; NMR Spectroscopy
ABSTRACT
In recent years, high yield expression of proteins in E. coli has witnessed rapid progress with developments of new methodologies and technologies. An important advancement has been the development of novel recombinant cloning approaches and protocols to express heterologous proteins for Nuclear Magnetic Resonance (NMR) studies and for isotopic enrichment. Isotope labeling in NMR is necessary for rapid acquisition of high dimensional spectra for structural studies. In addition, higher yield of proteins using various solubility and affinity tags has made protein overexpression cost-effective. Taken together, these methods have opened new avenues for structural studies of proteins and their interactions. This article deals with the different techniques that are employed for over-expression of proteins in E. coli and different methods used for isotope labeling of proteins vis-à-vis NMR spectroscopy.
1. INTRODUCTION
The two most popular methods today for structural studies of biomolecular systems are X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy. The former requires the preparation of single crystal of proteins whereas the latter involves the preparation of protein samples either in solution (for liquid state NMR) or in micro-crystalline form (for solid state NMR). In the case of NMR studies of proteins, homonuclear oneand two-dimensional (1D/2D) techniques are sufficient if the molecular mass of the system is less than 10 kDa (<90 - 100 amino acid residues) [1]. However, for larger proteins advanced NMR techniques combined with isotopic enrichment or isotope labeling of the molecule (with 13C/ 15N/2H) is required [2]. This is owing to the fact that with increase in size of the molecules the sensitivity of NMR experiments decrease and hence isotopic enrichment helps to alleviate to some extent the deleterious effects of large size.
Isotope labeling implies the replacement of 12C, 14N or 1H atoms of the backbone or side-chain of proteins by 13C, 15N or 2H, respectively, either uniformly throughout the protein (i.e., independent of the amino acid type) or in a selective manner (i.e., amino acid type dependent) [2]. This can be accomplished in one of the three ways: 1) over-expression of proteins in E. coli or higher organisms if required; 2) use of cell-free protein expression; and 3) chemical synthesis. Of the three, over-expression of proteins in E. coli is by far the most popular and costeffective method today. The protein to be expressed is first cloned using the recombinant DNA methods. The protein is either expressed as such or fused with a suitable tag for increasing the yield/solubility and/or facilitating easy purification. Starting from 1960s when the first isotope enrichment of proteins was carried out, isotope labeling has come a long way with various advancements. Today it is possible to express proteins with 13C and/or 15N at a fraction of cost and/or with an order of magnitude compared to a decade ago.
In this article, we focus on the different techniques used today for over-expression and isotope labeling of proteins in E. coli for NMR studies. The various methods proposed recently for high-yield expression are discussed with suitable examples. The article is divided into three sections. In the first section, the different general protocols for protein expression are described. In the second section, different methods for isotope labeling for NMR studies are discussed. In the last section, special protocols for expressing difficult proteins in E. coli for NMR are covered.
2. RECENT METHODS FOR HIGH YIELD PROTEIN EXPRESSION IN E. COLI
Purification of proteins in Escherichia coli at a very high yield requires over production by cells. A major impedement in this regard is that many proteins are poorly expressed especially those of eukaryotic origin, and sometimes in insoluble form which are prone to degradation by cellular proteases. In recent years, high level expression of proteins has been impelled by various developments in understanding and manipulating the biological processes of E. coli.
Paramount factors to obtain high yields of protein are gene of interest, expression vector, gene dosage, transcriptional regulation, codon usage, translational regulation, host design, growth media and culture conditions or fermentation conditions available for manipulating the expression conditions, specific activity or biological activity of the protein of interest, protein targeting, fusion proteins, molecular chaperones, protein degradation [3-6]. Recent methods to obtain high protein expression include high cell density shaking cultures, High cell density fermentation, fed batch based cultivation, cold shock induction, co-expression with chaperones or fusion tags, cell free protein synthesis among others. Figure 1 below depicts some of the methods used to achieve high yielding proteins. Each of these methods is described below in detail.
2.1. High Cell Density Shaking Cultures
High cell density shaking cultures based on IPTG induction reported by Qianqian Li and others used a regular incubator shaker to achieve a cell-density (measured as optical density at 600 nm; OD 600) of 10 - 20 in the normal laboratory setting instead of a fermenter [7]. Several parameters such as host strain selection, plasmid copy numbers, promoter selection, mRNA stability, and codon usage were optimized to attain a high cell density [3,7,8]. In this IPTG induction based method, rich media is used as starter culture, which is grown at an optimized temperature and time period, following which the cell pellet
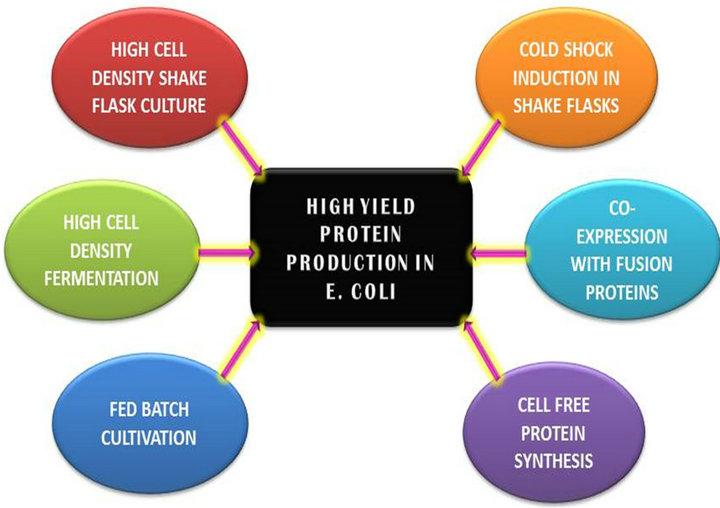
Figure 1. Depiction of different methods leading to high yield protein production in Escherichia coli.
is transferred into same volume of minimal media when the cells are in the middle of growing phase. Further, the cells are cultured for medium exchange at pre-determined, optimized temperature for the target protein production [7]. At this point, the bacterial culture is induced for target protein production by IPTG at the same temperature for an optimized time period. Bacterial expression parameters to be optimized in this method to attain a very high cell density in laboratory shaking cultures are: 1) double selection of colonies highly expressing the target protein; 2) optimization of temperature and time period for starter culture to avoid plasmid instability or loss; 3) optimizing induction temperature and time course; and 4) glucose optimization. These expression parameters must be optimized for every new target protein [7].
High cell density shaking cultures by auto-induction is an efficient cost effective method for high level protein production with glycerol as carbon source since glucose prevents auto-induction and lactose is necessary for autoinduction at previously optimized physiological conditions and optimized minimal medium [4,9]. Auto-induction method can be alternative to tedious fermenter cultures as the latter requires additional equipment to monitor various parameters which is economically prohibitive. Additionally, the smaller media volumes required make the production method ideal for selective side-chain or amino acid labeling procedures [10,11]. Auto-induction medium contains 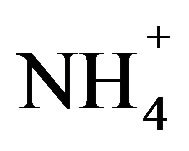 as nitrogen source, glycerol, lactose and glucose at optimized levels so that glycerol is used as carbon source. Lactose is metabolized for autoinduction once glucose is depleted, which is otherwise the most favorable carbon source. Individual isotopes (13C/15N) incorporation is slightly more efficient than 15N and 13C incorporated together. To overcome decreased rate of incorporation of appropriate isotopes, a longer period of clearance of unlabeled nutrients is allotted before induction. Also, it favors metabolic rates of E. coli in deuterated media [11] and high yield expression. Generally a lower temperature for prolonged duration of time is preferred to obtain soluble target protein at high yield without any additional requirement of amino acids and vitamin supplements [9].
as nitrogen source, glycerol, lactose and glucose at optimized levels so that glycerol is used as carbon source. Lactose is metabolized for autoinduction once glucose is depleted, which is otherwise the most favorable carbon source. Individual isotopes (13C/15N) incorporation is slightly more efficient than 15N and 13C incorporated together. To overcome decreased rate of incorporation of appropriate isotopes, a longer period of clearance of unlabeled nutrients is allotted before induction. Also, it favors metabolic rates of E. coli in deuterated media [11] and high yield expression. Generally a lower temperature for prolonged duration of time is preferred to obtain soluble target protein at high yield without any additional requirement of amino acids and vitamin supplements [9].
2.2. Fed Batch Cultivation Method
This refers to a fed batch liquid phase cultivation technology using enzymatic release of glucose for protein production in E. Coli in round-bottom Erlenmeyer shaken cultures [12]. Media comprising mixture of mineral salts and complex additives is optimized to provide high cell density and high protein yield. High cell density and favorable physiological conditions of the target protein are achieved in the shaken cultures with EnBase® Flo (glucose releasing polymer is in soluble form rather than a gel) throughout the protein expression period which is a day longer compared to other expression methods but reaches OD600 of 30 to 50 [12] yielding high amount of soluble protein in comparison to commonly used media. It can be carried out in lower volumes in 24-deep well plates as well as higher volumes in large shake flasks [12].
E. coli cultures induced with IPTG progressively in a fed-batch culture condition results in high level target protein production. Once the glucose from the batch phase has been depleted, an exponential substrate feed is used to provide a constant growth followed by continuous inducer feed in increasing linear fashion in a bioreactor maintaining the physiological conditions like pH, temperature and aeration [13]. Experiments show that lowering the induction/process temperature effectively increases the solubility/specific activity of the target protein in comparison to 37˚C achieving high cell density fed batch cultures with increased protein production [4, 7,13].
