Pharmacology & Pharmacy
Vol.5 No.2(2014), Article ID:43000,12 pages DOI:10.4236/pp.2014.52027
PGE2 Generation in Myocardium from Isolated Rat Atrium under Hypoxia and Reoxygenation Conditions. Effect of Anti-β1 IgG from Patients with Chronic Severe Periodontitis
![]()
1Pharmacology Unit, School of Dentistry, University of Buenos Aires, Buenos Aires, Argentina; 2National Research Council of Argentina (CONICET), Buenos Aires, Argentina; 3Institute of Cardiovascular Physiopathology, School of Medicine, University of Buenos Aires, Buenos Aires, Argentina.
Email: *enri@farmaco.odon.uba.ar
Copyright © 2014 Sabrina Ganzinelli et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Sabrina Ganzinelli et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received November 29th, 2013; revised January 12th, 2014; accepted February 8th, 2014
KEYWORDS
Myocardium; PGE2; Hypoxia; Histopathology; Periodontitis; Antibodies Anti-β1 Adrenoceptors; Xamoterol
ABSTRACT
Background: Hypoxia is one of the most frequently encountered stresses in health and disease. Methods: We compared the effects of an anti-β1 periodontal IgG (pIgG) and an authentic β1 adrenergic agonist, xamoterol, on isolated myocardium from rat atria contractility. We used an ELISA assay to measure the generation of PGE2 in vitro after the addition of either the antibody or the adrenergic agonist. We analyzed the myocardium histopathologically in the presence of both the antibody and/or the adrenergic agonist drug during normoxia, hypoxia and reperfusion conditions. Results: PGE2 generation increased during the hypoxia and was unchanged during reoxygenation period compared with the production of this prostanoid in atria during normoxia condition. A β1 specific adrenoceptor antagonist atenolol and the β1 synthetic peptide abrogated the increment of the prostanoid in the presence of pIgG but only atenolol due to it in the presence of xamoterol. The increment of PGE2 was dependent on the activation of cox-1 and cox-2 isoforms. Moreover, cox-2 was more active and produced more increments in the production of PGE2 in the presence of the pIgG than cox-1 activation. Histopathologically, studies of myocardium specimens during these different periods of the experimental protocol: basal (B), hypoxia (H) and reoxygenation (R), were also performed and showed tissue necrosis and edematization at the myocardium level. Conclusion: The phenomenon studied here supports the notion that PGE2 may be responsible for tissue edematization. PGE2 maybe acts as a beneficial modulator in the myocardium and prevents a major injury of it. The inflammation damage to the heart organ and cardiomyocytes caused by the actions of the antibodies in the course of heart lesions provoked by cardiovascular autoimmune disease, explains some of these results obtained in the present experiments. Further studies will be needed to establish the real role of PGE2 during hypoxia injury of the heart in the course of autoimmune diseases.
1. Introduction
Much research has been carried out into intermittent hypoxia and has shown that it may be imposed by physiological challenges, including strenuous exercise or spending time at high altitude, or by various diseases, including obstructive lung disease, asthma and systemic hypertension, myocardial infarction, stroke and cognitive dysfunction [1-4].
Intermittent hypoxia has been demonstrated to have powerful protective capabilities, and current studies are revealing important details regarding the mechanisms of hypoxia-induced cardiovascular protection [5]. Intermittent hypoxia produces a myriad of favorable effects in the cardiovascular system, brain and other organ systems. These effects can be grouped into five major categories: 1) adaptation of organs and tissues responsible for oxygen uptake and transport [6], 2) proliferation and increased density of vascular networks [7], and 3) increased mitochondrial density in the brain, liver and heart [8]. Consequently, adaptation to intermittent hypoxia has been used to treat patients with ischemic heart disease [9] and with post myocardial infarction heart failure [10].
Periodontitis is characterized by gingival inflammation, and periodontopathic bacteria are known to generate immunological inflammatory responses. Periodontitis is a key risk factor for the onset of cardiovascular disease [11-13]. Recently, we reported the identification of autoantibodies against atria cardiac β1-adrenoreceptors (AR), that were able to mimic the effect of an authentic β1-AR agonist acting on atria β1-AR [14,15] in the sera of patients with periodontitis. However, the release of host-derived inflammatory mediators, such as cytokines, into the circulation from chronically-inflamed periodontal tissues, together with the serum anti-β1-AR pIgG, may provide a link between periodontal disease and cardiovascular disease [12,13].
Cyclooxygenase (COX) is a key regulatory enzyme in eicosanoid metabolism, converting free arachidonic acid (AA) to PGH2 [16]. Two isoforms of COX have been identified, COX-1 and COX-2, which have both common and specific roles [17]. COX-1 is constitutively expressed, but in contrast, COX-2 is induced upon cell activation and its expression often is associated with inflammation and other pathophysiological states, and also, there is evidence suggesting a cardioprotective effect of COX-2 and potential detrimental effects of COX-2 inhibitors on the heart [18,19]. COX-2 appears to mediate the cardioprotective effects in the late phase of ischemic preconditioning [20]. COX-2 is an immediate early response gene that can be induced by direct hypoxia [21]. However, cardiac synthesis of prostanoids such as PGE2 [22] is enhanced by the induction of hypoxia in isolated heart preparations [23].
2. Aim
The aim of the present work was to examine the effect of anti β1 adrenergic IgG present in the sera of patients with chronic periodontitis (pIgG) and the authentic adrenergic β1 agonist xamoterol acting on β1-AR of rat isolated atria and the ability of both, the auto antibodies and the authentic agonist, to stimulate de synthesis of PGE2 under normoxia, hypoxia and reoxygenation conditions. Also, we study the histopathologically pattern of the atrium in these experimental conditions without additions (control) and in the presence of the pIgG and xamoterol. Our results suggested that the activation of COX with the subsequent generation of PGE2 may be involved in the pathological process during hypoxia in isolated rat atria.
3. Methodology
3.1. Animals
Adult male Wistar strain rats (250 - 300 g) were used. The animals were housed in standard environmental conditions and fed with a commercial pellet diet and water ad libitum. The experimental protocol followed the Guide to The Care and Use of Experimental Animals (DHEW Publication, NIH 80 - 23). Rats were anesthetized with a mixture of ketamine and xylazine (50 and 5 mg·kg−1 respectively) and killed by decapitation.
3.2. Preparation of Atria and Contractility Procedure
The atria were carefully dissected from the ventricles and immersed in a tissue bath containing Krebs Ringer Bicarbonate (KRB) solution gassed with 5% CO2 in oxygen and maintained at pH 7.4 and 37˚C. The composition of KRB solution was as described previously [24]. A preload tension of 750 mg was applied to the atria and tissues were allowed to equilibrate for 30 min. The initial values (control) for contractile tension of the isolated rat atria were recorder by use of a force transducer coupled to an ink writing oscillograph. Inotropic effects [dF/dt gram(g) per second(s)] were assessed by recording the maximal rate of isometric force development during electrical stimulation at a fixed frequency of 150 beats min−1. Control values (=100%) refer to the dF/dt g/s before the addition of xamoterol or pIgG or atenolol or β1 synthetic peptide. The absolute value for dF/dt g/s before any additions at the end of equilibrium period (30 min) was 4.6 ± 0.4 g/s, n = 45.
