Case Reports in Clinical Medicine
Vol.3 No.3(2014), Article ID:43706,6 pages DOI:10.4236/crcm.2014.33034
Ampullary Neuroendocrine Tumor: A Rare Cause of Recurrent Abdominal Pain
Andrew Ofosu1, Michael Taccone2, Laskhmi Potakamuri1, Sanjay Jagannath3
1Medstar Harbor Hospital, Department of Medicine, Baltimore, USA
2Saba University School of Medicine, Saba, Antilles, Netherlands
3Mercy Medical Center, Institute for Digestive Health and Liver Disease, Baltimore, USA
Email: andyofosu@hotmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 2 February 2014; revised 1 March 2014; accepted 11 March 2014
ABSTRACT
Ampullary Neuroendocrine tumor (ANET) is a rare GI malignancy, representing less than 1% of GI neuroendocrine tumors and less than 2% of ampullary tumors. Traditional treatment is often a pancreaticoduodenectomy; however, local and endoscopic resections have been successful. We report a rare case of ANET in a 21-year-old Burmese man who presented with a 6-year history of non-specific intermittent abdominal pain who was successfully managed through transduodenal ampullectomy. At 24 months postoperatively he remains disease and symptom free. ANET is a rare cause of recurrent abdominal pain, and local excision of small ANETs can be an alternative, less morbid treatment for young patients. We follow the case with a brief review of the literature.
Keywords:Ampullary Neuroendocrine Tumor; Pancreaticoduodenectomy; Endoscopic Resection; Abdominal Pain

1. Introduction
Ampullary Neuroendocrine tumors (ANETs), formerly carcinoids of the ampulla of Vater, are extremely rare neuroendocrine tumors accounting for less than 0.3% of gastrointestinal carcinoids [1] . ANETs are neoplasms that arise from cells of the endocrine and nervous systems possessing secretory granules and often producing biogenic amines and peptide hormones [2] . According to the most recent data from the Surveillance, Epidemiology and End-Results (SEER) of the National Cancer Institute only 139 cases of ANET (82 low and 57 highgrade) have been reported worldwide [3] making robust epidemiologic factors difficult to ascertain. With such little consensus available, management of ANET can pose a challenge for even the seasoned gastric surgeon.
Pancreaticoduodenectomy is often recommended as the treatment of choice for tumors of any size without distant spread. However procedures like local excision and endoscopic resections have also been successfully attempted, especially for ANET less than 2 cm size [4] [5] . Here, we demonstrate a case of low-grade, well-differentiated, ANET in a young patient who presented with non-specific abdominal pain who underwent a successful local resection at our institution followed by a review of the literature.
2. Case Presentation
A 21-year-old Burmese male immigrant was referred for evaluation of a 6-year history of non-specific upper abdominal pain that had worsened 8 months prior to presentation. He denied any prior history of nausea, vomiting, flushing, diarrhea or jaundice. He did not drink alcohol or take any medications and had no significant past medical or family history. Physical exam was notable for epigastric and right upper quadrant tenderness. There were no palpable abdominal masses or adenopathy. Skin was negative for café au lait spots and jaundice.
Initial lab workup including a complete blood count, liver function test, urine analysis, and serum electrolytes were within normal limits. A right upper quadrant ultrasonography revealed a thickened gallbladder wall without obvious cholelithiasis. He was therefore scheduled for a Hepatobiliary Iminodiacetic acid (HIDA) scan which showed normal gallbladder visualization, contraction and motility on cholecystokinin stimulation and the absence of obstruction. An upper endoscopy was performed at follow up appointment. This study revealed a normal exam up to the level of the second part of the duodenum. Within the ampulla however, a single large nodule was visualized at the area of the major papilla with normal appearing overlying mucosa (Figure 1).