2.3. High Cell Density Fermentation
Fermentation at lab scale normally results in high yield of proteins mostly by fed batch process. High yield protein production by fermentation is reported to give higher protein yield compared to traditional shake flask cultures [14]. High cell density fermentation enhances the overall yield of the protein [15]. Parameters such as feeding strategies, aeration, temperature, pH, media composition, expression strains, and plasmid stability need to be optimized [15]. High cell density fermentation is most preferred for large scale protein production. Detrimental fact is that fermentation is slightly expensive and thus not preferred over shake flask cultures. Achieving high cell density cultures in shake flasks is possible as highlighted in the above section.
2.4. Cold Shock Induced High Yield Protein Production
Cold shock along with IPTG induces protein production in E. coli transformed with pCold [16] vectors (cold shock expression vectors) under the control of cspA promoter at optimized growth conditions and cold temperature of 15˚C [16]. A unique feature of this technology developed by Inouye and others is the inhibition of non-target proteins at cold temperatures termed LACE effect [17] during which most of the translational machinery is dedicated for selectively isotope labeled target protein production termed lace effect [16,17] requiring no protein purification. In this method transformed cells are grown in rich medium starter culture at optimized growth temperature and then shifted to minimal medium and induced with IPTG at previously optimized cold temperature for prolonged hours. Thus obtained crude cell lysate is amenable for NMR studies making this a rapid high yield protein production method [16]. Enhanced protein expression is observed by cold shock induction in E. coli transformed with pCold-PST vectors in a similar fashion as described above with PrS2 tags having affinity to myxospores [18].
2.5. Co-Expression with Fusion/Solubility Tag
The application of tags has been highly effective in the structural studies of proteins previously thought unapproachable by solution NMR techniques. These tags are not only important for solubility and stability enhancement issues, but also for a favorable effect on the folding of their fusion protein partners [4]. Tags used to improve the yield of recombinant proteins can be roughly divided in two categories: 1) affinity tags for rapid and efficient purification of proteins; and 2) solubility tags to enhance the proper folding and solubility of the recombinant protein [5]. Multiple tags can be added together in different combination for a particular protein to get better result on these issues [7].
Various studies report high yield target protein expression with Maltose binding protein (MBP) [19], Glutathione-S transferase (GST) [20], Thioredoxin fusion (TRX) [21], Hexahistidine His6-Tag [19,22], Small ubiquitinmodifier fusion (SUMO) [21], N-utilizing substance A (NusA), Protein S tag (PrS2) [18], Solubility-enhancing tag (SET) [23], Disulfide bond C (DsbC), Seventeen kilodalton protein (Skp), Phage T7 protein kinase (T7PK), Protein G B1 domain (GB1), Protein A IgG ZZ repeat domain (ZZ) [24], histidine tagged Ubiquitin (Ub) fusion and histidine tagged deubiquitylating enzyme (DUB) fusion [25]. Recent studies propose the Outer membrane protein A (OmpA), Outer membrane protein F (OmpF) and Osmotically inducible protein Y (OsmY) as fusion partners secreting protein into the growth medium of E. coli depending on culture conditions via periplasm secretion. This is owing to the fact that the fusion partners are membrane proteins and there by render the purification procedure simple [26]. Recently, ELP (Elastin-like polypeptide) tags capable of self-cleavage and non-chromatographic protein purification in high cell density E. Coli fermentation has been reported [14]. Highly purified recombinant proteins obtained from E. Coli by fusion tags and co-expression are elaborated in the previous section.
Recent findings throw light on “rapid” expression among others [27] in which T7RNAP is controlled by arabinose promoter (ParabAD), while the target gene is controlled by the T7 lacpromoter (PT7lac) paving the way for dual induction by both arabinose and IPTG and increasing the yield of T7RNAP which in turn translates target gene resulting in rapid and high level protein expression [27]. Among other cost effective methods for high yield protein production include heat-cooling extraction coupled with ammonium precipitation followed by chromatography optimized based on amino acid composition of the target protein [28].
To choose an effective combination of the protein and tag, the advantages and disadvantages of various tags must be considered with respect to their yield, solubility, stability and easing the purification of the fusion partners [53] (Table 1). Additionally, as the affinity tag have the potential to interfere with structural or functional studies in some cases, provisions also be made for removing them after purification gets over [54]. Though these tags help in crystallization for some proteins, for solution NMR study it is often necessary to remove the tags before recording the spectra if the tag interferes with the NMR signals of the protein [33,55,56]. This is especially true for the large tags like GST and MBP. Endoproteases are widely used for the tag removal purpose [57] (Table 2).
An alternative strategy to endoprotease cleavage is the use of exopeptidases for the tag removal after protein purification. Several amino peptidases and carboxypeptidases are available from the natural resources like porcine kidney and bovine pancreas. Although the potential use of these enzyme for tag removal is somewhat limited because of the growing concern on contamination from animal sources also with the additional challenge for the purification of the target protein after tag cleavage [58].
There is no common affinity or, solubility tags that functions for all the proteins. Rather, the choice of a tag largely depends on the factors related to the protein being expressed [67]. The use of tags has been demonstrated to overcome mainly solubility and stability issues along with yield and proper folding of the recombinant protein in some cases. The recent development of NMR invisible tags promises the implementation of more such tags in the field of Bio-molecular NMR in near future for the structural study of proteins [54].
2.6. Cell Free Protein Synthesis
Ever since the advent of cell free expression systems in mid 90s [68] it has become a rapid, efficient method of protein synthesis [69] and recent advances in cell free systems has rendered it an even more powerful tool for simple, efficient and cost effective method of in-vitro protein synthesis [69], mainly for structural studies for
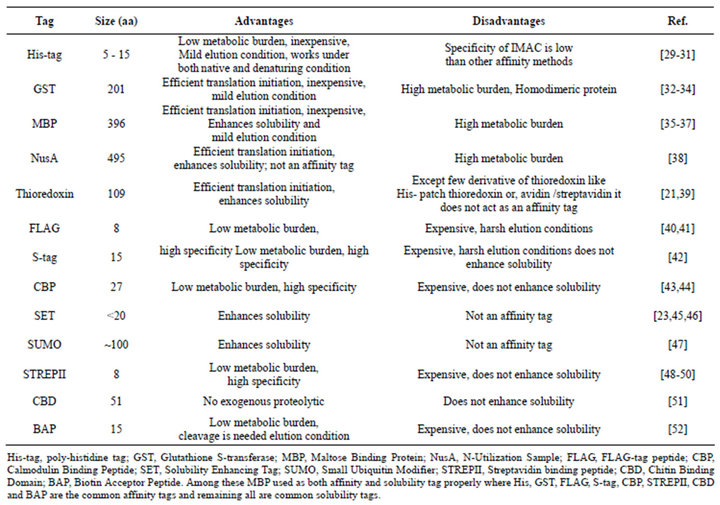
Table 1. Advantages and disadvantages of using different tags with their size.
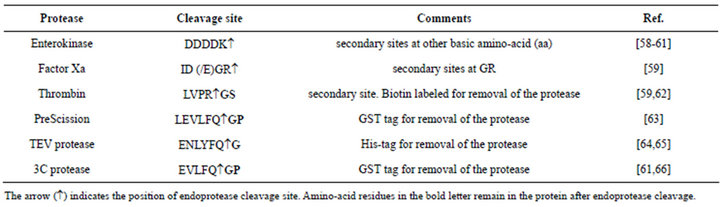
Table 2. Use of some familiar proteases for tag removal.
those proteins where purification is not feasible like-membrane proteins which pose obstacles to work with [70], viral proteins [69], incorporation of non-natural amino acids [70,71], in studying protein-interactions and also in high throughput proteomics [69,72]. In addition, cell free method provides a means by which target proteins can be decisively screened for NMR studies by means of multiple screening systems [73].
Highly condensed wheat-germ extract based cell free system condensed by polyethylene glycol precipitation is found to increase the protein yield [74] coupled with 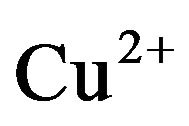 addition to the wheat germ extract to prevent mRNA destruction and decrease in ATP levels which are important parameters for cell free expression [74]. This method has shown to increase protein synthesis rate and thus the final protein concentration in comparison to uncondensed cell free extract [74]. Bernhard and others have reported a cell free protein synthesis of few integral membrane proteins at high yields by optimizing various parameters like macro nutrients, amino acids and modified the quality of S30 cell free extract by addition of detergents and lipids to obtain soluble IMP (avoiding precipitation) and monitoring the production by GFP expression [75]. NMR studies confirmed correct folded conformations of these proteins produced by cell-free method [75].
addition to the wheat germ extract to prevent mRNA destruction and decrease in ATP levels which are important parameters for cell free expression [74]. This method has shown to increase protein synthesis rate and thus the final protein concentration in comparison to uncondensed cell free extract [74]. Bernhard and others have reported a cell free protein synthesis of few integral membrane proteins at high yields by optimizing various parameters like macro nutrients, amino acids and modified the quality of S30 cell free extract by addition of detergents and lipids to obtain soluble IMP (avoiding precipitation) and monitoring the production by GFP expression [75]. NMR studies confirmed correct folded conformations of these proteins produced by cell-free method [75].
Major drawbacks of cell free system are the pH change and accumulation of phosphates which inhibit protein synthesis. Swartz and others have engineered the cell free system which successfully overcomes this problem by making use of pyruvate as energy source in batch reactions mimicking the cytoplasm of the cell [76]. This is found to bring down the cost of cell free system also cutting down the requirement of expensive high energy phosphate compounds [76].