3.3. Histopathology Study
In each group, four hearts were separated and processed for histological analysis. These hearts were fixed in 10% formaldehyde buffer for a minimum of 72 h. After fixation, they were cut from apex to base, and serial sections of 5 μM were prepared from each segment and stained with hematoxylin and eosine.
3.4. Patients
The study group consisted of 12 adult patients with periodontitis who were attending the Periodontology Clinic from the metropolitan area of Buenos Aires. The mean age was 41 (range, 32 - 50) years. Healthy subjects were used as controls (12 male subjects) with a mean age of 38 (range, 30 - 46) years. The assessment of clinical parameters was carried out by a trained periodontist following criteria based on clinical parameters and the severity of periodontal tissue destruction [14]. The characteristic clinical signs of periodontitis included loss of clinical attachment, horizontal and/or angular alveolar bone loss, periodontal pocket formation, and gingival inflammation. To be included in the study, at least six sites with ongoing periodontal disease were required. Clinical measurements in patients with periodontitis included sites with alveolar bone loss of >2 mm and a pocket depth >5 mm with bleeding and attachment loss of >3 mm. In healthy subjects (control group), the probing depth was <3 mm and the attachment loss was <2 mm. The pocket probe depth and clinical attachment level were assessed at six sites per tooth and bleeding on probing at four sites per tooth. All patients with periodontitis suffered periodontal infection about 5 five years ago. No subject (periodontal patient or healthy individual) had any systemic illness and they were all never-smokers. Patients with periodontitis had not received periodontal treatment or antibiotics within the preceding 5 months or any anti-inflammatory drug within the 3 weeks prior to the study. The clinical characteristics of the study population and the healthy subjects (controls) are shown in Table 1. Additionally, pocket probing depth (PPD ≥ 6 mm) and clinical attachment loss (CAL ≥ 6 mm) were used as the periodontitis selection index.
3.5. Human Sera and pIgG Purification
Sera and the corresponding IgG were obtained from patients with periodontitis and from healthy individuals. Blood (6 mL) was obtained by venipuncture and allowed to clot at room temperature. Serum was then separated by centrifugation (2000 × g) and stored at −80˚C until used in assays. IgG was obtained by precipitation with ammonium sulfate at 50%, followed by three washes and reprecipitation with 33% ammonium sulfate. The resulting precipitate was subjected to chromatography on DEAEcellulose, equilibrated with 10 mM phosphate buffer (pH 8). The eluted peaks were concentrated by ultra filtration to 10 mg protein mL−1. Control immune-electrophoresis using goat anti-human total serum and goat non-specific anti-human IgG showed only one precipitin line.
The IgG fraction of healthy subjects and patients with periodontitis was independently subjected to affinity chromatography on the synthesized peptide covalently linked to Affi-Gel 15 gel, Bio-Rad, Richmond, CA, USA) for purification of anti-peptide antibodies by affinity chromatography. The IgG fraction was loaded on the affinity column equilibrated with PBS, and the non-peptide frac-

Table 1. Characteristics of the study population.
tion was eluted with the same buffer. Specific anti-peptide auto-antibodies (anti β1-AR peptide IgG) were then eluted with 3 M KSCN and 1 M NaCl, followed by immediate extensive dialysis against PBS. The IgG concentration of both non-anti-peptide antibodies and specific anti β1-AR peptide antibodies were determined by radial immunodiffusion assay, and the immunologic reactivity against the β1-AR peptide was evaluated by ELISA [14].
3.6. Experimental Protocol
The isolated atria underwent a 30 min stabilization period under basal conditions, during which they were equilibrated in buffer gassed with 95% O2, 5% CO2 (37˚C, pH 7.4; pO2 > 600 mmHg); followed by 50 min of hypoxia when the atria were equilibrated in buffer gassed with 95% N2, 5% CO2 (37˚C, pH 7.4; pO2 > 100 mmHg) and then 50 min reoxygenation by re-equilibration with 95% O2, 5% CO2 (37˚C, pH 7.4; pO2 > 600 mmHg). The adrenergic agonist (xamoterol, 1 × 10−8 M) and the antibody (IgG: 1 × 10−7 M) were added in the last 10 min before beginning the hypoxic (H) period. The total duration of this experimental design was 130 min and at the end of this period atria samples were fixed in 10% formaldehyde buffer and then processed for histopathologically analysis, or processed for biochemical analysis to determine the production of PGE2. In the blocking experiments, atenolol (1 × 10−7 M, a β1-specific adrenergic antagonist), synthetic β1-adrenergic peptide (5 × 10−5 M) and an unrelated peptide (5 × 10−5 M) used as control, were added at the beginning of the stabilization period (0 min).
3.7. PGE2 Assays
Atria were incubated for 130 min in 0.50 mL KRB gassed with 5% CO2 in oxygen at 37˚C. Anti-β1 pIgG and xamoterol were added to the isolated atria 20 min before the end of the incubation period. Blockers were added 10 min before the addition of different concentrations of anti-β1 pIgG (1 × 10−7 M) and xamoterol (1 × 10−8 M). Atria were then homogenized and transferred into 1.5 mL polypropylene micro-centrifuge tubes. Thereafter, all samples were processed according to the protocol supplied with the Prostaglandin E2 Biotrak Enzyme Immunoassay (ELISA) System (Cayman Chemical, Ann Arbor, MI). The results are expressed as pg·mL−1.
3.8. Should Be Now mRNA Isolation and cDNA Synthesis and Semi-Quantitative RT-PCR
Total RNA was extracted from rat atria by homogenization using guanidinium isothiocyanate method. A 20 ml reaction mixture contained 2 ng of mRNA, 20 units of RNase inhibitor, 1 mM dNTPs and 50 units of Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI, USA). First strand cDNA was synthesized incubating rat atrium in KRB gassed with 5% CO2 in O2 pH 7.4 at 37˚C. In a selected tube, the reverse transcriptase was omitted to control for amplification from contaminating cDNA or genomic DNA. Semi-quantitation of cox isoforms (cox-1 and cox-2) was performed by a method that involves simultaneous coamplification of both the target cDNA and a reference template (MIMIC) with a single set of primers (Clontech Laboratories, Palo Alto, CA). The sequence of oligonucleotide primer pairs used for construction of MIMIC and amplification of cox-1 (sense 5’ TAAGT ACCAG TGCTG GATGG 3’, antisense 5’ AGATC GTCGA GAAGA GCATCA 3’), cox-2 (sense 5’ TCCAA TCGCT GTACA AGCAG 3’, antisense 5’ TCCCC AAAGA TAGCA TCTGG 3’) and g3pdh (sense 5’ ACCAC AGTCCA TGCCAT CAC 3’, antisense 5’ TCCAC CACCC TGTTG CTGTA 3’) mRNA. Aliquots were taken from pooled first-strand cDNA from the same group and constituted one sample for PCR. The internal control was the mRNA of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (g3pdh). PCR products were subjected to electrophoresis on ethidium bromide-stained gels. Band intensity was quantitated by densitometry using NIH Image software. Levels of mRNA were calculated from the point of equal density of the sample. cox-1, cox-2 mRNA levels were normalized with the levels of g3pdh mRNA present in each sample, which served to control for variations in RNA purification and cDNA synthesis. The relative mRNA expression of cox-1, cox-2 in each group was compared with those from the respective normal group and reported as a percentage of normal.