Subsequent endoscopic ultrasonography confirmed the presence of a hypoechoic heterogenous mass that measured 13.2 mm in maximal cross-sectional diameter with sonographic evidence of invasion into the deep mucosa without involvement of the pancreas (Figure 2). Intraprocedural fine-needle aspiration was undertaken. Biopsy results showed scattered loosely cohesive fragments and individual cells with uniform, rounded nuclei and speckled chromatin suggestive of a well differentiated neuroendocrine tumor (Figure 3). A subsequent computed tomography with intravenous contrast was obtained which was negative for both masses and metastatic disease. As well, serum chromogranin A (2.4 ng/mL, norm 1.9 - 15 ng/mL) and gastrin (175 pg/mL, norm 10 - 200 pg/mL) levels were acquired along with a 24-hour urine HIAA level (2.5 mg, norm < 6 mg/24hr) which were all within normal limits. Next, an octreotide scan was obtained over a 3-day period that yielded no abnormal findings. Magnetic resonance cholangiopancreatography showed findings suggesting a 13.2 mm polypoidmass which projected into the duodenal lumen on thin slices. Common bile and pancreatic duct dilatation were absent on our study, measured 5 mm and 2 mm respectively. Additionally, no abnormalities of the liver, pancreas or gallbladder were seen.
A resection of the ampulla of Vater was planned and performed with sphincteroplasty and cholecystectomy.
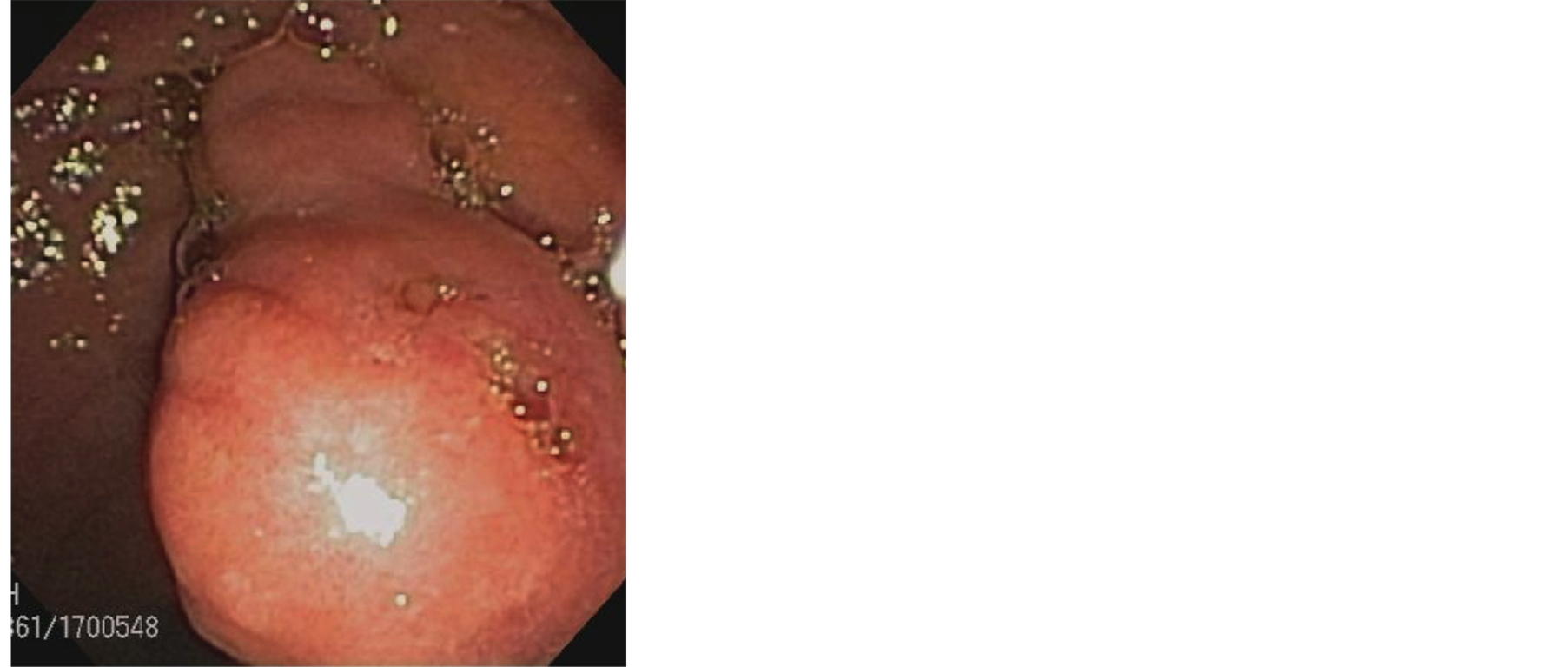
Figure 1. Esophagoduodenoscopy revealing a large nodule at the major papilla with normal appearing overlying mucosa.