2.7. Co-Expression with Molecular Chaperones
The high level expression of recombinant gene products in E. coli often results in misfolding of the protein of interest and its subsequent degradation by cellular proteases or the deposition of the protein into biologically inactive aggregates known as inclusion bodies [77]. It has been established that in vivo protein folding is an energy dependent process mediated by two classes folding modulators; molecular chaperones—DnaK-Dn-aJGrpE and GroEL-GroES systems, which suppress offpathway aggregation reaction and lead to the proper folding through ATP-coordinated cycle of binding and release of intermediates [78]. Additionally, it accelerates rate-limiting steps along the protein folding pathway such as cis-trans isomerization of peptide-prolyl bonds and formation/reshuffling of disulfide-bridges [79,80]. These two chaperone system holds great promise in facilitating the production, purification and proper folding of heterologous protein [81,82].
Molecular chaperone has three sub-classes based on their mechanism of actions [83]. “Folding” chaperones (e.g., DnaK and GroEL) as mentioned above, “Holding” chaperones (e.g. IbpB) maintain partially folded proteins on their surface to await availability of folding chaperones upon stress slack and the “Disaggregating” chaperone ClpB promotes the solubilization of the protein that became aggregated as a result of stress. The general mechanism of protein folding assisted by molecular chaperones is depicted in Figure 2 [84]. In Figure 2, A represents m-RNA which translates into unfolded-protein (U). In absence of any chaperone protein, U can fold back to its native form (N) along with the aggregationprocess which leads to inclusion-bodies (Z); but in presence of chaperone protein U solely folds back to its native form (N) via an intermediate (I) with chaperone protein without any aggregation path which leads to inclusion bodies. The path from I to N governs by the hydrolysis of ATP to ADP and Pi. The ktransl, kunfold and kfold represent the corresponding rate constants for the translational-process, unfolding and folding pathways.
A set of co-expression vectors with the different combination of DnaK-DnaJ-GrpE, GroEL-GroES and trigger-factor (tig) system is given below (Table 3):
Syntheses of DnaK-DnaJ-GrpE and GroEL-GroES are under positive control of a minor σ factor (σ32) encoded by the rpoH gene [77]. These folding chaperone plasmids
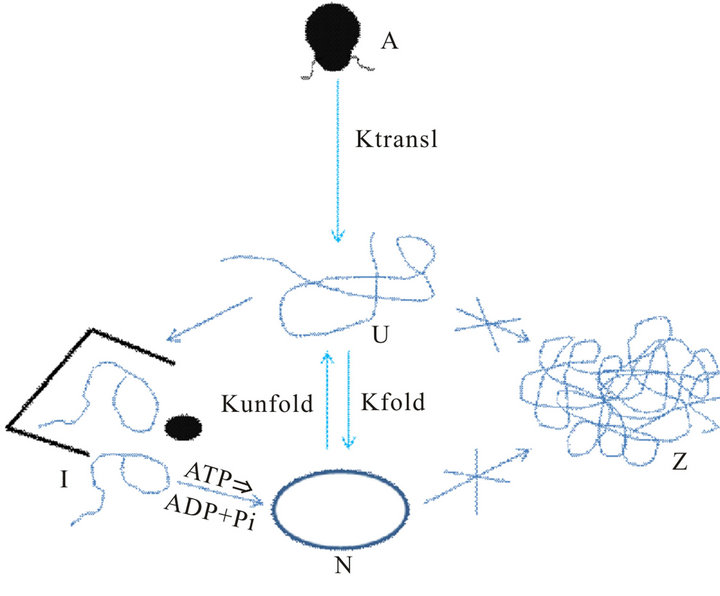
Figure 2. Schematic depiction of folding pathway of a protein in presence of chaperone proteins.
carry an origin of replication derived from pACYC and a chloramphenicol resistance gene (Cmr). This system allows their use with E. coli expression system that utilizes ColE1 type plasmids containing the ampicillin resistance gene as a marker. Expression of target proteins and chaperones can be induced individually, the chaperone plasmid contain either araC or, tetR for each promoter [88]. This is notable that this chaperone system cannot be used in combination with chloramphenicol resistant E. coli host strains or, expression that carry the chloramphenicol resistant gene. An effective method for constructing a system for co-expression of target proteins and chaperone involves transformation of E. coli with chaperone plasmid followed by expression with an expression plasmid for the target protein which results in high transformation efficiency than by doing it in any other way. In this regard, it is mentionable that E. coli Heat Shock Protein40 (HSP40) and E. coli Heat Shock Protein 70 (HSP70) are also known as DnaJ and DnaK and also widely used for differently or, in combination with the GroEl-GroEs chaperones [83,89-91] (Table 4).
Molecular chaperones were constitutively expressed and play an important role in the synthesis of properly folded proteins. In addition to protein folding molecular chaperones control wide range of cell function such as transcription, protein assembly and membrane translocation [92-94]. Bacterial chaperonins have cylindrical structure composed of two stacked 7-fold rotational symmetric rings of cpn60 subunit and the co-chaperonins has the dome-shaped structure composed of 7 fold rotational symmetric rings of cpn10 subunit [95]. These chaperonins assist the protein folding in an ATP dependent manner and are expected to improve the refolding yields from inclusion body [96,97].
The demand of pure, soluble and properly folded
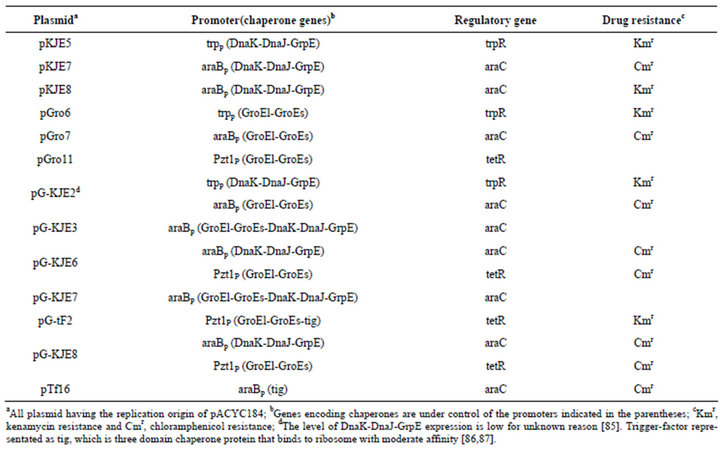
Table 3. Details of the chaperone expression systems.
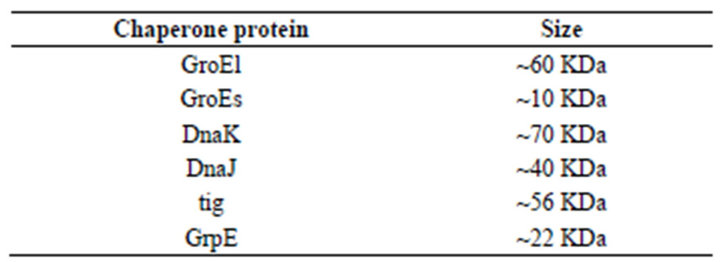
Table 4. Sizes of the different chaperone protein are as follows.
functional protein is very high in modern technology as well as in the drug delivery purpose. E. coli is frequently used host for the purification of recombinant protein [98- 101]. But the problem of having non-functional and insoluble protein expressed by E. coli can be overcome by using the chaperon systems. This is a promising way to get back the protein in its proper functional form with higher yield, which is also a cheaper and easy to do approach [102]. The typical chaperone target is a short unstructured stretch of hydrophobic amino acids covered on either side by basic residues (lacking acidic residues). In addition to proper de novo folding, GroEl and DnaK refold host protein that become unfolded under the stressed condition of cell [103]. Holding chaperones assist in this process that stabilizes partially folded protein rather promoting them to their native states generally. In addition to this, generic chaperones GroEl and DnaK assist in the incorporation of the synthesized protein in the cytoplasm to inner-membrane or, translocation to ![]() the periplasm.
the periplasm.
3. EXPRESSION OF ISOTOPE LABELED PROTEINS FOR NMR STUDIES
NMR studies for analyzing structures of proteins are performed with isotopically labeled samples enriched with 13C, 15N or 2H by growing E. coli cultures in a medium containing the appropriate isotope [104]. The various isotope labeling schemes for NMR is categorized in Figure 3 [2]. The choice of a particular isotope labeling scheme depends on what kind of information we want by using particular NMR experiments for that sample (Table 5).
The Bacterial expression vector BL21, producing high yields of recombinant proteins are frequently used expression strain of E. coli which responds differently to different gluconeogenic carbon sources and salt contents [104]. The most frequently used medium is the M9 medium used for growing E. coli BL21 did not support the optimum growth lacking ferrous sulphate. The addition of bivalent iron to M9 medium inoculated with E. coli increases the growth rate and the cell density in the stationary phase (iron is required by the enzymes of the tricarboxylic acid cycle and aerobic respiration chain) [105].