3.9. Ethical Approval of the Study Protocol
The study protocol complied with the tenets of the Declaration of Helsinki and the rules established by the Ethics Committee of the University of Buenos Aires (Buenos Aires, Argentina). All subjects provided written informed consent.
3.10. Drugs
Xamoterol (specific β1-adrenoceptor agonist) and atenolol (β1-adrenoceptor antagonist) were obtained from Sigma Chemical Company (St. Louis, MO). Stock solutions were freshly prepared in the appropriate buffers. The vehicles used for both xamoterol and atenolol were distilled water. The drugs were then diluted in the buffer to achieve the final concentration stated in the text. The β1-synthetic peptide corresponded to the sequence of the second extra cellular loop of the human β1-AR (HWWRA ESDEA RRCYN DPKCC DFVTN RC). A 27-mer unrelated peptide SGSGS GSGSG SGSGS GSGSG SGSGS GS was also synthesized as a negative control.
3.11. Statistical Analysis
Student’s t test for unpaired values was used to determine the levels of significance. When multiple comparisons were necessary, after analysis of variance, the StudentNewman-Keuls test was applied. Differences between means were considered significant if P < 0.05.
4. Results
To determine the cardiac β1 adrenoceptor involved in the biological effect of anti β1 adrenergic antibody from patients with periodontitis (pIgG) upon rat atria contractility in normoxia, hypoxia and reperfusion conditions, was studied. The effect of the authentic β1 adrenoceptor agonist xamoterol also was assayed. Both, pIgG and xamoterol effects were compared with dF/dt obtained from rat contractility without additions taken as control experiments.
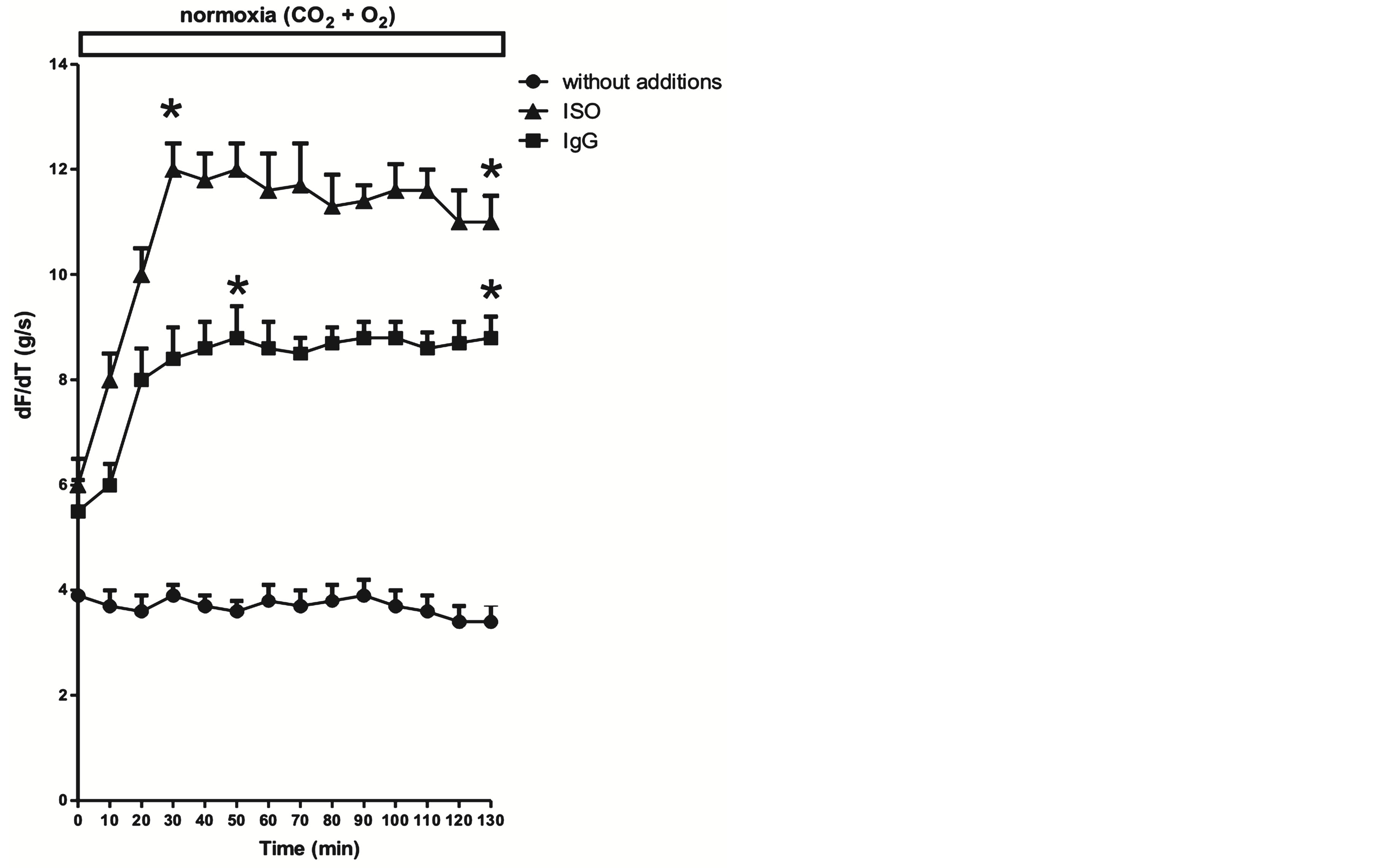
It can be seen in Figure 1(a) in normoxia conditions, that the pIgG increased dF/dt in a time-dependent manner. Normal (nIgG) is without effect (control). Xamoterol a specific β1 adrenoceptor agonist, also, as expected, increase dF/dt in a time-dependent manner.
