Advances in Chemical Engineering and Science
Vol. 2 No. 3 (2012) , Article ID: 20834 , 5 pages DOI:10.4236/aces.2012.23045
Synthesis of Vildagliptin-β-O-Glucuronide
Chemical and Analytical Development, Novartis Pharmaceuticals Corporation, One Health Plaza, East Hanover, USA
Email: *wen.shieh@Novartis.com
Received April 13, 2012; revised May 6, 2012; accepted May 17, 2012
Keywords: Vildagliptin; O-Glycosylation; DPP-4 Inhibitors; Type-2 Diabetes
ABSTRACT
A linear 7-step synthesis of vildagliptin-β-O-glucuronide (2) starting from commercially available D-glucurono-6,3- lactone (3) was herein achieved with 11.3% overall yield. Efficient preparation of compound 6 in pure α form was obtained, which was proved critical to achieve high anomeric selectivity in β-O-glycosylation later. The direct β-O-glycosylation of vildagliptin (1) containing both a tertiary alcohol and a secondary amine was studied and achieved in good yield. The deprotection step to afford product was delicately executed to avoid hydrolysis of nitrile group. The target compound 2 was obtained after purification by reversed-phase C18 chromatography.
1. Introduction
Vildagliptin (Galvus®) (1, Scheme 1) is a member of a new class of DPP-4 (dipeptidyl peptidase-4) inhibitors for the treatment of type-2 diabetes [1]. Vildagliptin has been approved for use in more than 70 countries including Japan and the European Union.
During early tox studies in monkeys, several vildagliptin metabolites were found. One of them was vildagliptin-β-O-glucuronide (2) [2]. To support further tox evaluation of this metabolite, our objective was to develop a practical route for the preparation of gram quantities of this compound. Therefore, a multi-step synthesis of 2 was designed as shown below. Based on a retro-synthetic analysis (Scheme 1), 2 could be prepared via O-glycosylation of 1 with the corresponding glucuronic acid derivative 7, which could in turn be prepared from D-(+)-glucurono- 6,3-lactone (3) using a published procedure [3]. The successful synthesis of 2 critically would rely on how the O-glycosylation of 1 with 7 is taken. Herein, we disclose an efficient synthesis of 2, which is the first chemical synthesis of 2 ever reported.
O-glycosylation methodologies have been well documented [4,5]. Reactivity of the glycosyl donor can be tuned by putting on a different leaving group (X) to meet the needs for O-glycosylation of different glycosyl acceptors (Scheme 1). Acetoxy group at 5-position of a glycosyl donor has been reported to play a role in high stereoselective O-glycosylations. Besides, the selection of coupling reagents (activators) is critical. Stereoselective β-O-glycosylations of 1-adamantanol (tertiary alcohol) with different glycosyl donors have been reported [6,7]. Stereoselective β-O-glycosylation of morphine that contains secondary alcohol and tertiary amine has also been reported under Koenigs-Knorr conditions [8]. Stereoselective β-O-glycosylation of a phenol containing a tertiary amine with α-O-trichloroacetimidate glycosyl donor has also been reported [3]. However, stereoselective β-O-glycosylation of a substrate that contains a tertiary alcohol and a secondary amine has not been reported in literature to the best of our knowledge.
2. Results and Discussion
Synthesis of 5 from 3 through 4 (Scheme 2) was based on a reported procedure [3]. Acylation of 4 was completed within 14 h at rt, and 5 was precipitated by using brine/water in 76% yield as a mixture of anomers (35/65, α/β). Regio-selective deprotection of 5 leading to 6 could be achieved with either NH4OAc/DMF at room temperature [9] or tributyltin methoxide (Bu3SnOMe) in 1,2- dichloroethane at 90˚C [3]. While applying NH4OAc/ THF conditions, we found that the presence of sodium 2- ethyl-hexanoate was critical to drive the reaction to completion. A crystallization procedure was developed to afford 6 (pure α form) as pale solid in 53% yield.