Two of the popular isotope labeling schemes (apart from uniform 13C/15N labeling) is selective labeling and unlabeling [2]. In the selective labeling approach, the bacteria are grown in the M9 minimal medium supplemented with the amino acid to be selective labeled in 13C/15N. In the method of selective unlabeling, the cells are grown in M9 minimal medium supplemented with the unlabeled amino acid which is to be selective unlabeled. The biosynthesis of the different amino acids in E. coli is well known and depicted in Figure 4. Selective amino acid labeling or unlabeling aids in sequence specific resonance assignment by helping to identify resonance which are otherwise buried in the crowded regions
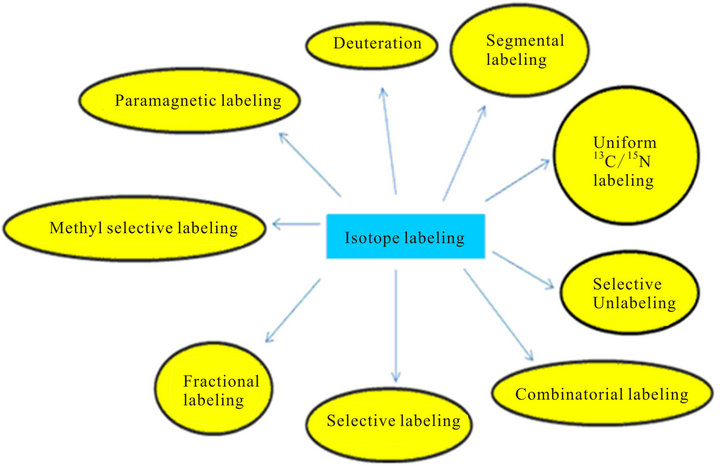
Figure 3. Various isotope-labeling schemes in proteins for NMR studies.
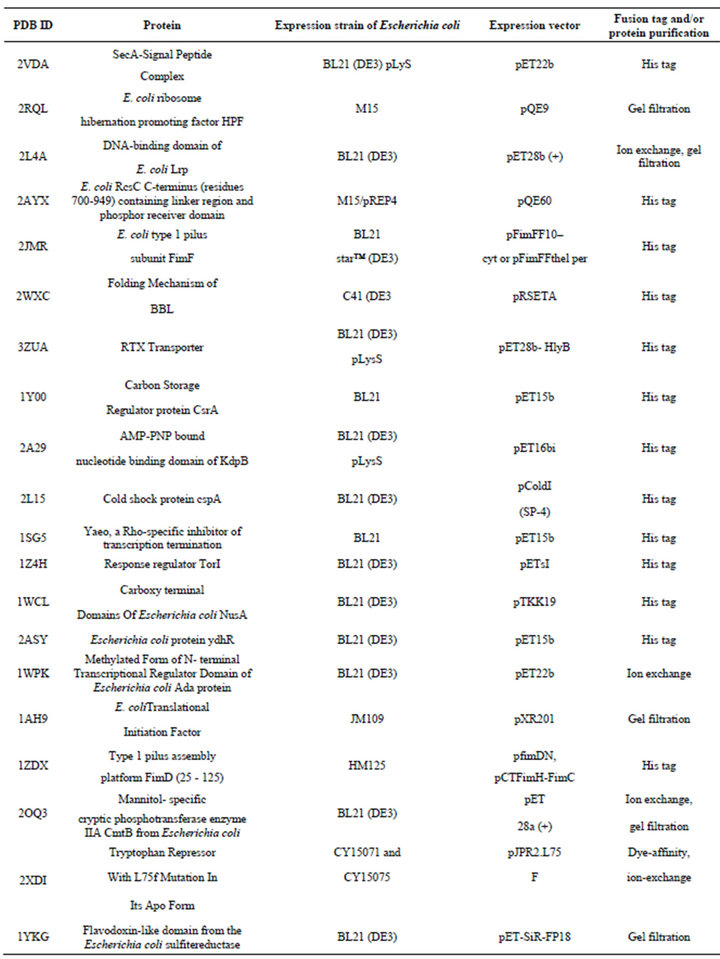
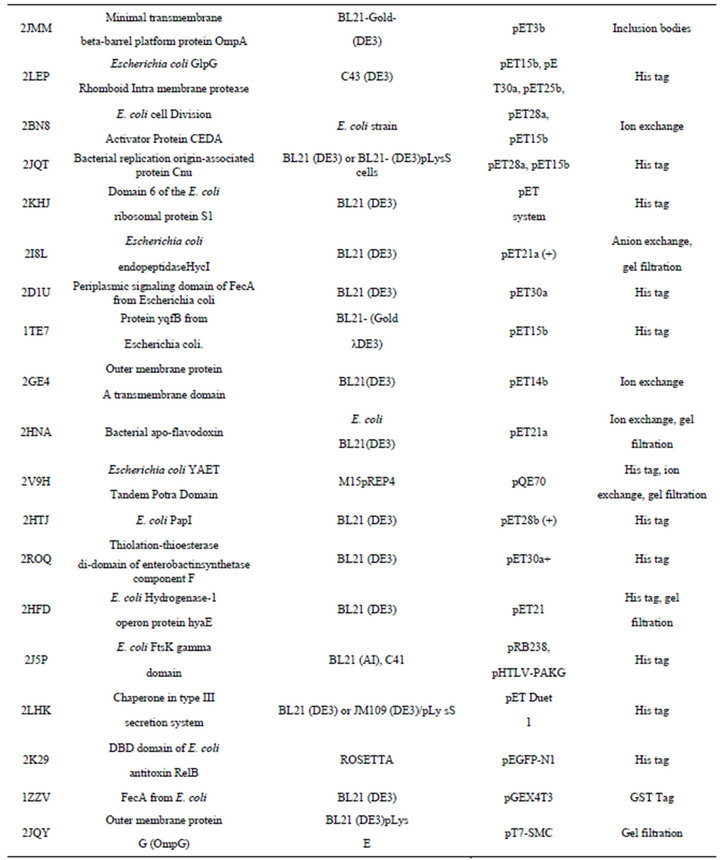
Table 5. Representative examples of E. coli expressed proteins studied by NMR Spectroscopy.
of 2D and 3D NMR spectra. However, a disadvantage of this method is “Isotope-scrambling” [106], which leads to mis-incorporation of 15N (for selective labeling) or 14N (for selective unlabeling) in undesired amino-acid. This happens due to metabolic conversion of one amino acid to the other in the bio-synthetic pathway of the cell, which is shown below in Figure 4. For Asp, Glu and Gln isotopic scrambling is maximum as they higher up the
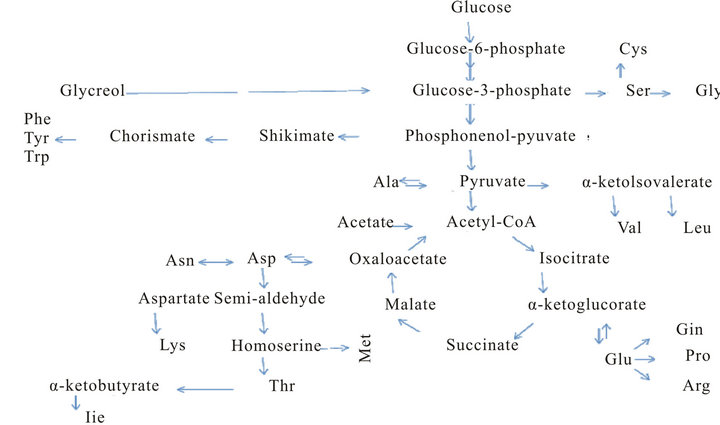
Figure 4. Pathways of amino acid biosynthesis in E. coli.
intermediates in the metabolic pathway. Isotope-scrambling in E. coli can be reduced by reducing the activity of the enzyme(s) catalyzing the inter-conversion of amino acids using specific (auxotrophic) strains [107] or, enzyme inhibitors [108] or, by doing cell-free synthesis with one more alternative of using in-vitro systems that lack these enzymes [109].
Over the last few decades deuterium labeling have played a vital role in solution NMR studies of proteins, in by most cases improving the quality of spectra by both including reduction in the number of peaks and narrowing of line widths [110]. To reduce the complexity of one-dimensional (1D) 1H spectra of proteins, Crespi HL and Jardetzky O. initially used the deuteration method in a set of refined experiments [111-113]. Since then, amino-acid selective labeling in deuterated environment or, selective deuteration in an otherwise protonated molecule has been regularly used for spectral simplification and residue type assignment [113]. As the gyromagnetio-ratio of deuterium (2H) is significantly lower than the proton (1H), replacement of 1H by 2H removes contribution to proton line widths from proton-proton dipolar relaxation and 1H-1H scalar coupling. Gain in sensitivity was initially demonstrated in 1D 1H spectra of 43 kDa E. coli EF-Tu protein [114] and afterwards in 2D homonuclear spectra of E. coli thioredoxin used for chemical-shift assignment [115]; significant improvements have also been noted in many heteronuclear NMR experiments later on it has also been reported that substitution of aliphatic/aromatic protons with deuterons result in impressive sensitivity gain in NOESY spectra that record NH-NH correlations [116].
The optimal level of deuteration, i.e. uniform or, fractional labeling of deuterium depends on the experiments that are planned on that particular protein. Uniformly labeled deuterium has been used to eliminate signal from one component in a macromolecular complex [117]. Although fractionally labeled deuterium samples also have several applications like improvement of sensitivity of 2D 1H-1H homonuclear spectra by reducing dipolar relaxation pathways, spin-diffusion and passive scalar couplings; fractional deuteration also significantly improves the sensitivity of many triple resonance experiments (15N, 13C, 1H) and side-chain dynamics of protein prepared in this manner can be studied using a number of new 13C and 2H relaxation experiments [118,119]. Now a days, combination of isotopic labeling and multidimensional multinuclear experiments is used which has significantly expanded the range of problems in structural biology amenable to NMR.