During hypoxia conditions (Figure 1(b)) both pIgG and xamoterol increased dF/dt peaking at 30 min the
 (a)
(a)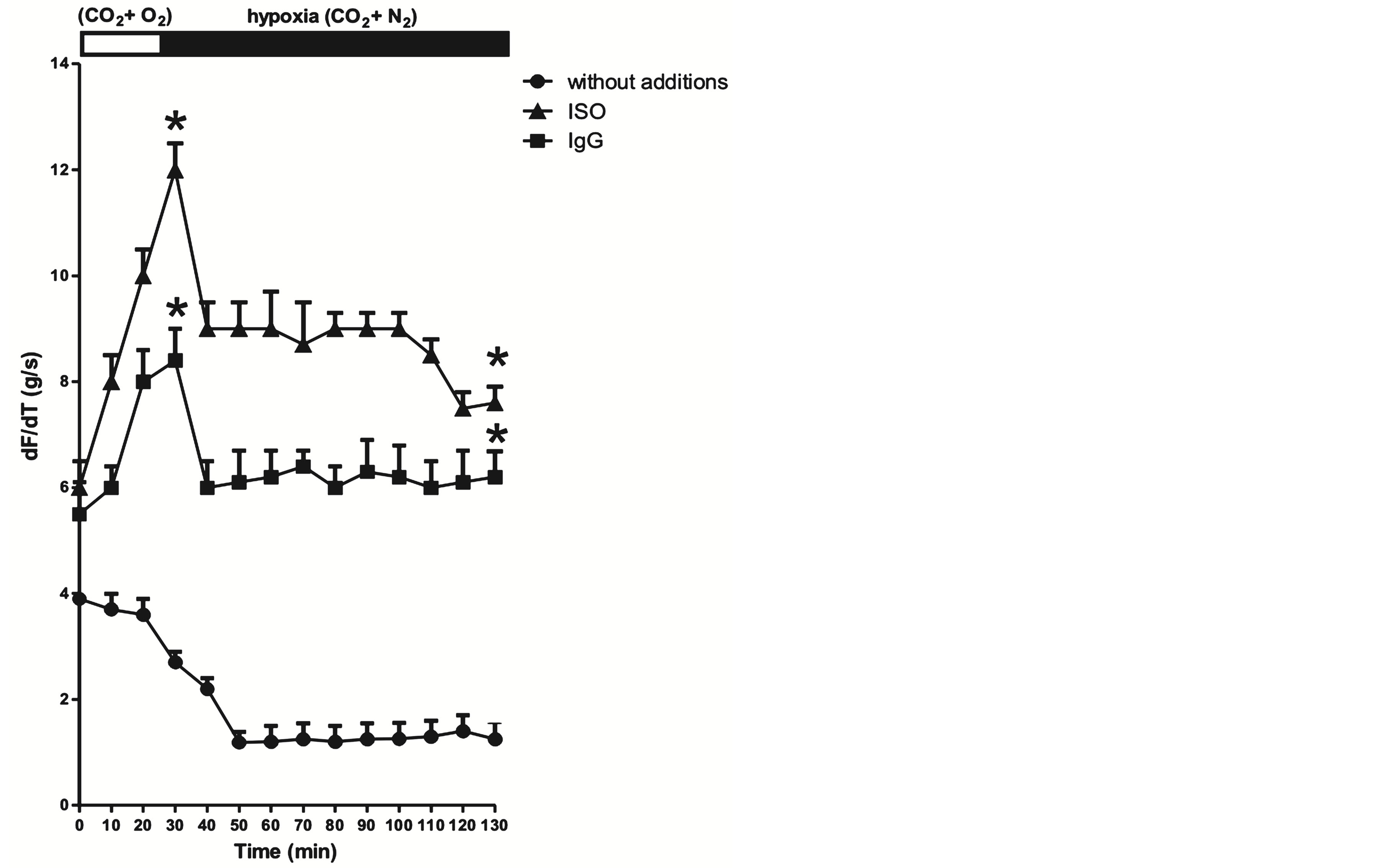 (b)
(b)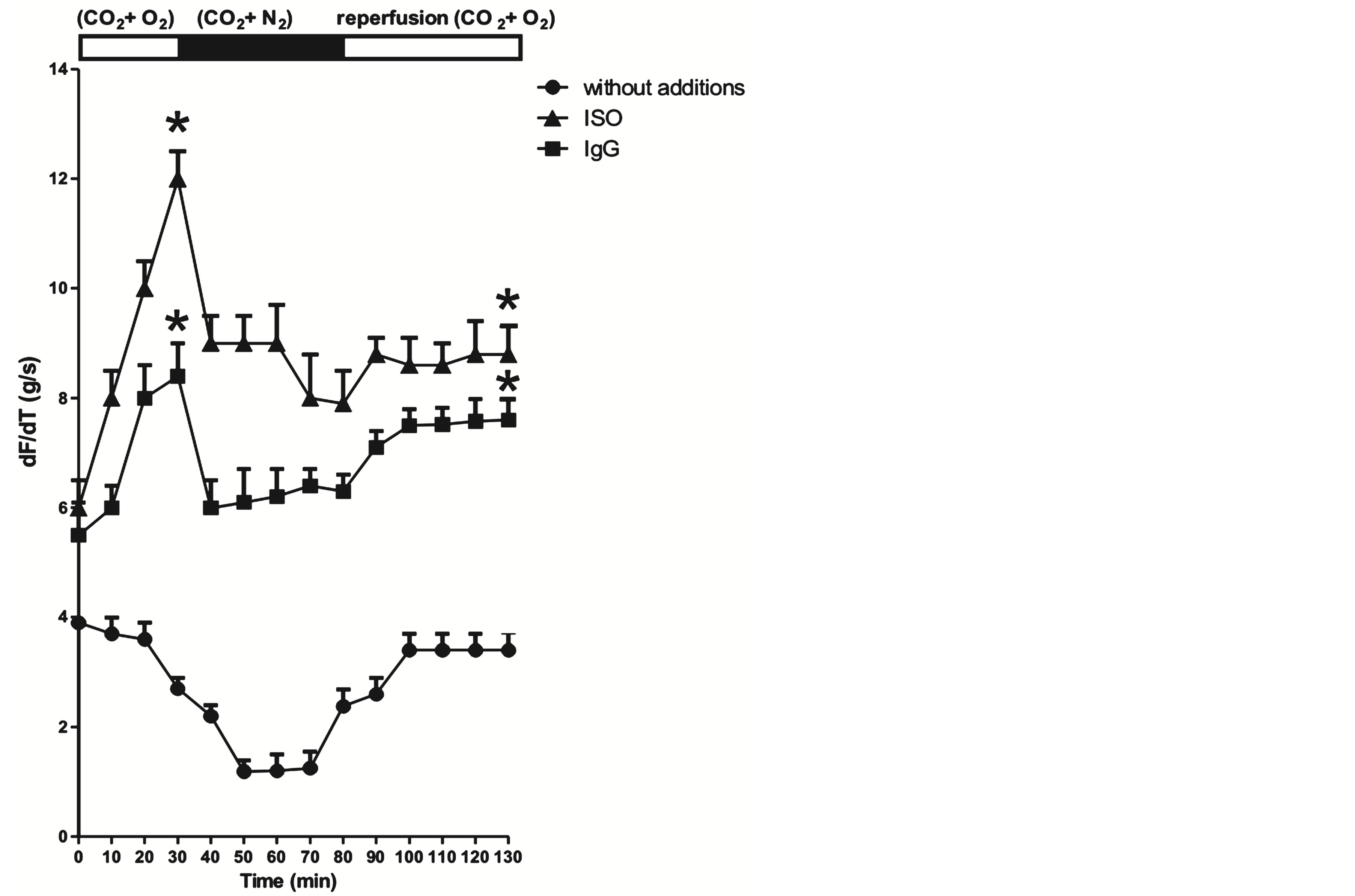 (c)
(c)
Figure 1. Effect of xamoterol 1 × 10−8 M and pIgG 1 × 10−7 M on contractility (dF/dt g/s) in isolated rat atria in a time-dependent manner from rats under normoxia (a), hypoxia (b) and reoxygenation (c) experimental conditions. In all cases, basal values of dF/dt without additions are also shown. Values are mean ± SEM of five experiments in each group by duplicate. *P < 0.001 xamoterol and pIgG versus without additions (control).
maximal increase IgG: (increment 311%); xamoterol (increment 444%) and had both a rapid although less pronounced increase in dF/dt (IgG: increment 496% and xamoterol: increment 608%) at 130 min. Figure 1(b) also shows that rat atria contractility without additions (basal values) decreased in a time-dependent manner, reaching at 68% above dF/dt initial values (3.9 ± 0.2 g/s, n = 12).