The imidate 7 was synthesized from 6 using a reported method [3] with some optimization. The uses of molecular sieves during the reaction and flash chromatography for purification were eliminated. A crystallization procedure was developed to afford 7 in 78% yield. Since the C-O bond was intact in this reaction, the anomeric purity of 7 was unchanged from that of 6. As crystallization of a mixture (α/β) of 7 provided no enrichment of the desired α anomer, preparing anomerically pure 6 (α) was crucial
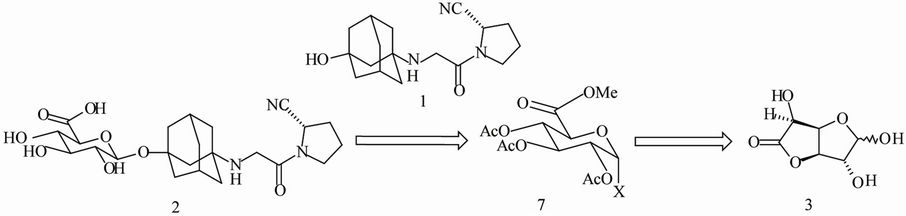
Scheme 1. Retro-synthetic analysis.
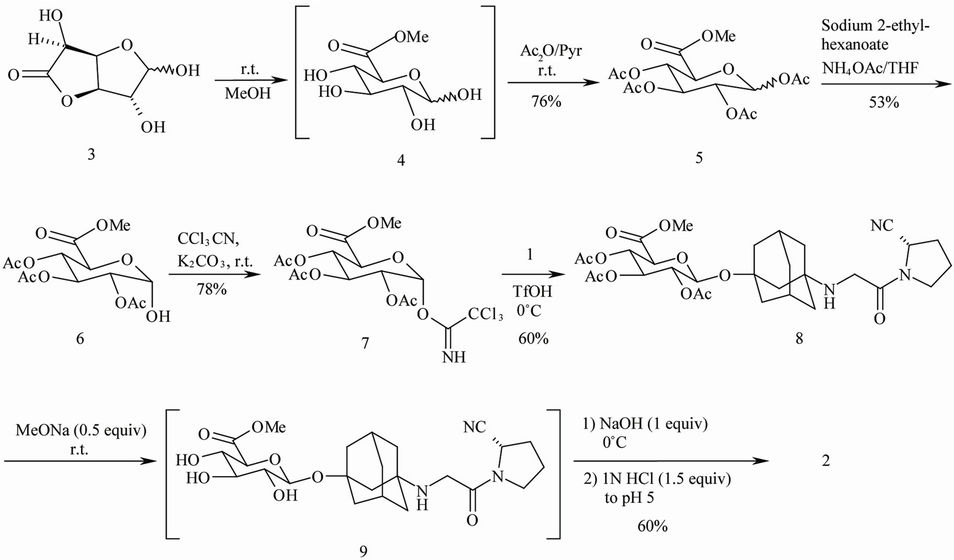
Scheme 2. Synthesis of target compound 2.
in preparing anomerically pure 7. We later found that pure α form of 7 was essential to give the β form of 8.
Stereoselective β-O-glycosylation of 1 with imidate 7 was a challenging step. Lewis acids have been known as activators in promoting this O-glycosylation. However, several Lewis acids such as TMSOTf, BF3 etherate, (nBu)2BOTf, (nBu)2Sn(OAc)2 and Yb(OTf)3, typically used for O-glycosylations were tried in the current study but gave 8 in only low yield (Table 1). The best Lewis acid for this glycosylation was BF3 etherate, which afforded a 25% isolated yield of 8. Since the secondary amine in vildagliptin is extremely difficult to protect, it may not be a competitive nucleophile. But it may pose as a base and impact or nullify the function of the Lewis acid. Thus stronger acids, such as protic acids were considered. More than 1 equiv of Lewis acid or protic acid was necessary for the reaction to take place. Unlike benzenesulfonic acid, methanesulfonic acid, or sulfuric acid,
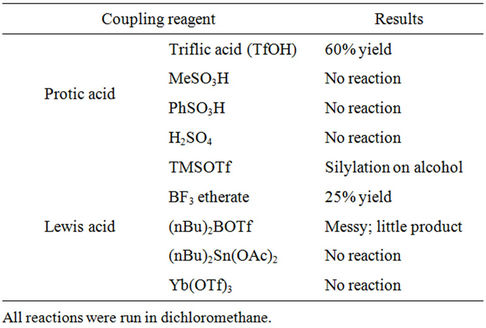
Table 1. Screening of coupling reagents.
triflic acid (TfOH) was effective for this stereoselective β-O-glycosylation. After work-up, crude 8 (15/85, α/β) was purified using silica gel chromatography to afford 8 (β) as a foamy solid in 60% yield. Further purification by re-crystallization from methanol/water afforded 8 as an off-white solid.