4. EXPRESSION OF HETEROLOGOUS PROTEINS IN E. COLI FOR NMR STUDIES
The high yield expression of heterologous protein in E. coli requires modification of rare codons in the host according to its usage frequency. The codon usage frequency refers to t-RNA levels proportional to 61 amino acid codons within a functional mRNA molecule [120]. The improvement thus attained does not alter the amino acid sequence of the encoded protein but codons which otherwise would exacerbate the degeneracy property of the genetic code. As reported by J F Kane 1995, the rare codon clusters of AGG/AGA, CUA, AUA, CGA, CCC causes translational errors. The greater problems are frame shifts and low level expression of heterologous protein; a mistranslation goes unrecognized [120]. Therefore, gene synthesis performed by monitoring E. coli codon usage frequency, GC content and unfavorable codon pairs eliminates codon biases [121,122] which favors over expression of proteins [36,123].
Improving the solubility of recombinant proteins in E. coli commonly involves changing some of the expression factors like reduced temperature, changes in the E. coli expression strain, different promoters or induction conditions, reduced translational rates and co-expression of molecular chaperones and folding modulators. Heat shock can enhance protein solubility without affecting induction [124]. All have been examined to show increased chances of folding into a native state prior to aggregating with folding intermediates leading to enhancement of soluble protein production.
There are many factors involved in expression of target proteins but codon bias plays an important role. Considering other factors like selection of expression vectors and transcriptional promoters are equally important. The codon biases vary in the same operon or during recombinant expression produced at high or low levels within same or different organisms [125]. The expression of target proteins can be increased in the host by manipulating over-expression of genes coding for rare tRNAs which masks the associated problem of low expression which might have been because of rare codons appearing as clusters or in the amino terminus of target protein [123].
When a heterologous protein is over-expressed in E. coli misfolding and aggregation occur frequently, resulting in the aggregation of proteins into inclusion bodies. Difficulty to express proteins of higher organisms or of eukaryotic origin in E. coli is due to an order of magnitude faster rate of translation and protein folding in latter compared to the former system [24,124]. Incubating inclusion bodies with 10% sarkosyl effectively yields > 95% of solubilized proteins. Using specific ratio of Triton X-100 and CHAPS, a high yield recovery of protein is possible from sarkosyl-solubilized fusion proteins. A combination of these three detergents significantly improves binding efficiency of GST and GST fusion proteins to glutathione (GSH) Sepharose. It is postulated that the sarkosyl molecules encapsulate proteins and disrupt aggregates, while Triton X-100 and CHAPS, with a critical micelle concentrations of 0.25 mM and 6 - 10 mM, respectively, forms mixed micelle or bicelle structures that incorporate sarkosyl molecules from the solution. This in turn facilitates proper protein refolding [126].
5. CONCLUSION
The development of new methods and technologies to over-express proteins in E. coli has opened up new avenues for structural studies of proteins. It is expected that this trend will continue and many new vectors/methods will be developed for high yield expression of eukaryotic proteins (Table 6).
6. ACKNOWLEDGEMENTS
The facilities provided by NMR Research Centre at IISc supported by Department of Science and Technology (DST), India is gratefully ac
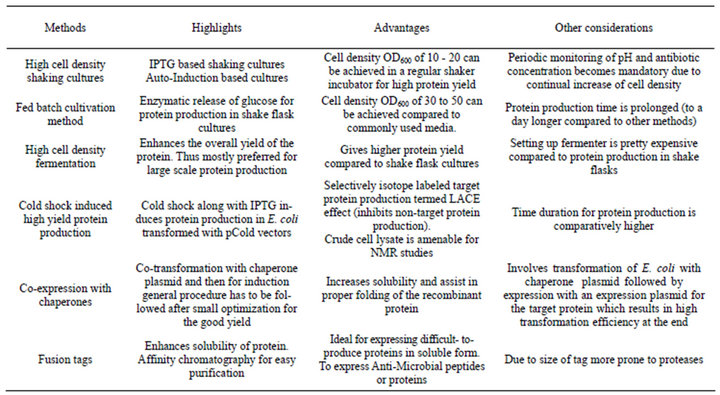
Table 6. Comparison of different methods for high yield protein expression.
.
REFERENCES
- Wüthrich, K. (1986) NMR of proteins and nucleic acids, Wiley, New York.
- Atreya, H.S. (2012) Isotope labeling in biomolecular NMR. Springer, Netherlands. doi:10.1007/978-94-007-4954-2
- Jana, S. and Deb, J.K. (2005) Strategies for efficient production of heterologous proteins in Escherichia coli. Applied Microbiology and Biotechnology, 67, 289-298. doi:10.1007/s00253-004-1814-0
- Studier, F.W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expression and Purification, 41, 207-234. doi:10.1016/j.pep.2005.01.016
- Makrides, S.C. (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiological Reviews, 60, 512-538.
- Burgess-Brown, N.A., Sharma, S., Sobott, F., Loenarz, C., Oppermann, U. and Gileadi, O. (2008) Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expression and Purification, 59, 94-102. doi:10.1016/j.pep.2008.01.008
- Sivashanmugam, A., Murray, V., Cui, C.X., Zhang, Y.H., Wang, J.J. and Li, Q.Q. (2009) Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Science, 18, 936-948. doi:10.1002/pro.102
- Graslund, S., Nordlund, P., Weigelt, J., Bray, J., Hallberg, B.M., Gileadi, O., Knapp, S., Oppermann, U., Arrowsmith, C., Hui, R., Ming, J., Dhe-Paganon, S., Park, H.W., Savchenko, A., Yee, A., Edwards, A., Vincentelli, R., Cambillau, C., Kim, R., Kim, S.H., Rao, Z., Shi, Y., Terwilliger, T.C., Kim, C.Y., Hung, L.W., Waldo, G.S., Peleg, Y., Albeck, S., Unger, T., Dym, O., Prilusky, J., Sussman, J.L., Stevens, R.C., Lesley, S.A., Wilson, I.A., Joachimiak, A., Collart, F., Dementieva, I., Donnelly, M.I., Eschenfeldt, W.H., Kim, Y., Stols, L., Wu, R., Zhou, M., Burley, S.K., Emtage, J.S., Sauder, J. M.,Thompson, D., Bain, K., Luz, J., Gheyi, T., Zhang, F., Atwell, S., Almo, S.C., Bonanno, J.B., Fiser, A., Swaminathan, S., Studier, F.W., Chance, M.R., Sali, A., Acton, T.B., Xiao, R., Zhao, L., Ma, L.C., Hunt, J.F., Tong, L., Cunningham, K., Inouye, M., Anderson, S., Janjua, H., Shastry, R., Ho, C.K., Wang, D.Y., Wang, H., Jiang, M., Montelione, G.T., Stuart, D.I., Owens, R.J., Daenke, S., Schutz, A., Heinemann, U., Yokoyama, S., Bussow, K. and Gunsalus, K.C., (2008) Protein production and purification. Nature Methods, 5, 135-146. doi:10.1038/nmeth.f.202
- Li, Z.P., Kessler, W., van den Heuvel, J. and Rinas, U. (2011) Simple defined autoinduction medium for highlevel recombinant protein production using T7-based Escherichia coli expression systems. Applied Microbiology and Biotechnology, 91, 1203-1213. doi:10.1007/s00253-011-3407-z
- Goto, N.K. and Kay, L.E. (2000) New developments in isotope labeling strategies for protein solution NMR spectroscopy. Current Opinion in Structural Biology, 10, 585-592. doi:10.1016/S0959-440X(00)00135-4
- Marley, J., Lu, M. and Bracken, C. (2001) A method for efficient isotopic labeling of recombinant proteins. Journal of Biomolecular NMR, 20, 71-75. doi:10.1023/A:1011254402785
- Krause, M., Ukkonen, K., Haataja, T., Ruottinen, M., Glumoff, T., Neubauer, A., Neubauer, P. and Vasala, A. (2010) A novel fed-batch based cultivation method provides high cell-density and improves yield of soluble recombinant proteins in shaken cultures. Microbial Cell Factories, 9, 1-11.
- Pinsach, J., de Mas, C., Lopez-Santin, J., Striedner, G. and Bayer, K. (2008) Influence of process temperature on recombinant enzyme activity in Escherichia coli fed-batch Cultures. Enzyme and Microbial Technology, 43, 507-512. doi:10.1016/j.enzmictec.2008.08.007
- Fong, B.A. and Wood, D.W. (2010) Expression and purification of ELP-intein-tagged target proteins in high cell density E. coli fermentation. Microbial Cell Factories, 9, 1-11.
- Tripathi, N.K., Sathyaseelan, K., Jana, A.M. and Rao, P.V.L. (2009) High Yield production of heterologous proteins with Escherichia coli. Defence Science Journal, 59, 137- 146.
- Qing, G.L., Ma, L.C., Khorchid, A., Swapna, G.V.T., Mal, T.K., Takayama, M.M., Xia, B., Phadtare, S., Ke, H.P., Acton, T., Montelione, G.T., Ikura, M. and Inouye, M. (2004) Cold-shock induced high-yield protein production in Escherichia coli. Nature Biotechnology, 22, 877- 882. doi:10.1038/nbt984
- Bae, W., Jones, P.G. and Inouye, M. (1997) CspA, the major cold shock protein of Escherichia coli, negatively regulates its own gene expression. Journal of Bacteriology, 179, 7081-7088.