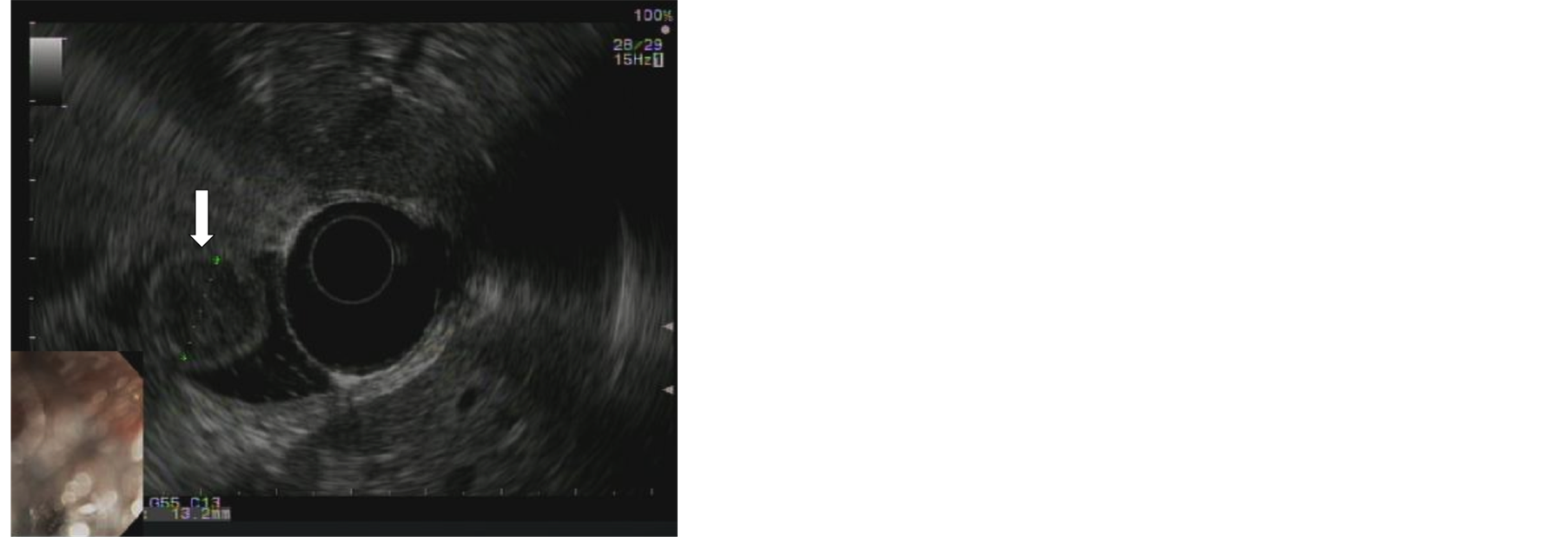
Figure 2. Endoscopic ultrasound of hypoechoic heterogeneous mass measuring 13.2mm in maximal cross-sectional diameter (white arrow).