Deprotection of 8 to the target compound 2 was initially accomplished by heating a mixture of 8, MeOH, water and triethylamine at 50˚C for 3 h. However, under these conditions, an amide (formed by hydrolysis of the nitrile group) and an unknown byproduct was observed. Thus, a modified approach was used to deprotect 8. The acetyl groups were removed via transesterification at rt with MeOH/MeONa (0.5 equiv). The resulting methyl acetate byproduct was removed under reduced pressure. The residual methyl ester was hydrolyzed with MeOH/ NaOH (1 M in water, 1 equiv) at 0˚C. Finally, the reaction mixture was adjusted to pH 5 with 1 N HCl (1.5 equiv) at 0˚C. After removal of MeOH under reduced pressure, the residue was subjected to reversed-phase C18 column chromatography to afford 2 in 60% yield. Compound 2 was found to be identical to the metabolite isolated from Vildagliptin in all respects.
3. Experimental Section
Thin-layer chromatography (TLC) was performed on Kiesegel 60 F254 (Merck) plates and visualized by staining with iodine. Column chromatography utilized silica gel grade 60 (Fisher Chemical, 170 - 230 mesh). Reversed-phase chromatography was performed with C18 HS cartridge (Biotage). Melting points were uncorrected. NMR spectra were recorded in CDCl3 or DMSO-d6 using Bruker 500 or Bruker 400 as indicated. Anomeric ratios were determined by 1H NMR analysis.
(2S,3S,4S,5R)-3,4,5,6-Tetraacetoxytetrahydropyran- 2-carboxy-lic acid methyl ester (5). A 2-L, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer, was charged with the lactone (3, 177 g, 1.0 mol), methanol (1 L) and triethylamine (7.3 g, 70 mmol). The resulting suspension was stirred at 21˚C for 2 h. It gradually became a brown solution with a bit of undissolved solid. The reaction mixture was filtered over a pad of Celite. The flask and the filter cake were rinsed with MeOH (2 × 0.2 L). The filtrate was concentrated on a rotavapor (bath temperature 25˚C) to give 249 g of crude 4 as a foamy solid. Crude 4 was dissolved in warm pyridine (450 mL including rinse, 5.5 mol) and the resulting solution was transferred to another 2-L, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer. The brown solution was cooled in an ice bath under nitrogen. Acetic anhydride (663.6 g, 6.5 mol) was added via an addition funnel at <22˚C over 1 h. The dark solution was stirred at 21˚C overnight (14 h). The reaction mixture was cooled in an ice bath and quenched by adding water (540 g) via an addition funnel at <22˚C over 20 min. Thereafter, the mixture was stirred at 21˚C for 2 h. The mixture was poured into 4 L of brine in a 12-L, 4-necked, roundbottomed flask with stirring. The 2-L flask was rinsed with water (1.5 L) which was added to the mixture. The resulting suspension was stirred at 21˚C for 2 h and filtered over a polypropylene pad. The flask was rinsed with water (0.5 L), which was filtered through the cake. The filter cake was washed with water (2 × 0.5 L). The solid (425 g, wet) was dried in an oven (45˚C, 1 in Hg) for 20 h to offer 287 g of 5, as a sea-sand-like solid, in 76% yield over two steps from 3 as a mixture of α/β (35/65) anomers.