In reoxygenation condition (Figure 1(c)) the effect of pIgG and xamoterol are similar to that showed in Figure 1(b), but atria contractility without additions, revealed only slightly increased (39%) in the net atria dF/dt at 80 min but did not reached at the level of baseline values obtained at 0 time (3.9 ± 0.12 g/s, n = 11).
Table 2 show the influence of β1 adrenoceptor antagonist (atenolol) and β1 synthetic peptide (β1 peptide) upon rat atria contractility (dF/dt g/s) alone or in the presence of pIgG and xamoterol in normoxia, hypoxia and reperfusion conditions and also, the action of nIgG was stated.
It is important to note that the loss of contractility during hypoxia conditions remaining after reperfusion condition, showing that the capacity of the machinery contractile of the atria, is able to maintain normal myocardium contractile during pIgG and xamoterol stimulation compared with the values obtained in atria without additions, in which the atria contractility decreased. Normal IgG (nIgG) is ineffective in our studied system (Figure 2).
Figure 3 shows the effect of pIgG and xamoterol on rat atria comparing with auricles without any additions (control) in normoxia, hypoxia and reperfusion conditions, and its capacity to provoke a generation of PGE2. Both, pIgG and xamoterol, are able to stimulate the production of PGE2 and this production is significantly higher than those observed in control experiments in the three experimental conditions. Also, Figure 3, shows that the inhibitory action of the COX-1 (FR-122047, 5 × 10−8 M) and COX-2 (DuP 697, 5 × 10−8 M) by a specific enzymatic inhibitors in the three experimental conditions mentioned above, impaired the enhancement in myocardium PGE2 provoking by pIgG and xamoterol.
In the restitution experiments with the addition of exogenous PGE2 (1 × 10−11 M), the production of PGE2 in the presence of COX antagonistic drugs was reversed at the basal values, pointing that COX-1 and COX-2 pharmacologically participate in this phenomenon (Figure 3). Table 3 shows the influence of atenolol, synthetic β1 peptide and an unrelated peptide on the production of PGE2 in different experimental conditions.
In order to define the role of different COX isoforms due by pIgG and xamoterol, both with the capacity to activate and stimulate the β1 atria adrenoceptor, the cox-1 and cox-2 gene expression was determined by RT-PCR amplification.
Using specific primers for cox-1 and cox-2, the product of RT-PCR amplification showed single clear bands of the predicted size (Figure 4 lower panel). Semiquantitative RT-PCR demonstrated that stimulation with pIgG and xamoterol in normoxia appears a major activation in cox-1 than in cox-2 but in hypoxic condition, the size of the band is significantly higher for cox-2 component than that of cox-1; while in reperfusion conditions the values and the size of the both bands observed, are not significantly different from those obtained in hypoxic conditions (histogram of the Figure 4 upper panel).

Table 2. Influence of β1 adrenoceptor antagonist (atenolol) and β1 synthetic peptide (β1 peptide) upon rat atria contractility (dF/dt g/s) alone or in the presence of pIgG and xamoterol in normoxia, hypoxia and reperfusion conditions.

Figure 2. Rat atria contractility (dF/dt g/s) in a time dependent-manner under different experimental conditions: normoxia (●), hypoxia (■) and reperfusion (▲) alone or in the presence of normal IgG (nIgG) taken as control: normoxia (○), hypoxia (□) and reperfusion (∆). Values are mean ± SEM of five rat atria in each group.
Figure 5 shows the histological pattern of the rat atria in different experimental conditions such as normoxia, hypoxia and reoxygenation. In normoxia Figure 5(a): without any additions it can be seen that contiguous areas of viable myocardium, confluent areas of necrosis surrounded by viable myocardium and diffuse edema were observed; in Figure 5(b): in the presence of xamoterol the “wave fibers” and edema were more pronounced and in Figure 5(c): in the presence of pIgG both wave fibers and edema affected the majority of the tissue. In hypoxia Figure 5(d): without any additions it can be seen that contiguous areas of viable myocardium, confluent areas of necrosis surrounded by viable myocardium and diffuse interstitial edema were observed; in Figure 5(e): in the presence of xamoterol the “wave fibers” and edema were more pronounced and lesions affected the entire myocardium and in Figure 5(f): in the presence of pIgG both wave fibers and edema affect a large area and this was the worst lesion. Finally, in Figures 5(g)-(i): we do not observed any changes as view in hypoxic conditions during reperfusion.
5. Discussion
At present, virtually nothing is known regarding the mechanism of the final stage of late ischemic preconditioning after applications of anti-β1 adrenoceptor IgG or in the presence of a specific β1 adrenoceptor agonist in isolated atrium. The majority of investigators have reached a tacit assumption that the late phase of ischemic preconditioning is underlain by the same mechanism throughout its duration [5,22,23].
The salient findings of this study can be summarized as follows: even in the presence of the autoantibody obtained in the sera of patients with chronic periodontitis, or in the presence of a specific β1 adrenoceptor [14,15] agonist stimulus, a powerful protection against myocardial lesions is still present. Experimental hypoxia and subsequent reoxygenation are associated with no significant differences in the production of PGE2 remained its production elevated during these experimental conditions [23]. The present work confirms the changes of the COX-2/PGE2 systems during hypoxia/reoxygenation conditions, supporting the idea of regional variability in this system during hypoxia, considering that PGE2 was located mainly in the cytosolic supernatant fraction of the heart atria homogenates and its concentrations (ng/mg protein) were significantly higher in atria tissue than those of ventricular tissue [25,26].
Alterations in arachidonic acid metabolism are often involved in the myocardial disturbances associated with hypoxia and ischemia [26]. Additionally, Peredo et al. [27] demonstrated that the prostanoid production pattern changes in hypoxic rat atria, generating a cardioprotective action involving the prevention of increases of lactate and cAMP during ischemia [27,28]. Moreover, PGE2 would be expected to have an important physiological role in the hypoxic rat heart. In this sense, a possible mechanism could be that just after the hypoxic stimulus, COX-2-derived PGE2 generation rises to exert a vasodilatory effect in order to regulate and/or modulate the maintenance of blood flow through the hypoxic tissues. Thus, the maintained levels observed in the COX-2-derived PGE2 system during hypoxia/reoxygenation could be involved in adaptation of the heart to such situations. This implies a crucial moment in the changes occurring in the rat atria in response to hypoxia/reoxygenation [23]. Therefore, hypoxia seems to directly stimulate atria cox-2 enzyme expression, provoking a specific enhanced in PGE2 synthesis, resulting in vasodilatation and decreased vascular resistance of the ischemic bed, improving blood flow that in turn, this improves cardiac function.