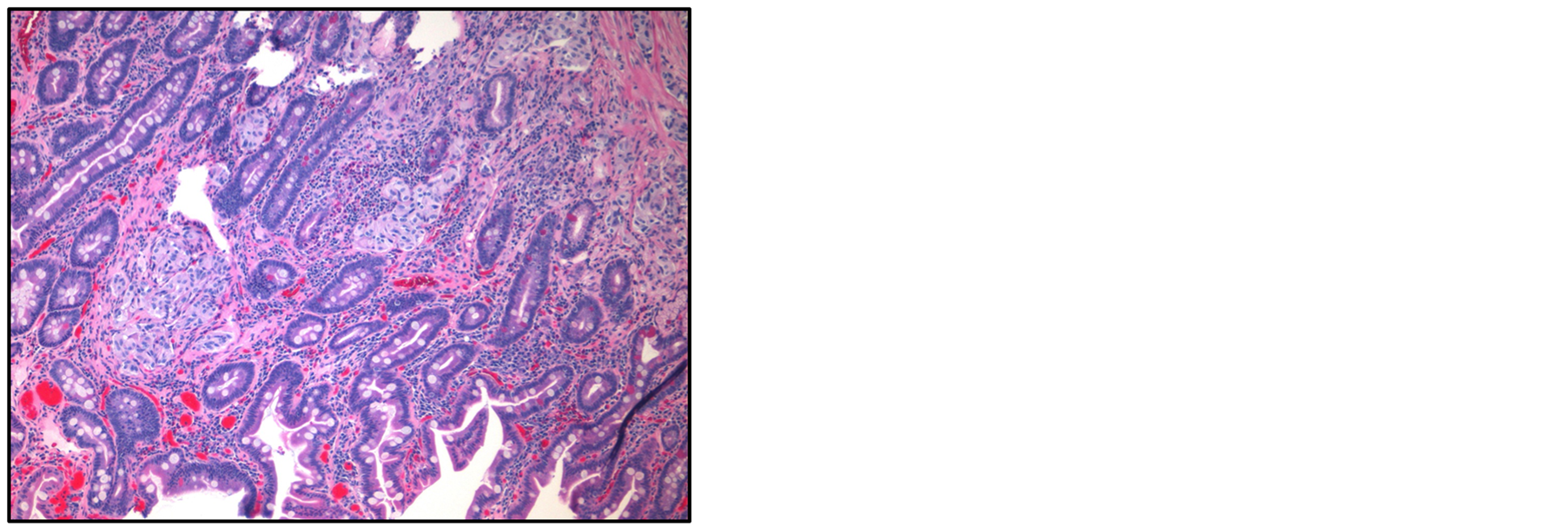
Figure 3. Microscopic examination of excisional biopsy of ampullary tumor (H&E stain). Note cells with uniform rounded nuclei and speckled chromatin.
Surgery revealed a 13.2 mm mass at the level of the ampulla of Vater. Pathology confirmed well-differentiated neuroendocrine tumor (WHO grade 1) with clear margins of 2 mm, with no lymphvascular and perinueral invasion. Further characterization included the presence of psammoma calcifications, positive immunoreactivity for somatostatin, CD8/18, chromogranin A, synaptophysin, and focal positivity of s100 and Ki67 (<1%). Of note, the sample was negative for c-KIT, gastrin, CD34 and CD68.
The patient made an uneventful recovery and was discharged home on post-operative day four. He is currently disease free at 24 months from his date of surgery. He returns to our department for follow up on a regular basis.
3. Discussion
Historically, any and all neuroendocrine tumors that arose within the gastrointestinal tract were called carcinoid tumors. However, recently this terminology has been abandoned and now the term carcinoid is reserved for only low-grade, well differentiated, neuroendocrine tumors. ANET are extremely rare and account for less then 1% of all gastrointestinal neuroendocrine tumors and less than 2% of all tumors of the ampullary region [3] [6] . Carcinoids involving the ampulla of Vater can be derived from cell types of the duodenum, common bile duct or the pancreas. Two main categories of ANET exist: low-grade ANET and high-grade ANET which are distinguished on the basis of their proliferative rates assessed by amount of necrosis, mitotic figures per high power field or Ki67 labeling index [7] [8] .