(2S,3S,4S,5R,6S)-3,4,5-Triacetoxy-6-hydroxytetrahydropyran-2-carboxylic acid methyl ester (6). A 1-L, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer, was charged with 5 (113 g, 300 mmol), sodium 2-ethyl-hexanoate (75 g, 450 mmol), ammonium acetate (46.2 g, 600 mmol) and THF (0.6 L). The resulting suspension was stirred at 21˚C for 24 h. THF was removed on a rotavapor (bath temperature 25˚C) to give a dark syrup. Isopropyl acetate (0.8 L) and water (0.6 L) were added. The two layers were separated. The aqueous layer was extracted with isopropyl acetate (1 × 0.35 L). The combined organic layers were washed with water (2 × 0.5 L). The organic solvent was removed on a rotavapor (bath temperature 25˚C) to give 150 g of gummy residue plus some pale liquid. The whole residue was dissolved in warm isopropyl acetate (200 mL including rinse) and the resulting solution was transferred to another 1-L, 3-necked, roundbottomed flask equipped with a mechanical stirrer and a digital thermometer. The brown solution was heated to 60˚C. Heptane (375 mL) was added via an addition funnel at 60˚C ± 3˚C over 0.5 h. The hazy solution was cooled. Seeds were added (0.2 g) at 55˚C and solids appeared at 45˚C - 50˚C. Additional heptane (75 mL) was added at 35˚C. The suspension was cooled to 21˚C and stirred at 21˚C for 2 h. The suspension was then filtered and the filter cake was washed with a 1:2 (v/v) mixture of isopropyl acetate/heptane (2 × 110 mL), via re-slurry. The solid (54 g) was dried in an oven (50˚C, 1 in Hg) for 14 h to afford 53.2 g of 6 (α), as a pale solid, in 53% yield. mp: 97.5˚C - 99.5˚C. 1H NMR (CDCl3, 500 MHz) δ: 5.58 (t, J = 10.0 Hz, 1H), 5.54 (bt, J = 3.15 Hz, 1H), 5.18 (t, J = 10.0 Hz, 1H), 4.91 (dd, J = 10.0, 3.45 Hz, 1H), 4.59 (d, J = 10.0 Hz, 1H), 3.83 (s, 1H), 3.75 (s, 3H), 2.09 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ: 170.13, 170.02, 169.66, 168.24, 90.28, 70.74, 69.52, 69.11, 68.07, 52.90, 20.65 (2C), 20.51.
(2S,3S,4S,5R,6R)-3,4,5-Triacetoxy-6-(2,2,2-trichloro-acetimidoyloxy)-tetrahydropyran-2-carboxylic acid methyl ester (7). A 250-mL, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer, was charged with 6 (20 g, 60 mmol), potassium carbonate (14.1 g, 102 mmol) and dichloromethane (80 mL, anhydrous) under nitrogen. The suspension was cooled in an ice bath. Trichloroacetonitrile (26 g, 180 mmol) was added via an addition funnel at 5˚C ± 3˚C over 5 min. Then, the reaction mixture was stirred at 21˚C for 16 h. The reaction mixture was poured into ice (70 g). Brine (50 mL) was added and the two layers were separated. The aqueous layer was extracted with dichloromethane (1 × 0.15 L). The combined organic extracts were washed with 10% NaCl (1 × 0.1 L) and dried over sodium sulfate. The solvent was removed on a rotavapor (bath temperature 25˚C) to give 27 g of a foamy syrup. The foamy syrup was dissolved in warm isopropyl acetate (35 mL including rinse) and the resulting solution was transferred to another 250-mL, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer. The brown solution was heated to 60˚C. Heptane (80 mL) was added via an addition funnel at 60˚C ± 3˚C over 0.5 h. The hazy solution was cooled. Seeds (0.2 g) were added at 50˚C and solids appeared at 30˚C. More heptane (70 mL) was added over 40 min. The suspension was then cooled to 0˚C ± 3˚C and stirred at 0˚C ± 3˚C for 1 h. The suspension was filtered and the filter cake was washed with a 1:4 (v/v) mixture of isopropyl acetate/heptane (2 × 38 mL). The solid (24 g) was dried in an oven (45˚C, 1 in Hg) for 18 h to offer 22.4 g of 7 (α), as a pale powder, in 78% yield. mp: 78.5˚C - 80.5˚C. 1H NMR (CDCl3, 500 MHz) δ: 8.73 (s, 1H), 6.64 (d, J = 3.25 Hz, 1H), 5.63 (t, J = 9.78 Hz, 1H), 5.27 (t, J = 9.93 Hz, 1H), 5.15 (dd, J = 10.1, 3.45 Hz, 1H), 4.50 (d, J = 10.4 Hz, 1H), 3.75 (s, 3H), 2.051 (s, 3H), 2.046 (s, 3H), 2,01 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ: 169.78, 169.71, 169.46, 167.14, 160.61, 92.66, 90.55, 70.51, 69.49, 69.12, 68.99, 53.03, 20.66, 20.48, 20.39.