- Kobayashi, H., Yoshida, T. and Inouye, M. (2009) Significant enhanced expression and solubility of human proteins in Escherichia coli by fusion with protein S from Myxococcus xanthus. Applied and Environmental Microbiology, 75, 5356-5362. doi:10.1128/AEM.00691-09
- Ducka, P., Eckhard, U., Schonauer, E., Kofler, S., Gottschalk, G., Brandstetter, H. and Nuss, D. (2009) A universal strategy for high-yield production of soluble and functional clostridial collagenases in E. coli. Applied Microbiology and Biotechnology, 83, 1055-1065. doi:10.1007/s00253-009-1953-4
- Lee, M.S., Hseu, Y.C., Lai, G.H., Chang, W.T., Chen, H.J., Huang, C.H., Wang, M.Y., Kao, J.Y., You, B.J., Lin, W.H., Lien, Y.Y. and Lin, M.K. (2011) High yield expression in a recombinant E. coli of a codon optimized chicken anemia virus capsid protein VP1 useful for vaccine development. Microbial Cell Factories, 10, 1-11.
- Das, K.M.P., Banerjee, S., Shekhar, N., Damodaran, K., Nair, R., Somani, S., Raiker, V.P., Jain, S. and Padmanabhan, S. (2011) Cloning, soluble expression and purification of high yield recombinant hGMCSF in Escherichia coli. International Journal of Molecular Sciences, 12, 2064-2076. doi:10.3390/ijms12032064
- Swain, M., Slomiany, M.G., Rosenweig, S.A. and Atreya, H.S. (2010) High-yield bacterial expression and structural characterization of recombinant human insulin-like growth factor binding protein-2. Archives of Biochemistry and Biophysics, 501, 195-200. doi:10.1016/j.abb.2010.06.006
- Zhou, P. and Wagner, G. (2010) Overcoming the solubility limit with solubility-enhancement tags: Successful applications in biomolecular NMR studies. Journal of Biomolecular NMR, 46, 23-31. doi:10.1007/s10858-009-9371-6
- Esposito, D. and Chatterjee, D.K. (2006) Enhancement of soluble protein expression through the use of fusion tags. Current Opinion in Biotechnology, 17, 353-358. doi:10.1016/j.copbio.2006.06.003
- Catanzariti, A.M., Soboleva, T.A., Jans, D.A., Board, P.G. and Baker, R.T. (2004) An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Science, 13, 1331-1339. doi:10.1110/ps.04618904
- Kotzsch, A., Vernet, E. Hammarstrom, M., Berthelsen, J., Weigelt, J., Graslund, S. and Sundstrom, M. (2011) A secretory system for bacterial production of high-profile protein targets. Protein Science, 20, 597-609. doi:10.1002/pro.593
- Narayanan, A., Ridilla, M. and Yernool, D.A. (2011) Restrained expression, a method to overproduce toxic membrane proteins by exploiting operator-repressor interactions. Protein Science, 20, 51-61. doi:10.1002/pro.535
- Yi, S.L., Brickenden, A. and Choy, W.Y. (2008) A new protocol for high-yield purification of recombinant human prothymosin alpha expressed in Escherichia coli for NMR studies. Protein Expression and Purification, 57, 1-8. doi:10.1016/j.pep.2007.09.005
- Chaga, G., Bochkariov, D.E., Jokhadze, G.G., Hopp, J. and Nelson, P. (1999) Natural poly-histidine affinity tag for purification of recombinant proteins on cobalt (II)- carboxymethylaspartate crosslinked agarose. Journal of Chromatography A, 864, 247-256. doi:10.1016/S0021-9673(99)01008-0
- Chatterjee, S., Schoepe, J., Lohmer, S. and Schomburg, D. (2005) High level expression and single-step purification of hexahistidine-tagged L-2 hydroxyisocaproate dehydrogenase making use of a versatile expression vector set. Protein Expression and Purification, 39, 137-143. doi:10.1016/j.pep.2004.08.019
- Ratnala, V.R.P., Swarts, H.G.P., VanOostrum, J., Leurs, R., DeGroot, H.J.M., Bakker, R.A. and DeGrip, W.J. (2004) Large-scale overproduction, functional purification and ligand affinities of the His-tagged human histamine H1 receptor. European Journal of Biochemistry, 271, 2636- 2646. doi:10.1111/j.1432-1033.2004.04192.x
- Scheich, C., Sievert, V. and Bussow, K. (2003) An automated method for high-throughput protein purification applied to a comparison of His-tag and GST-tag affinity chromatography. BMC Biotechnology, 3, 1-8.
- Smyth, D.R., Mrozkiewicz, M.K., McGrath, W.J., Listwan, P. and Kobe, B. (2003) Crystal structures of fusion proteins with large-affinity tags. Protein Science, 12, 1313- 1322. doi:10.1110/ps.0243403
- Purbey, P.K., Jayakumar, P.C., Deepalakshmi, P.D., Patole, M.S. and Galande, S. (2005) GST fusion vector with caspase-6 cleavage site for removal of fusion tag during column purification. BioTechniques, 38, 360-366. doi:10.2144/05383BM03
- Fox, J.D., Routzahn, K.M., Bucher, M.H. and Waugh, D.S. (2003) Maltodextrin-binding proteins from diverse bacteria and archaea are potent solubility enhancers. FEBS Letters, 537, 53-57. doi:10.1016/S0014-5793(03)00070-X
- Dyson, M.R., Shadbolt, S.P., Vincent, K.J., Perera, R.L. and McCafferty, J. (2004) Production of soluble mammalian proteins in Escherichia coli: Identification of protein features that correlate with successful expression. BMC Biotechnology, 4, 32.
- Feher, A., Boross, P., Sperka, T., Oroszlan, S. and Tozser, J. (2004) Expression of the murine leukemia virus protease in fusion with maltose-binding protein in Escherichia coli. Protein Expression and Purification, 35, 62-68. doi:10.1016/j.pep.2004.01.008
- Nallamsetty, S. and Waugh, D.S. (2006) Solubility-enhancing proteins MBP and NusA play a passive role in the folding of their fusion partners. Protein Expression and Purification, 45, 175-182. doi:10.1016/j.pep.2005.06.012
- Kim, S. and Lee, S.B. (2008) Soluble expression of archaeal proteins in Escherichia coli by using fusion-partners. Protein Expression and Purification, 62, 116-119. doi:10.1016/j.pep.2008.06.015
- Einhauer, A. and Jungbauer, A. (2001) The FLAGTM peptide, a versatile fusion tag for the purification of recombinant proteins. Journal of Biochemical and Biophysical Methods, 49, 455-465. doi:10.1016/S0165-022X(01)00213-5
- Knappik, A. and Pluckthun, A. (1994) An improved affinity tag based on the FLAG (R) peptide for the detection and purification of recombinant antibody fragments. Biotechniques, 17, 754-761.
- Slootstra, J.W., Kuperus, D., Pluckthun, A. and Meloen, R.H. (1997) Identification of new tag sequences with differential and selective recognition properties for the antiFLAG monoclonal antibodies M1, M2 and M5. Molecular Diversity, 2, 156-164. doi:10.1007/BF01682203
- Terpe, K. (2003) Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Applied Microbiology and Biotechnology, 60, 523-533.
- Vaillancourt, P., Zheng, C.F., Hoang, D.Q. and Briester, L. (2000) Affinity purification of recombinant proteins fuse to calmodulin or to calmodulin-binding peptides. Applications of Chimeric Genes and Hybrid Proteins, Pt. A, 326, 340-362.
- Kato, A., Maki, K., Ebina, T., Kuwajima, K., Soda, K. and Kuroda, Y. (2007) Mutational analysis of protein solubility enhancement using short peptide tags. Biopolymers, 85, 12-18. doi:10.1002/bip.20596
- Zhang, Y.B., Howitt, J., McCorkle, S., Lawrence, P., Springer, K. and Freimuth, P. (2004) Protein aggregation during over expression limited by peptide extensions with large net negative charge. Protein Expression and Purification, 36, 207-216.
- Marblestone, J.G., Edavettal, S.C., Lim, Y., Lim, P., Zuo, X. and Butt, T.R. (2006) Comparison of SUMO fusion technology with traditional gene fusion systems: Enhanced expression and solubility with SUMO. Protein Science, 15, 182-189. doi:10.1110/ps.051812706
- Skerra, A. and Schmidt, T.G.M. (2000) Use of the streptag and streptavidin for detection and purification of recombinant proteins. Applications of Chimeric Genes and Hybrid Proteins, Pt. A, 326, 271-304.
- Witte, C.P., Noel, L.D., Gielbert, J., Parker, J.E. and Romeis, T. (2004) Rapid one-step protein purification from plant material using the eight-amino acid StrepII epitope. Plant Molecular Biology, 55, 135-147. doi:10.1007/s11103-004-0501-y
- Junttila, M.R., Saarinen, S., Schmidt, T., Kast, J. and Westermarck, J. (2005) Single-step Strep-tag (R) purifycation for the isolation and identification of protein complexes from mammalian cells. Proteomics, 5, 1199-1203. doi:10.1002/pmic.200400991
- Chong, S.R., Mersha, F.B., Comb, D.G., Scott, M.E., Landry, D., Vence, L.M., Perler, F.B., Benner, J., Kucera, R.B., Hirvonen, C.A., Pelletier, J.J., Paulus, H. and Xu, M.Q. (1997) Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene, 192, 271-281. doi:10.1016/S0378-1119(97)00105-4
- Predonzani, A., Arnoldi, F., Lopez-Requena, A. and Burrone, O.R. (2008) In vivo site-specific biotinylation of proteins within the secretory pathway using a single vector system. BMC Biotechnology, 8, 41.