Atenolol abrogated the effects of both xamoterol and the adrenergic autoantibodies but the synthetic β1 peptide only abrogated the effect of the autoantibodies demonstrating that these autoantibodies recognize cardiac β1 adrenoceptor from sarcolemma of rat atria and its capacity to interact with, acting as inducer of cox-2 mRNA levels with an increase in the cardiac proinflammatory substances PGE2.
The major new finding of this work was the demonstration that an anti-β1 adrenoceptor IgG behaving as adrenergic agonist, have the capacity to alter the rate of
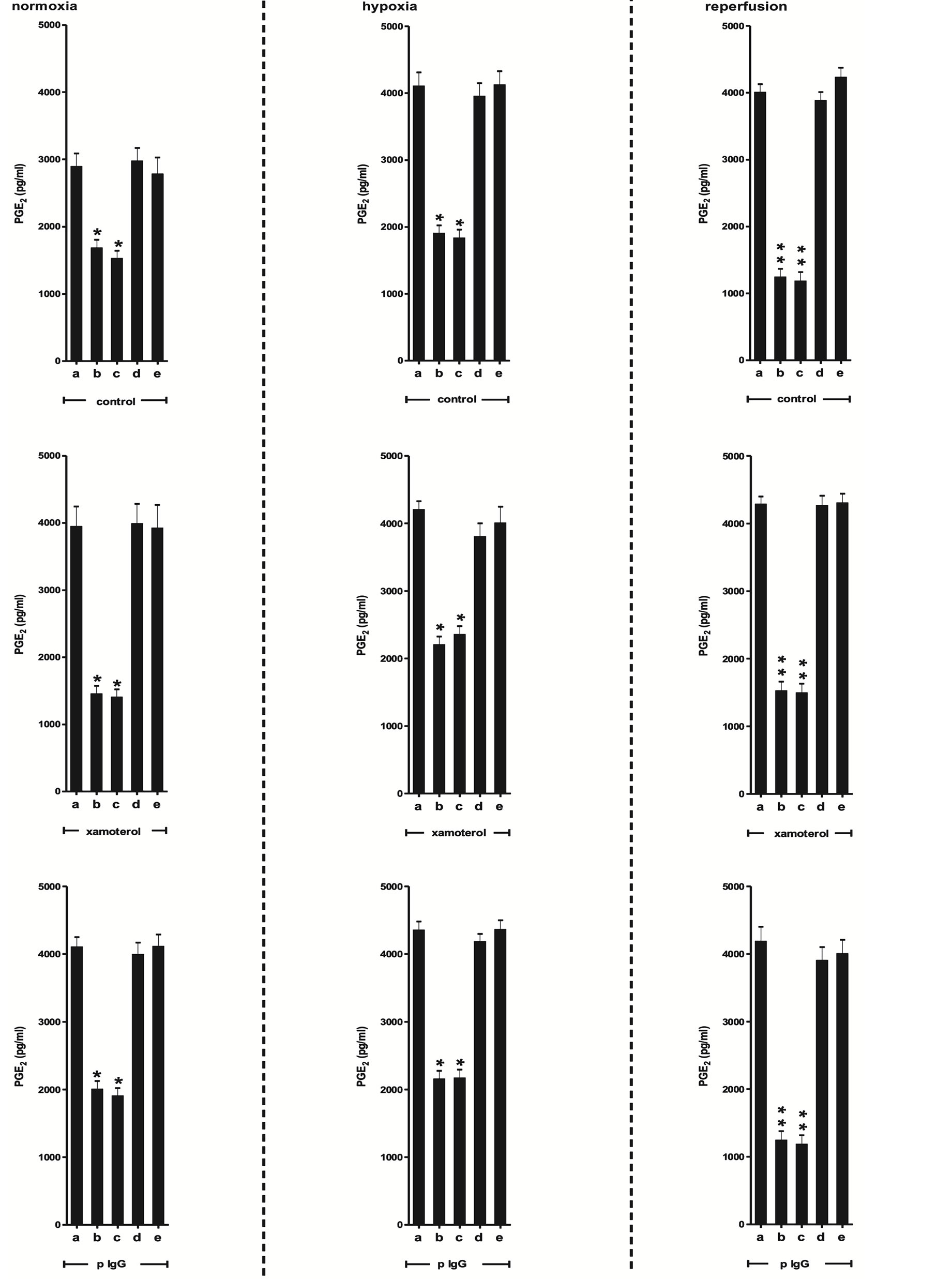
Figure 3. PGE2 production in isolated rat atria in absence (control) or in the presence of xamoterol 1 × 10−8 M and pIgG 1 × 10−7 M alone (a) or in the presence of COX-1 antagonistic drug FR-122047 5 × 10−8 M (b) or COX-2 antagonistic drug DuP 697 5 × 10−8 M (c) and COX-1 antagonistic drug + exogenous PGE2 1 × 10−11 M (d) or COX-2 antagonistic drug + exogenous PGE2 1 × 10−11 M (e) during normoxia, hypoxia and reperfusion experimental conditions. Values are mean ± SEM of seven experiments in each group. *P < 0.001 b and c versus a; **P < 0.0001 b and c versus a.

Table 3. Influence of the β1 adrenoceptor antagonist, human β1 synthetic peptide and an unrelated peptide on basal values in rat isolated atria on the production of PGE2.

Figure 4. Xamoterol and pIgG action on semi-quantitative RT-PCR analysis for cox-1 and cox-2 in normoxia, hypoxia and reperfusion experimental conditions mRNA expression. Also, g3pdh are shown as control (lower panel). Rat atria were incubated during 2 hours in absence or in presence of xamoterol 1 × 10−8 M and pIgG 1 × 10−7 M (upper panel). Values are mean ± SEM of six experiments in each group RT-PCR products obtained from xamoterol and pIgG on cox-1 and cox-2. *P < 0.001 between cox-1 and cox-2 mRNA levels in the presence of xamoterol or pIgG.