The SEER database reported 76 males and 63 females out of a total of 139 cases of ANET, with an average age at presentation of 61 years for low grade and 62 years for high-grade neuroendocrine tumors [3] .
ANET does not show any sex predilection although data suggests that females typically present younger than their male counterparts. To our knowledge this patient is the youngest case in the medical literature to be diagnosed with this tumor.
3.1. Presenting Symptoms and Diagnosis
The most common presenting symptom in patients with ANET is jaundice (53% - 62%). Non-specific abdominal discomfort (24%), as in our patient, is the second most common symptom. Less frequently, gastrointestinal bleeding or pancreatitis (6%) may be present. Weight loss has been seen in 4% - 10% of patients at the time of presentation [5] [9] . ANET have been found to occur with higher frequency in patients with neurofibromatosis type 1 and multiple endocrine neoplasia type 1 syndrome, along with other rare cancers such as gastrointestinal stromal cell tumors and periampullary carcinomas. Unlike extraintestinal carcinoids, findings of true carcinoid syndrome are rare in patients with carcinoid of the ampulla of Vater without liver metastasis because most hormones produced by functional tumors are eliminated by first-pass metabolism in the portal system.
The incidence and prevalence of neuroendocrine tumors appears to have increased in recent years likely due to an improvement in diagnostic techniques. The most helpful diagnostic techniques are esophagogastroduodenoscopy (EGD), endoscopic retrograde cholangiopancreatography (ERCP) and endosopic ultrasound. The finding of a submucosal bulge at the ampulla on endoscopic examination should raise clinical suspicion of ANET [5] [10] . Despite these studies being key, they only rarely correctly confirm the diagnosis preoperatively. This fact can be partly explained by the tendency for ampullary carcinoid to proliferate under intact normal epithelium, making visualization and biopsy sampling difficult.
ERCP is commonly used to diagnose ampullary tumors as well as to obtain tissue for histopathology. Endoscopic ultrasound is helpful in detecting the depth of invasion and the presence of lymph node metastasis. Computed tomography and octreotide scan are helpful in metastatic workup once, and only when, the diagnosis of ANET has been established [10] [11] because there are no specific features on imaging that can help in distinguishing ANET from the more commonly occurring adenocarcinoma.
Certain markers may be useful in diagnosis. Ampullary carcinoid cells express chromogranin A in 92% of cases and neuron specific enolase, synaptophysin and cytokeratin in 100%. Somatostatin is found in 58 to 67% of cases. Serotonin is secreted in 17% of cases as well as CCK. Insulin will stain positive in 25% of tumors. However, 13% are negative for any hormonal staining [10] .
Tumor size has been regarded as a prognostic marker for adenocarcinoma of the periampullary region. However, tumor size does not predict the metastatic potential in ANET. In the case review by Hatzitheoklitoset al. [11] metastasis was present in 46% of ampullary carcinoids > 2 cm, in 50% of tumors between 1 - 2 cm, and in 66% of tumors < 1 cm. Makhlouf et al. [8] have reported two tumors measuring less than 2 cm demonstrating metastases, as well as a 5 cm tumor without any evidence of metastatic disease. Together, their findings demonstrate that ANET metastasize approximately 50% of the time irrespective of the size of the primary.
3.2. Therapeutic Options
The treatment protocol for ANET remains controversial, as they are rare tumors with an unpredictable biological behavior and prognosis [7] . Since tumor size has not been clearly established to correlate with lymph nodal positivity status, pancreaticoduodenectomy is often recommended as the treatment of choice for tumors of any size with no distant spread [9] [12] [13] . However, less invasive procedures like local excision and endoscopic resections have also been successfully attempted, especially for ANET less than 2 cm size or in high risk surgical candidates [4] [5] [14] . Extensive debulking surgery should be considered in patients with hormonal hypersecretion, even in the presence of advanced disease (extensive local or distant metastasis) since it has been shown to offer survival rates of up to 80% at 5 years [7] . Our patient had a successful local resection of his tumor, and has been disease free for the past twenty-four months.