- Lichty, J.J., Malecki, J.L., Agnew, H.D., MichelsonHorowitz, D.J. and Tan, S. (2005) Comparison of affinity tags for protein purification. Protein Expression and Purification, 41, 98-105. doi:10.1016/j.pep.2005.01.019
- Durst, F.G., Ou, H.D., Lohr, F., Dotsch, V. and Straub, W.E. (2008) The better tag remains unseen. Journal of the American Chemical Society, 130, 14932-14933. doi:10.1021/ja806212j
- Smits, S.H.J., Mueller, A., Grieshaber, M.K. and Schmitt, L. (2008) Coenzymeand His-tag-induced crystallization of octopine dehydrogenase. Acta Crystallographica Section F-Structural Biology and Crystallization Communications, 64, 836-839. doi:10.1107/S1744309108025487
- Carson, M., Johnson, D.H., McDonald, H., Brouillette, C. and DeLucas, L.J. (2007) His-tag impact on structure. Acta Crystallographica Section D-Biological Crystallography, 63, 295-301. doi:10.1107/S0907444906052024
- Walker, P.A., Leong, L.E.C., Ng, P.W.P., Tan, S.H., Waller, S., Murphy, D. and Porter, A.G. (1994) Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Bio-Technology, 12, 601-605. doi:10.1038/nbt0694-601
- Arnau, J., Lauritzen, C., Petersen, G.E. and Pedersen, J. (2006) Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expression and Purification, 48, 1-13. doi:10.1016/j.pep.2005.12.002
- Jenny, R.J., Mann, K.G. and Lundblad, R.L. (2003) A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expression and Purification, 31, 1-11. doi:10.1016/S1046-5928(03)00168-2
- [61] Liew, O.W., Chong, J.P.C., Yandle, T.G. and Brennan, S.O. (2005) Preparation of recombinant thioredoxin fused N-terminal proCNP: Analysis of enterokinase cleavage products reveals new enterokinase cleavage sites. Protein Expression and Purification, 41, 332-340. doi:10.1016/j.pep.2005.03.006
- [62] Leong, L.E.C. (1999) The use of recombinant fusion proteases in the affinity purification of recombinant proteins. Molecular Biotechnology, 12, 269-274. doi:10.1385/MB:12:3:269
- [63] Hefti, M.H., Van Vugt-Van der Toorn, C.J.G., Dixon, R. and Vervoort, J. (2001) A novel purification method for histidine-tagged proteins containing a thrombin cleavage site. Analytical Biochemistry, 295, 180-185. doi:10.1006/abio.2001.5214
- [64] Waugh, D.S. (2011) An overview of enzymatic reagents for the removal of affinity tags. Protein Expression and Purification, 80, 283-293. doi:10.1016/j.pep.2011.08.005
- [65] Kwon, S.Y., Choi, Y.J., Kang, T.H., Lee, K.H., Cha, S.S., Kim, G.H., Lee, H.S., Kim, K.T. and Kim, K.J. (2005) Highly efficient protein expression and purification using bacterial hemoglobin fusion vector. Plasmid, 53, 274-282. doi:10.1016/j.plasmid.2004.11.006
- [66] de Marco, A., Casatta, E., Savaresi, S. and Geerlof, A. (2004) Recombinant proteins fused to thermostable partners can be purified by heat incubation. Journal of Biotechnology, 107, 125-133. doi:10.1016/j.jbiotec.2003.10.008
- [67] Rubio, V., Shen, Y.P., Saijo, Y., Liu, Y.L., Gusmaroli, G., Dinesh-Kumar, S.P. and Deng, X.W. (2005) An alternative tandem affinity purification strategy applied to Arabidopsis protein complex isolation. Plant Journal, 41, 767-778. doi:10.1111/j.1365-313X.2004.02328.x
- [68] Waugh, D.S. (2005) Making the most of affinity tags. Trends in Biotechnology, 23, 316-320. doi:10.1016/j.tibtech.2005.03.012
- [69] Nirenberg, M. and Matthaei, J.H. (1961) Dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proceedings of the National Academy of Sciences of the United States of America, 47, 1588-1602. doi:10.1073/pnas.47.10.1588
- [70] Carlson, E.D., Gan, R., Hodgman, C.E. and Jewett, M.C. (2012) Cell-free protein synthesis: Applications come of age. Biotechnology Advances, 30, 1185-1194. doi:10.1016/j.biotechadv.2011.09.016
- [71] Savage, D.F., Anderson, C.L., Robles-Colmenares, Y., Newby, Z.E. and Stroud, R.M. (2007) Cell-free complements in vivo expression of the E. coli membrane proteome. Protein Science, 16, 966-976. doi:10.1110/ps.062696307
- [72] Ozawa, K., Dixon, N.E. and Otting, G. (2005) Cell-free synthesis of N-15-labeled proteins for NMR studies. Iubmb Life, 57, 615-622. doi:10.1080/15216540500217859
- [73] Sawasaki, T., Ogasawara, T., Morishita, R. and Endo, Y. (2002) A cell-free protein synthesis system for highthroughput proteomics. Proceedings of the National Academy of Sciences of the United States of America, 99, 14652-14657. doi:10.1073/pnas.232580399
- [74] Vinarov, D.A., Newman, C.L.L. and Markley, J.L. (2006) Wheat germ cell-free platform for eukaryotic protein production. FEBS Journal, 273, 4160-4169. doi:10.1111/j.1742-4658.2006.05434.x
- [75] Nakano, H., Tanaka, T., Kawarasaki, Y. and Yamane, T. (1996) Highly productive cell-free protein synthesis system using condensed wheat-germ extract. Journal of Biotechnology, 46, 275-282. doi:10.1016/0168-1656(96)00022-3
- [76] Klammt, C., Lohr, F., Schafer, B., Haase, W., Dotsch, V., Ruterjans, H., Glaubitz, C. and Bernhard, F. (2004) High level cell-free expression and specific labeling of integral membrane proteins. European Journal of Biochemistry, 271, 568-580. doi:10.1111/j.1432-1033.2003.03959.x
- [77] Jewett, M.C. and Swartz, J.R. (2004) Mimicking the Escherichia coli cytoplasmic environment activates longlived and efficient cell-free protein synthesis. Biotechnology and Bioengineering, 86, 19-26. doi:10.1002/bit.20026
- [78] Thomas, J.G., Ayling, A. and Baneyx, F. (1997) Molecular chaperones, folding catalysts, and the recovery of active recombinant proteins from E-coli—To fold or to refold. Applied Biochemistry and Biotechnology, 66, 197- 238. doi:10.1007/BF02785589
- [79] Brinker, A., Pfeifer, G., Kerner, M.J., Naylor, D.J., Hartl, F.U. and Hayer-Hartl, M. (2001) Dual function of protein confinement in chaperonin-assisted protein folding. Cell, 107, 223-233. doi:10.1016/S0092-8674(01)00517-7
- [80] Gierasch, L.M., Wang, Z., Hunt, J., Landry, S.J., Weaver, A. and Deisenhofer, J. (1995) Chaperone-substrate interactions. Protein Engineering, 8, 14.
- [81] Hayer-Hartl, M.K., Ewalt, K.L. and Hartl, F.U. (1999) On the role of symmetrical and asymmetrical chaperonin complexes in assisted protein folding. Biological Chemistry, 380, 531-540. doi:10.1515/BC.1999.068
- [82] Tanaka, S., Kawata, Y., Otting, G., Dixon, N.E., Matsuzaki, K. and Hoshino, M. (2010) Chaperonin-encapsulation of proteins for NMR. Biochimica Et Biophysica ActaProteins and Proteomics, 1804, 866-871. doi:10.1016/j.bbapap.2009.12.016
- [83] Weissman, J.S., Rye, H.S., Fenton, W.A., Beechem, J.M. and Horwich, A.L. (1996) Characterization of the active intermediate of a GroEL-GroES-mediated protein folding reaction. Cell, 84, 481-490. doi:10.1016/S0092-8674(00)81293-3
- [84] Baneyx, F. and Mujacic, M. (2004) Recombinant protein folding and misfolding in Escherichia coli. Nature Biotechnology, 22, 1399-1408. doi:10.1038/nbt1029
- [85] Minton, A.P. (2005) Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: Macromolecular crowding and protein stability revisited. Biophysical Journal, 88, 971-985. doi:10.1529/biophysj.104.050351
- [86] Nishihara, K., Kanemori, M., Yanagi, H. and Yura, T. (2000) Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli. Applied and Environmental Microbiology, 66, 884-889. doi:10.1128/AEM.66.3.884-889.2000
- [87] Crooke, E. and Wickner, W. (1987) Trigger factor: A soluble-protein that folds pro-OmpA into a membraneassembly-competent form. Proceedings of the National Academy of Sciences of the United States of America, 84, 5216-5220. doi:10.1073/pnas.84.15.5216
- [88] Hesterkamp, T., Hauser, S., Lutcke, H. and Bukau, B. (1996) Escherichia coli trigger factor is a prolyl isomerase that associates with nascent polypeptide chains. Proceedings of the National Academy of Sciences of the United States of America, 93, 4437-4441. doi:10.1073/pnas.93.9.4437
- [89] Hartl, F.U. and Hayer-Hartl, M. (2002) Protein folding— Molecular chaperones in the cytosol: From nascent chain to folded protein. Science, 295, 1852-1858. doi:10.1126/science.1068408
- [90] Horwich, A.L., Fenton, W.A., Weissman, J.S., Boisvert, D.C., Braig, K. and Sigler, P.B. (1995) Structure and functional-analysis of the chaperonin GroEL. FASEB Journal, 9, A1251.