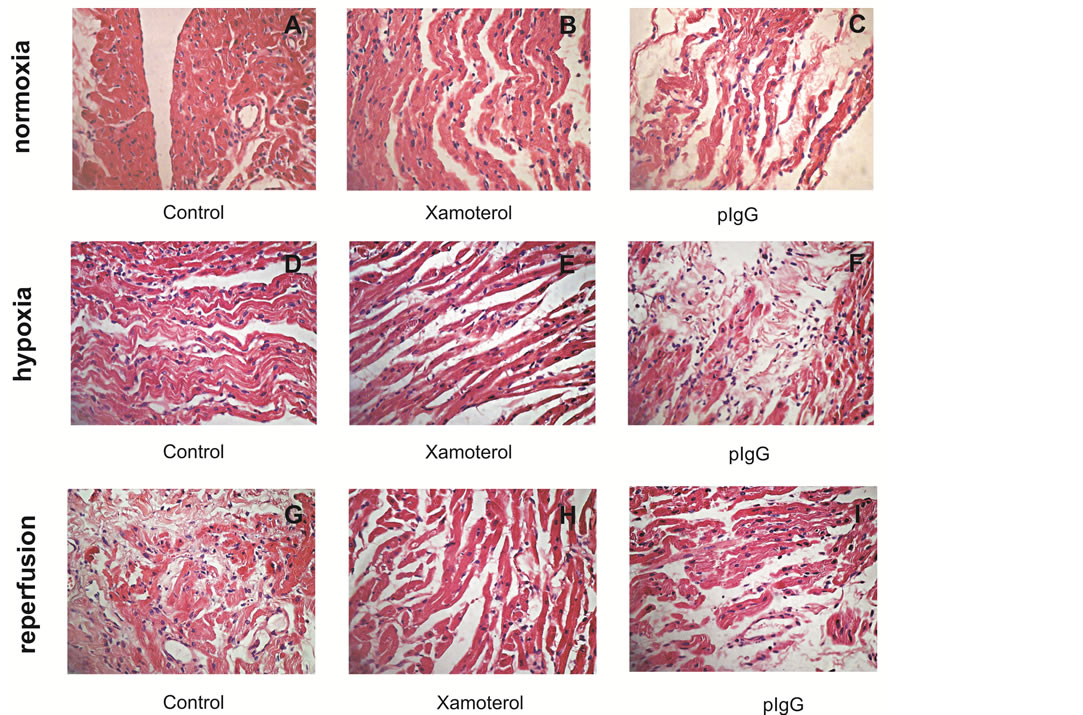
Figure 5. Histological pattern of isolated rat atria under different experimental conditions. In normoxia: contiguous areas of viable myocardium, confluent areas of necrosis surrounded by viable cardiac tissue and diffuse interstitial edema (A); in the presence of xamoterol wave fibers and edema were more pronounced (B) and in the presence of pIgG wave fibers and edema affected the majority of the myocardium (C). In hypoxia: contiguous areas of viable myocardium, confluent areas of necrosis surrounded by viable cardiac tissue and diffuse interstitial edema (D); in the presence of xamoterol wave fibers and edema were more pronounced than those observed in normoxia (E) and in the presence of pIgG wave fibers and edema affected a large areas of the myocardium and was the worst lesion (F). In reperfusion: we observed the same pattern as seen in hypoxia without any recuperation at the level of myocardium tissue (G, H, I).
transcription of specific proinflammatory target genes, triggering the cardiac production of PGE2 in response to receptor-mediated signaling events at the receptor cell cardiac atrium. The autoantibodies might play a role in the heart preconditioning phenomenon underlying the relevant inflammatory process during hypoxia/reoxygenation conditions in the course of autoimmune disease associated with ischemic heart and in heart failure contributing to inflammatory cell infiltration [29].
6. Conclusion
The present observations expand our understating involved during hypoxia and reoxygenation and the effect of PGE2 on atria myocardium associated with and/or dependent on upregulation of COX-2. Thereafter, by an adaptive mechanism, activation and an increment of mRNA levels of cox-2 with a subsequent increment of PGE2 generation occur. The inflammatory response after myocardial ischemia or in preconditioning in vivo will require further investigation to determine the role of PGE2 as a cytoprotective heart prostanoid in hypoxic conditions. On the whole, the cardiac prostanoid’s effect is, at least in part, due to their regulatory actions on the cardiomyocyte beating rate.
7. Acknowledgements
This work was supported by grants from Buenos Aires University (UBACyT O 003) and the Argentine National Research Council (CONICET, PIP 11220110100019). The authors thank Mr. Alejandro Thornton for his expert technical assistance.
Statement of Conflicts of Interest
There are no competing interests.
REFERENCES
- T. V. Serebrovskaya, R. J. Swanson and E. E. Kolesnikova, “Intermittent Hypoxia: Mechanisms of Action and Some Applications to Bronchial Asthma Treatment,” Journal of Physiology and Pharmacology, Vol. 54, No. Suppl. 1, 2003, pp. S35-S41.
- J. A. Neubauer, “Physiological and Pathophysiological Responses to Intermittent Hypoxia,” Journal of Apply Physiology, Vol. 90, No. 4, 2001, pp. 1593-1599.
- N. R. Prabhakar and G. K. Kumar, “Oxidative Stress in the Systemic and Cellular Responses to Intermittent Hypoxia,” Biological Chemistry, Vol. 385, No. 3-4, 2004, pp. 217-221. http://dx.doi.org/10.1515/BC.2004.015
- B. W. Row, L. Kheirandish, J. J. Neville and D. Gozal, “Impaired Spatial Learning and Hyperactivity in Developing Rats Exposed to Intermittent Hypoxia,” Pediatric Research, Vol. 52, No. 3, 2002, pp. 449-453. http://dx.doi.org/10.1203/00006450-200209000-00024
- Z. Cai, D. J. Manalo, G. Wei, E. R. Rodriguez, K. FoxTalbot, J. L. Lu, H. Zweier and G. L. Semenza, “Hearts from Rodents Exposed to Intermittent Hypoxia or Erythropoietin Are Protected against Ischemia-Reperfusion Injury,” Circulation, Vol. 108, No. 1, 2003, pp. 79-85. http://dx.doi.org/10.1161/01.CIR.0000078635.89229.8A
- N. A. Agadzhanyan and A. Y. Katkov, “Ways of Increasing Human Resistance to Acute Hypoxia,” Human Physiology, Vol. 9, No. 4, 1983, pp. 217-223.
- J. C. LaManna, J. C. Chavez and P. Pichiule, “Structural and Functional Adaptation to Hypoxia in the Rat Brain,” Journal of Experimental Biology, Vol. 207, No. 18, 2004, pp. 3163-3169. http://dx.doi.org/10.1242/jeb.00976
- L. D. Lukyanova, “Novel Approach to Understanding of Molecular Mechanisms of Adaptation to Hypoxia,” In: A. Hargens, N. Takeda and P. K. Singal, Eds., Adaptation Biology and Medicine, New Deli, Vol. 4, 2005, pp. 1-19.
- M. Burtscher, O. Pachinger, I. Ehrenbourg, G. Mitterbauer, M. Faulhaber, R. Pühringer and E. Tkatchouk, “Intermittent Hypoxia Increases Exercise Tolerance in Elderly Men with and without Coronary Artery Disease,” International Journal of Cardiology, Vol. 96, No. 2, 2004, pp. 247-254. http://dx.doi.org/10.1016/j.ijcard.2003.07.021
- T. D. Sabdanbekov, “Effect of Hypoxic Training on Clinico-Hemodynamic Indices and Physical Work Capacity of Patients with Large-Focus Myocardial Infarct,” Kardiologiia, Vol. 27, No. 12, 1987, pp. 59-62.