In cases of liver metastases, surgical resection or other cyto-reductive techniques, such as radiofrequency ablation and chemoembolization, have been shown to improve hormone-mediated symptoms, quality of life and survival in certain groups of patients [15] . Patients with slowly growing carcinoid tumors do not generally benefit from cytotoxic chemotherapy. Somatostatin analogues can induce a symptomatic and biochemical response, but more recent studies have also indicated a cytostatic effect [16] . Tumor-targeted radioactive treatment with 90yttrium and 177lutetium coupled to a somatostatin analogue is currently under clinical evaluation [16] . Preliminary data indicate promising clinical potentials.
3.3. Survival and Long-Term Follow up
Low-grade tumors show a 5 and 10-year survival rate to the tune of 80% and 71%, respectively, whereas high-grade neuroendocrine tumors have abysmal 5- and 10-year survival rates of about 15% [3] [17] . Tumor grade and distant metastasis are the most important prognostic factors in determining survival in ANET. Other tumor properties, like nodal involvement, tumor size and resection margins, appear to be of lesser significance in predicting long-term survival [18] -[20] .
Patients undergoing curative surgery should be followed every 3 - 6 months for at least 5 years in order to detect eventual surgically removable recurrences. Examination should include biochemical markers depending on the associated clinical syndrome. Imaging by CT or MRI should be completed every 6 months.
4. Conclusion
ANETs are rare GI tumors but nevertheless should be included in the differential list for recurrent abdominal pain of unknown cause. Though pancreatiduodenectomy has been standard practice, there is increasing evidence that local resection could be an alternative treatment modality with much less morbidity—an important consideration when deciding management options in younger patients.
References
- Ricci, J.L. (1993) Carcinoid of the Ampulla of Vater: Local Resection or Pancreaticoduodenectomy. Cancer, 71, 686- 690. http://dx.doi.org/10.1002/1097-0142(19930201)71:3<686::AID-CNCR2820710306>3.0.CO;2-Z
- Ramage, J.K., Davies, A.H.G., Ardill, J., Bax, N., Caplin, M., Grossman, A., et al. (2005) Guidelines for the Management of Gastroenteropancreatic Neuroendocrine (Including Carcinoid) Tumours. Gut, 54, iv1-iv16. http://dx.doi.org/10.1136/gut.2004.053314
- Albores-Saavedra, J., Hart, A., Chablé-Montero, F. and Henson, D.E. (2010) Carcinoids and High-Grade Neuroendocrine Carcinomas of the Ampulla of Vater: A Comparative Analysis of 139 Cases from the Surveillance, Epidemiology, and End-Results Program—A Population Based Study. Archives of Pathology & Laboratory Medicine, 134, 1692- 1696. http://dx.doi.org/10.1043/2009-0697-OAR.1
- Gilani, N. and Ramirez, F.C. (2007) Endoscopic Resection of an Ampullary Carcinoid Presenting with Upper Gastrointestinal Bleeding: A Case Report and Review of the Literature. World Journal of Gastroenterology, 13, 1268- 1270.
- Senda, E., Fujimoto, K., Ohnishi, K., Higashida, A., Ashida, C., Okutani, T., et al. (2009) Minute Ampullary Carcinoid Tumor with Lymph Node Metastases: A Case Report and Review of Literature. World Journal of Surgical Oncology, 7, 9. http://dx.doi.org/10.1186/1477-7819-7-9
- Jaoude, W.A., Lau, C., Sugiyama, G. and Duncan, A. (2010) Management of Ampullary Carcinoid Tumors with Pancreaticoduodenectomy. Journal of Surgical Case Reports, 8, 4. http://dx.doi.org/10.1093/jscr/2010.8.4
- Klimstra, D., Modlin, I.R., Coppola, D., Lloyd, R.V. and Suster, S. (2010) The Pathologic Classification of Neuroendocrine Tumors: A Review of Nomenclature, Grading and Staging Systems. Pancreas, 39, 707-712. http://dx.doi.org/10.1097/MPA.0b013e3181ec124e
- Bosman, F., Carneiro, F., Hruban, R. and Theise, N. (2010) WHO Classification of Tumors of the Digestive System. IARC Press, Lyon.