- [91] Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D.C., Joachimiak, A., Horwich, A.L. and Sigler, P.B. (1994) The crystal-structure of the bacterial chaperonin GroEL at 2.8-angstrom. Nature, 371, 578-586. doi:10.1038/371578a0
- [92] Hunt, J.F., Weaver, A.J., Landry, S.J., Gierasch, L. and Deisenhofer, J. (1996) The crystal structure of the GroES co-chaperonin at 2.8 angstrom resolution. Nature, 379, 37-45. doi:10.1038/379037a0
- [93] Schroder, H., Langer, T., Hartl, F.U. and Bukau, B. (1993) Dnak, dnaj and grpe form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO Journal, 12, 4137-4144.
- [94] Laufen, T., Mayer, M.P., Beisel, C., Klostermeier, D., Mogk, A., Reinstein, J. and Bukau, B. (1999) Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proceedings of the National Academy of Sciences of the United States of America, 96, 5452-5457. doi:10.1073/pnas.96.10.5452
- [95] Teter, S.A., Houry, W.A., Ang, D., Tradler, T., Rockabrand, D., Fischer, G., Blum, P., Georgopoulos, C. and Hartl, F.U. (1999) Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell, 97, 755-765. doi:10.1016/S0092-8674(00)80787-4
- [96] Bukau, B., Weissman, J. and Horwich, A. (2006) Molecular chaperones and protein quality control. Cell, 125, 443-451. doi:10.1016/j.cell.2006.04.014
- [97] Martin, J., Mayhew, M., Langer, T. and Hartl, F.U. (1993) The reaction cycle of GroEL and GroES in chaperoninassisted protein-folding. Nature, 366, 228-233. doi:10.1038/366228a0
- [98] Inobe, T., Makio, T., Takasu-Ishikawa, E., Terada, T.P. and Kuwajima, K. (2001) Nucleotide binding to the chaperonin GroEL: Non-cooperative binding of ATP analogs and ADP, and cooperative effect of ATP. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology, 1545, 160-173. doi:10.1016/S0167-4838(00)00274-0
- [99] Hayerhartl, M.K., Ewbank, J.J., Creighton, T.E. and Hartl, F.U. (1994) Conformational specificity of the chaperonin GroEL for the compact folding intermediates of alpha-lactalbumin. EMBO Journal, 13, 3192-3202.
- [100] Buchberger, A., Schroder, H., Hesterkamp, T., Schonfeld, H.J. and Bukau, B. (1996) Substrate shuttling between the DnaK and GroEL systems indicates a chaperone network promoting protein folding. Journal of Molecular Biology, 261, 328-333. doi:10.1006/jmbi.1996.0465
- [101] Mogk, A., Mayer, M.P. and Deuerling, E. (2002) Mechanisms of protein folding: Molecular chaperones and their application in biotechnology. Chembiochem, 3, 807-814. doi:10.1002/1439-7633(20020902)3:9<807::AID-CBIC807>3.0.CO;2-A
- [102] Fenton, W.A. and Horwich, A.L. (1997) GroEL-mediated protein folding. Protein Science, 6, 743-760. doi:10.1002/pro.5560060401
- [103] Langer, T., Lu, C., Echols, H., Flanagan, J., Hayer, M.K. and Hartl, F.U. (1992) Successive action of dnak, dnaj and GroEL along the pathway of chaperone-mediated protein folding. Nature, 356, 683-689. doi:10.1038/356683a0
- [104] Baneyx, F. and Nannenga, B.L. (2010) Chaperones: A story of thrift unfolds. Nature Chemical Biology, 6, 880- 881. doi:10.1038/nchembio.468
- [105] Paliy, O. and Gunasekera, T.S. (2007) Growth of E-coli BL21 in minimal media with different gluconeogenic carbon sources and salt contents. Applied Microbiology and Biotechnology, 73, 1169-1172. doi:10.1007/s00253-006-0554-8
- [106] Zhang, Z.G., Gosset, G., Barabote, R., Gonzalez, C.S., Cuevas, W.A. and Saier, M.H. (2005) Functional interacttions between the carbon and iron utilization regulators, Crp and Fur, in Escherichia coli. Journal of Bacteriology, 187, 980-990. doi:10.1128/JB.187.3.980-990.2005
- [107] Muchmore, D.D., McIntosh, L.P., Russell, C.B., Anderson, D.E. and Dahlquist, F.W. (1989) Expression and nitrogen-15 labelling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods in Enzymology, 177, 44-73. doi:10.1016/0076-6879(89)77005-1
- [108] Muchmore, D.C., McIntosh, L.P., Russell, C.B., Anderson, D.E. and Dahlquist, F.W. (1989) Expression and n- 15 labeling of proteins for proton and n-15 nuclear-magnetic-resonance. Methods in Enzymology, 177, 44-73. doi:10.1016/0076-6879(89)77005-1
- [109] Tong, K.I., Yamamoto, M. and Tanaka, T. (2008) A simple method for amino acid selective isotope labeling of recombinant proteins in E-coli. Journal of Biomolecular NMR, 42, 59-67. doi:10.1007/s10858-008-9264-0
- [110] Ohki, S. and Kainosho, M. (2008) Stable isotope labeling methods for protein NMR. Progress in Nuclear Magnetic Resonance Spectroscopy, 53, 208-226. doi:10.1016/j.pnmrs.2008.01.003
- [111] Gardner, K.H. and Kay, L.E. (1998) The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annual Review of Biophysics and Biomolecular Structure, 27, 357-406. doi:10.1146/annurev.biophys.27.1.357
- [112] Crespi, H.L. and Katz, J.J. (1969) High resolution proton magnetic resonance studies of fully deuterated and isotope hybrid proteins. Nature, 224, 560-562. doi:10.1038/224560a0
- [113] Katz, J.J. and Crespi, H.L. (1966) Deuterated organisms: Cultivation and uses. Science, 151, 1187-1194. doi:10.1126/science.151.3715.1187
- [114] Pachter, R., Arrowsmith, C.H. and Jardetzky, O. (1992) The effect of selective deuteration on magnetization transfers in larger proteins. Journal of Biomolecular NMR, 2, 183-194. doi:10.1007/BF01875529
- [115] Kalbitzer, H.R., Leberman, R. and Wittinghofer, A. (1985) 1H-NMR spectroscopy on elongation-factor Tu from escherichia-coli. Resolution enhancement by perdeuteration. FEBS Letters, 180, 40-42. doi:10.1016/0014-5793(85)80226-X
- [116] Lemaster, D.M. and Richards, F.M. (1988) NMR sequential assignment of escherichia-coli thioredoxin utilizing random fractional deuteriation. Biochemistry, 27, 142- 150. doi:10.1021/bi00401a022
- [117] Torchia, D.A., Sparks, S.W. and Bax, A. (1988) Delineation of alpha-helical domains in deuteriated staphylococcal nuclease by 2D Noe NMR spectroscopy. Journal of the American Chemical Society, 110, 2320-2321. doi:10.1021/ja00215a063
- [118] Batey, R.T., Cloutier, N., Mao, H.Y. and Williamson, J.R. (1996) Improved large scale culture of Methylophilus methylotrophus for 13C/15N labeling and random fractional deuteration of ribonucleotides. Nucleic Acids Research, 24, 4836-4837. doi:10.1093/nar/24.23.4836
- [119] Agback, P., Maltseva, T.V., Yamakage, S.I., Nilson, F.P.R., Foldesi, A. and Chattopadhyaya, J. (1994) The differences in the T2 relaxation rates of the protons in the partially-deuteriated and fully protonated sugar residues in a large oligo-DNA (NMR-window) gives complementary structural information. Nucleic Acids Research, 22, 1404-1412. doi:10.1093/nar/22.8.1404
- [120] Foldesi, A., Yamakage, S.I., Nilsson, F.P.R., Maltseva, T.V. and Chattopadhyaya, J. (1996) The use of non-uniform deuterium labelling ‘NMR-window’ to study the NMR structure of a 21mer RNA hairpin. Nucleic Acids Research, 24, 1187-1194. doi:10.1093/nar/24.7.1187
- [121] Kane, J.F. (1995) Effects of rare codon clusters on highlevel expression of heterologous proteins in Escherichiacoli. Current Opinion in Biotechnology, 6, 494-500. doi:10.1016/0958-1669(95)80082-4
- [122] Knight, R.D., Freeland, S.J. and Landweber, L.F. (2001) A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biology, 2, 1- 13.
- [123] Boycheva, S., Chkodrov, G. and Ivanov, I. (2003) Codon pairs in the genome of Escherichia coli. Bioinformatics, 19, 987-998. doi:10.1093/bioinformatics/btg082
- [124] Gustafsson, C., Govindarajan, S. and Minshull, J. (2004) Codon bias and heterologous protein expression. Trends in Biotechnology, 22, 346-353. doi:10.1016/j.tibtech.2004.04.006
- [125] Chen, J.Q., Acton, T.B., Basu, S.K., Montelione, G.T. and Inouye, M. (2002) Enhancement of the solubility of proteins overexpressed in Escherichia coli by heat shock. Journal of Molecular Microbiology and Biotechnology, 4, 519-524.
- [126] Gouy, M. and Gautier, C. (1982) Codon usage in bacteria: Correlation with gene expressivity. Nucleic Acids Research, 10, 7055-7074. doi:10.1093/nar/10.22.7055
- [127] Tao, H., Liu, W.J., Simmons, B.N., Harris, H.K., Cox, T.C. and Massiah, M.A. (2010) Purifying natively folded proteins from inclusion bodies using sarkosyl, triton X-100, and CHAPS. Biotechniques, 48, 61-64. doi:10.2144/000113304
NOTES
*Corresponding author.