(2S,3S,4S,5R,6S)-3,4,5-Triacetoxy-6-{3-[2-((S)-2-cy-anopyrrolidin-1-yl)-2-oxo-ethylamino]adamantan-1- yloxy}tetrahydropyran-2-carboxylic acid methyl ester (8). A 1-L, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer, was charged with 7 (20 g, 41.8 mmol), 1 (vildagliptin, 25.4 g, 83.6 1mmol) and dichloromethane (260 mL, anhydrous) under nitrogen. The suspension was cooled in an ice/ ethanol bath. Triflic acid (13.8 g, 91.9 mmol) was added via an addition funnel at 0˚C ± 5˚C over 5 min. Then, the reaction mixture was warmed to 21˚C over 1 h. The reaction mixture was cooled in an ice bath and quenched by adding saturated NaHCO3 (150 mL) at <5˚C, over 10 min. Brine (0.1 L) was added and the two layers were separated. The aqueous layer was extracted with dichloromethane (1 × 0.1 L). The combined organic layers were washed with brine (1 × 50 mL) and dried over sodium sulfate. The solvent was removed on a rotavapor (bath temperature 25˚C) to give 60 g of a foamy syrup (α/β, 15/85), which was then purified by silica gel chromatography (5% MeOH/iPrOAc, then 3% iPrNH2/iPrOAc) to give 25 g of 8 (β) as a foamy solid. Further purification of 8 could be done by recrystallization from MeOH/ H2O, in 60% yield (15.5 g). mp: 82˚C - 92˚C (decomposed). 1H NMR (CDCl3, 500 MHz) δ: 5.26 (t, J = 9.45 Hz, 1H), 5.18 (t, J = 9.45 Hz, 1H), 4.94 (t, J = 9.45 Hz, 1H), 4.79 - 4.85 (m, 1H), 4.77 (dd, J = 8.0, 2.0 Hz, 1H), 4.01 (d, J = 9.9 Hz, 1H), 3.74 (s, 3H), 3.55 - 3.65 (m, 1H), 3.41 - 3.49 (m, 1H), 3.39 (bd, J = 1.5 Hz, 1H), 2.24 - 2.35 (m, 4H), 2.12 - 2.22 (m, 2H), 2.03 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 1.72 - 1.83 (m, 4H), 1.47 - 1.72 (m, 10H); 13C NMR (CDCl3, 125 MHz) δ: 170.50, 170.21, 169.39, 169.13, 167.23, 118.22, 94.24, 76.80, 72.37, 72.34, 71.44, 69.57, 53.34, 52.84, 47.07, 46.55, 45.49, 43.45, 41.57, 41.43, 41.32, 41.01, 35.17, 30.57, 30.49, 29.92, 25.07, 20.77, 20.64, 20.53.
(2S,3S,4S,5R,6S)-6-{3-[2-((S)-2-Cyanopyrrolidin-1-yl)-2-oxo-ethylamino]-adamantan-1-yloxy}-3,4,5-trihydroxytetrahydropyran-2-carboxylic acid (2). A 250- mL, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a digital thermometer, was charged with 8 (6.5 g, 10.5 mmol), MeOH (100 mL, anhydrous, 2.47 mol) and MeONa (0.3 g, 5.28 mmol) under nitrogen. The suspension was stirred at 21˚C for 2 h. Volatiles were removed on a rotavapor. The residue was dissolved in MeOH (15 mL). The solution was cooled in an ice bath and 1 N NaOH (11 mL) was added at 0˚C. The reaction mixture was stirred at 0˚C for 1 - 2 h and 1.0 N HCl (17 mL) was added at 0˚C (pH 5.0). The solvent was removed on a rotavapor (bath temperature 37˚C) to give 10 g of a brown liquid which was purified by reversedphase C18 chromatography (water, then 1.5% MeOH/ water, then 2.5% MeOH/water, then 4% MeOH/water) to afford 2 (β) as a pale powder (3.0 g) in 60% yield. mp: 183˚C (decomposed). 1H NMR (DMSO-d6, 400 MHz) δ: 4.76 (dd, J = 7.3, 3.8 Hz, 1H), 4.42 (d, J = 7.8 Hz, 1H), 3.58 - 3.70 (m, 1H), 3.40 - 3.58 (m, 3H), 3.08 - 3.25 (m, 2H), 2.88 (t, J = 8.1 Hz, 1H), 1.93 - 2.28 (m, 6H), 1.35 - 1.80 (m, 12H); 13C NMR (DMSO-d6, 100 MHz) δ: 171.55, 168.31, 119.16, 95.99, 76.45, 74.92, 74.72, 73.15, 71.81, 54.95, 46.16, 45.28, 45.11, 42.07, 40.82, 40.67, 34.48, 29.81, 29.79, 29.45 (2C), 24.64 (2C).