- O. M. Andriankaja, R. J. Genco, J. Dorn, J. Dmochowski, K. Hovey, K. L. Falkner and M. Trevisan, “Periodontal Disease and Risk of Myocardial Infarction: The Role of Gender and Smoking,” European Journal of Epidemiology, Vol. 22, No. 10, 2007, pp. 699-705. http://dx.doi.org/10.1007/s10654-007-9166-6
- F. Cairo, S. Castellani, A. M. Gori, M. Nieri, G. Baldelli, R. Abbate and G. P. Pini-Prato, “Severe Periodontitis in Young Adults Is Associated with Sub-Clinical Atherosclerosis,” Journal of Clinical of Periodontology, Vol. 35, No. 6, 2008, pp. 465-472. http://dx.doi.org/10.1111/j.1600-051X.2008.01228.x
- L. L. Humphrey, R. Fu, D. I. Buckley, M. Freeman and M. Helfand, “Periodontal Disease and Coronary Heart Disease Incidence: A Systematic Review and Meta-Analysis,” Journal of General Internal Medicine, Vol. 23, No. 12, 2008, pp. 2079-2086. http://dx.doi.org/10.1007/s11606-008-0787-6
- M. Segovia, S. Ganzinelli, S. Reina, E. Borda and L. Sterin-Borda, “Role of Anti-β1 Adrenergic Antibodies from Patients with Periodontitis in Cardiac Dysfunction,” Journal of Oral Patholology & Medicine, Vol. 41, No. 3, 2012, pp. 242-248. http://dx.doi.org/10.1111/j.1600-0714.2011.01090.x
- M. Segovia, S. Reina, E. Borda and L. Sterin-Borda, “Autoantibodies to the β1-Adrenoceptor from Patients with Periodontitis as a Risk Factor for Cardiac Dysfunction,” International Scholarly Research Network Dentistry, 2011, Article ID: 791393.
- M. Goppelt-Struebe, “Regulation of Prostaglandin Endoperoxide Synthase (Cyclooxygenase) Isozyme Expression,” Prostaglandins, Leukotrienes and Essential Fatty Acids, Vol. 52, No. 6, 1995, pp. 213-222. http://dx.doi.org/10.1016/0952-3278(95)90039-X
- W. L. Smith, and R. Langenbach, “Why There Are Two Cyclooxygenase Isozymes,” Journal Clinical Investigation, Vol. 107, No. 6, 2001, pp. 1491-1495. http://dx.doi.org/10.1172/JCI13271
- S. Gottlieb, “COX 2 Inhibitors May Increase Risk of Heart Attack,” British Medical Journal, Vol. 323, No. 7311, 2001, pp. 471-472. http://dx.doi.org/10.1136/bmj.323.7311.471a
- K. H. Pitkala, T. E. Strandberg and R. S. Tilvis, “Worsening Heart Failure Associated with COX-2 Inhibitors,” American Journal of Medicine, Vol. 112, No. 5, 2002, pp. 424-426. http://dx.doi.org/10.1016/S0002-9343(01)01107-X
- K. Shinmura, X. L. Tang, Y. Wang, Y. T. Xuan, S. Q. Liu, H. Takano, A. Bhatnagar and R. Bolli, “Cyclooxygenase- 2 Mediates the Cardioprotective Effects of the Late Phase of Ischemic Preconditioning in Conscious Rabbits,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 97, No. 18, 2000, pp. 10197-10202. http://dx.doi.org/10.1073/pnas.97.18.10197
- X. Yang, K. K. Sheares, N. Davie, P. D. Upton, G. W. Taylor, J. Horsley, J. Wharton and N. W. Morrell, “Hypoxic Induction of Cox-2 Regulates Proliferation of Human Pulmonary Artery Smooth Muscle Cells,” American Journal of Respiratory Cell and Molecular Biology, Vol. 27, No. 6, 2002, pp. 688-696. http://dx.doi.org/10.1165/rcmb.2002-0067OC
- R. Bolli, K. Shinmura, X. L. Tang, E. Kodani, Y. T. Xuan, Y. Guo and B. Dawn, “Discovery of a New Function of Cyclooxygenase (COX)-2: COX-2 Is a Cardioprotective Protein That Alleviates Ischemia/Reperfusion Injury and Mediates the Late Phase of Preconditioning,” Cardiovascular Research, Vol. 55, No. 3, 2002, pp. 506-519. http://dx.doi.org/10.1016/S0008-6363(02)00414-5
- G. Wu G, A. P. Mannam, J. Wu, S. Kirbis, J. L. Shie, C. Chen, R. J. Laham, F. W. Sellke and J. Li, “Hypoxia Induces Myocyte-Dependent COX-2 Regulation in Endothelial Cells: Role of VEGF,” American Journal of Physiology Heart Circulation Physiology, Vol. 285, No. 6, 2003, pp. H2420-H2429.
- L. Sterin-Borda, P. M. Cossio, M. F. Gimeno, A. L. Gimeno, C. Diez, R. P. Laguens, P. C. Meckert and R. M. Arana, “Effect of Chagasic Sera on the Rat Isolated Atrial Preparation: Immunological, Morphological and Function Aspects,” Cardiovascular Research, Vol. 10, No. 6, 1976, pp. 613-622. http://dx.doi.org/10.1093/cvr/10.6.613
- P. Zong, S. Setty, W. Sun, R. Martinez, J. D. Tune, I. V. Ehrenbourg, E. N. Tkatchouk, R. T. Mallet and H. F. Downey, “Intermittent Hypoxic Training Protects Canine Myocardium from Infarction,” Experimental Biology and Medicine (Maywood, N.J.), Vol. 229, No. 8, 2004, pp. 806-812.
- B. Zamorano, “Subcellular Localization of ProstaglandinE3 in Rat Heart Tissue,” Cardiovascular Drugs & Therapeutics, Vol. 5, No. 3, 1991, pp. 655-657. http://dx.doi.org/10.1007/BF03029735
- H. A. Peredo, E. J. Filinger, S. Sanguinetti, P. S. Lorenzo and E. Adler-Graschinsky, “Prostanoid Production in Hypoxic Rat Isolated Atria: Influence of Acute Diabetes,” Prostaglandins Leukot Essent Fatty Acids, Vol. 51, No. 4, 1994, pp. 231-234. http://dx.doi.org/10.1016/0952-3278(94)90184-8
- R. Rösen, P. Rösen, R. Ohlendorf and K. Schrör, “Prostacyclin Prevents Ischemia-Induced Increase of Lactate and Cyclic AMP in Ischemic Myocardium,” European Journal of Pharmacology, Vol. 69, No. 4, 1981, pp. 489- 491. http://dx.doi.org/10.1016/0014-2999(81)90454-4
- S. C. Wong, M. Fukuchi, P. Melnyk, I. Rodger and A. Giaid, “Induction of Ciclooxygenase 2 and Activation of Nuclear Factor-kappa B in Myocardium of Patients with Congestive Heart Failure,” Circulation, Vol. 98, No. 2, 1998, pp. 100-103. http://dx.doi.org/10.1161/01.CIR.98.2.100
NOTES
*Corresponding author.