- Carter, J.T., Grenert, J.P., Rubenstein, L., Stewart, L. and Way, L.W. (2009) Neuroendocine Tumors of the Ampulla of Vater: Biological Behavior and Surgical Management. Archives of Surgery, 144, 527-531. http://dx.doi.org/10.1001/archsurg.2009.80
- Singhal, D., Vasdev, N., Soin, A., Gupta, S. and Nundy, S. (2006) Distinguishing between Periampullary Carcinoid and Carcinomas—Is This Possible Preoperatively? Indian Journal of Gastroenterology, 25, 206-207.
- Hatzitheoklitos, E., Buchler, M.W., Friess, H., Poch, B., Ebert, M., Mohr, W., et al. (1994) Carcinoid of the Ampulla of Vater. Clinical Characteristics and Morphologic Features. Cancer, 73, 1580-1588. http://dx.doi.org/10.1002/1097-0142(19940315)73:6<1580::AID-CNCR2820730608>3.0.CO;2-0
- Norton, J.A.,Kivlen, M., Li, M., Schneider, D., Chuter, T. and Jensen, R.T. (2003) Morbidity and Mortality of Aggressive Resection in Patients with Advanced Neuroendocrine Tumors. Archives of Surgery, 138, 859-866. http://dx.doi.org/10.1001/archsurg.138.8.859
- Poultides, G.A. and Frederick, W.A. (2006) Carcinoid of the Ampulla of Vater: Morphologic Features and Clinical Implications. World Journal of Gastroenterology, 12, 7058-7060.
- Hwang, S., Lee, S.G., Lee, Y.J., Han, D.J., Kim, S.C., Kwon, S.H., et al. (2008) Radical Surgical Resection for Carcinoid Tumors of the Ampulla. Journal of Gastrointestinal Surgery, 12, 713-717. http://dx.doi.org/10.1007/s11605-007-0389-3
- Oberg, K. (2004) Management of Neuroendocrine Tumours. Annals of Oncology, 15, iv293-iv298.
- Oberg, K. and Eriksson, B. (2005) Nuclear Medicine in the Detection, Staging and Treatment of Gastrointestinal Carcinoid Tumours. Best Practice & Research. Clinical Endocrinology & Metabolism, 19, 265-276. http://dx.doi.org/10.1016/j.beem.2004.11.016
- Hochwald, S.N., Zee, S., Conlon, K.C., Colleoni, R., Louie, O., Brennan, M.F., et al. (2002) Prognostic Factors in Pancreatic Endocrine Neoplasms: An Analysis of 136 Cases with a Proposal for Low-Grade and Intermediate-Grade Groups. Journal of Clinical Oncology, 20, 2633-2642. http://dx.doi.org/10.1200/JCO.2002.10.030
- Pyun, D.K., Moon, G., Han, J., Kim, M.H., Lee, S.S., Seo, D.W., et al. (2004) A Carcinoid Tumor of the Ampulla of Vater Treated by Endoscopic Snare Papillectomy. Korean Journal of Internal Medicine, 19, 257-260.
- Sakka, N., Smith, R.A., Whelan, P., Ghaneh, P., Sutton, R., Raraty, M., et al. (2009) A Preoperative Score for Resected Pancreatic and Periampullary Neuroendocrine Tumors. Pancreatology, 9, 670-676. http://dx.doi.org/10.1159/000181179
- Jarufe, N.P., Coldham, C., Orug, T., Mayer, A.D., Mirza, D.F., Buckels, J.A., et al. (2005) Neuroendocrine Tumors of the Pancreas: Predictors of Survival after Surgical Treatment. Digestive Surgery, 22, 157-162. http://dx.doi.org/10.1159/000087148