4. Conclusions
We disclosed herein a practical synthesis of vildagliptin- β-O-glucuronide (2), a metabolite of vildagliptin, in six steps and 11.3% overall yield. Some notable highlights are: 1) Pure alpha form of 6 was obtained via crystallization; 2) Stereoselective β-O-glycosylation of vildagliptin (1), and thus the first O-glycosylation of a tertiary alcohol in the presence of an unprotected secondary amine was achieved; 3) Triflic acid (TfOH) as a promotor for O-glycosylation is new to our knowledge; d) Deprotection of 8 to 2 was precisely designed and carried out; and (6) the target compound was successfully isolated and purified.
5. Acknowledgements
We are grateful to Oreste Ghisalba of Novartis Biotransformation Lab in Basel for the analytical data and an authentic sample of the target compound. We thank Dr. Prasad Kapa for his help in preparing this manuscript.
REFERENCES
- E. B. Villhauer, J. A. Brinkman, G. B. Naderi, B. F. Burkey, B. E. Dunning, P. Kapa, B. L. Mangold, M. E. Russell and T. E. Hughes, “1-[[(3-Hydroxy-1-adamantyl) amino] acetyl]-2-cyano-(S)-pyrrolidine: A Potent, Selective, and Orally Bioavailable Dipeptidyl Peptidase IV Inhibitor with Antihyperglycemic Properties,” Journal of Medicinal Chemistry, Vol. 46, No. 13, 2003, pp. 2774-2789.
- U. Hassiepen and M. Kittelmann, “Adamantyl O-Glucuronide Derivatives as Inhibitors of Dipeptidyl Peptidase IV for the Treatment of Diabetes,” PCT International Application, 2009, Article ID: 068531.
- R. Nakajima, M. Ono, S. Aiso and H. Akita, “Synthesis of Methyl 1-O-(4-Hydroxymethamphetaminyl)-α-D-Glucopyranouronate,” Chemical and Pharmaceutical Bulletin, Vol. 53, No. 6, 2005, pp. 684-687. doi:10.1248/cpb.53.684
- K. Toshima and K. Tatsuta, “Recent Progress in O-Glycosylation Methods and Its pplication to Natural Products Synthesis,” Chemical Review, Vol. 93, No. 4, 1993, pp. 1503-1531.
- H. Pellissier, “Use of O-Glycosylation in Total Synthesis,” Tetrahedron, Vol. 61, No. 12, 2005, pp. 2947-2993. doi:10.1016/j.tet.2005.01.070
- Y. Okada, T. Mukae, K. Okajima, M. Taira, M. Fujita and H. Yamada, “Highly β-Selective O-Glucosidation Due to the Restricted Twist-Boat Conformation,” Organic Letters, Vol. 9, No. 8, 2007, pp. 1573-1576. doi:10.1021/ol070427b
- K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee and J. Park, “2- (Hydroxycarbonyl)benzyl Glycosides: A Novel Type of Glycosyl Donors for Highly Efficient β-Mannopyranosylation and Oligosaccharide Synthesis by Latent-Active Glycosylation,” Journal of the American Chemical Society, Vol. 123, No. 35, 2001, pp. 8477-8481. doi:10.1021/ja015842s
- C. Lacy and M. Sainsbury, “A Synthesis of Morphin-6- Glucuronide,” Tetrahedron Letters, Vol. 36, No. 22, 1995, pp. 3949-3950. doi:10.1016/0040-4039(95)00649-W
- S. Chittaboina, B. Hodges and Q. Wang, “A Facile Route for the Regioselective Deacetylation of Peracetylated Carbohydrates at Anomeric Position,” Letters in Organic Chemistry, Vol. 3, No. 1, 2006, pp. 35-38. doi:10.2174/157017806774964521
NOTES
*Corresponding author.