Pharmacology & Pharmacy
Vol.4 No.2(2013), Article ID:29749,10 pages DOI:10.4236/pp.2013.42027
Determination of Amlodipine in Human Plasma by LC-MS/MS and Its Bioequivalence Study in Healthy Chinese Subjects*
![]()
1School of Pharmaceutical Sciences, Shandong University, Jinan, China; 2Institute of Clinical Pharmacology, Qilu Hospital of Shandong University, Jinan, China.
Email: #grc7636@126.com
Copyright © 2013 Chan-Mei Lv et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 19th, 2013; revised February 25th, 2013; accepted April 2nd, 2013
Keywords: Amlodipine; LC-MS/MS; Bioequivalence; Pharmacokinetics; Gliclazide
ABSTRACT
A sensitive and reproducible liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-MS/MS) method was developed and validated for the quantification of amlodipine in human plasma, with gliclazide as an internal standard (IS). The analyte was extracted with ethyl acetate and analyzed on a Diamond C18 (150 mm × 4.6 mm, 5 μm) column. The mobile phase was composed of methanol −10 mM ammonium acetate with gradient flow rates and gradient conditions. Amlodipine and IS were ionized by positive ion pneumatically assisted electrospray and detected in the multi-reaction monitoring (MRM) mode using precursor→productions of m/z 409.2→238.1, 294.1 and m/z 324.2→127.3, respectively. The specificity, matrix effect, recovery, sensitivity, linearity, accuracy, precision, and stabilities were all validated over the concentration range of 0.05 - 12 ng/mL. The 90% confidence interval (CI) for AUC0-t, AUC0-∞ and Cmax ratios (test: reference) were all within 80% - 125% interval proposed by FDA. It is concluded that the validated method can successfully fulfill the requirement of clinical bioequivalence study of amlodipine in healthy Chinese subjects after administration of amlodipine/losartan combination tablets and amlodipine tablets, and the test and reference tablets were bioequivalent.
1. Introduction
Calcium channel blockers have been widely used in the treatment of hypertension and angina pectoris, and combination therapy with an angiotensin II receptor blocker would enhance antihypertensive activity with greater efficacy and better tolerability, which maximize the blood pressure-lowering effects and minimize the severity of their side effects of each component [1-3].
Losartan is an active and highly specific non-peptide angiotensin II receptor blocker for the treatment of hypertension and heart failure. Its major active metabolite, EXP3174 is 10 to 40 times more potent than parent losartan in blocking the angiotensin II [4,5].
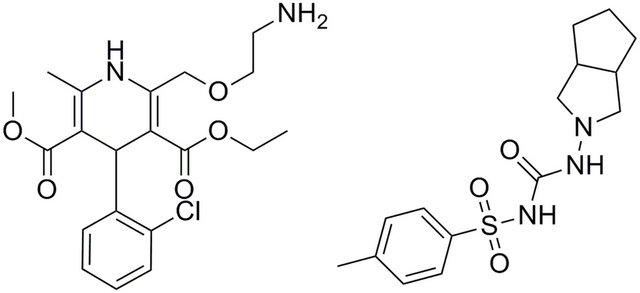
Amlodipine, (R,S)-2-[(2-aminoethoxy) methyl]-4-(2- chlorophenyl)-3-ethoxycarbonyl-5-methoxycarbonyl-6methyl-1,4-dihydropyridine (Figure 1(a)), is a potent calcium channel blocker used for the treatment of hypertension and angina pectoris. It has high bioavailability, large volume of distribution and long elimination halflife (t1/2) ranging from 35 to 45 h [6,7]. It has been reported that low plasma amlodipine concentrations are achieved after oral administration [8], therefore, a sensitive and specific analytical method is essential for determination of amlodipine in human plasma.
Numerous analytical methods have been reported for the quantification of amlodipine in plasma, such as gas chromategraphy (GC) with electron capture detection [9, 10], high performance liquid chromatography (HPLC) with fluorimetric detection [7,11] or with UV detection [12], or with electrochemical detection [13,14], highperformance thin-layer chromatography-densitometry [15] and liquid chromatography-tandem mass spectrometry [6, 16-19]. However, the sensitivity of the HPLC-UV method is inadequate due to the low absorbance of the analytes, and the sensitivity of the gas chromatography (GC) methods was improved, but the major disadvantage is the thermal decomposition of amlodipine to the pyridine analogue under GC conditions, which is already present in plasma as a metabolite. HPTLC procedure requires 2 mL of human plasma at sensitivity of 2 ng/mL [15], and the linearity range from 0.1 - 20 ng/mL did not fulfill the requirement for a much low limit of quantitation (LLOQ) of amlodipine pharmacokinetic studies. Liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-MS/MS) methods with the LLOQ of 0.1 ng/mL by Shentu et al. [18] and Carvalho et al. [19] for amlodipine and by Sarkar et al. [20] for simultaneous determination of metoprolol and amlodipine in human plasma, all couldn’t meet the requirements amlodipine human pharmacokinetic studies.
Sample pretreatment has also been an item of interest for the analysis of amlodipine in biological samples. Previous methods included solid-phase extraction (SPE), protein precipitation and Liquid-liquid extraction (LLE). SPE is relatively expensive, time-consuming and complex for a large number of samples collected and treated. X-Terra (C18) [21], HLB cartridges [6,22], silica cartridges [7] and online solid-phase [18,23] have been studied as well, but a relatively complicated procedure for elution of amldoipine from the column is required. After protein precipitation, the samples remained impure and were with the risk of column blocking and poor response [12]. LLE with diethylether/hexane (80:20, v/v) [19], methyl tert-butyl ether [24] or diethylether [25] has been reported, but hexane was toxic and expensive, methyl tert-butyl ether and diethylether were easy to evaporate and mixed solvent required complicated preparation procedures.
In this study, a simple, sensitive, precise, economical and less toxic LC-MS/MS method based on the LLE with ethyl acetate for the determination of amlodipine in plasma was established and applied to bioequivalence study of amlodipine in healthy Chinese subjects. Gliclazide (Figure 1(b)) was used as an internal standard (IS).
2. Experimental
2.1. Reagents and Materials
The standards of amlodipine besylate (lot: 100,374 - 200,602) and gliclazide (IS, lot: 100,269 - 200,402) were obtained from the National Institute for the Control of Pharmaceutical and Biological Products, Beijing China. Methanol (lot: K50E30) and ethyl acetate (lot: H39B05) were both of HPLC grade, from J. T. Baker. Ammonium acetate (lot: 10,133,751) was analytical grade, from A
 (a) (b)
(a) (b)
Figure 1. The chemical structures of amlodipine (a) and gliclazide (b).
Jonson Mattery Company. Blank human plasma was provided by Shandong Blood Center. Purified water was obtained from the Company of Wahaha. The Compound Amlodipine Tablets (lot: 100,901), 5 mg amlodipine and 50 mg losartan per tablet, was provided by Shangrui Pharmaceutical (Si-Chun) Co., Ltd, China. Amlodipine Besylate Tablet (5 mg per tablet, lot: 1,005,061), was provided by Pfizer Inc.
2.2. Instrumentation
Agilent 1200 series HPLC system (equipped with a G1312B duplex pump, a G1379B vacuum degasser, a G1316B thermostated column compartment and a G1367C autosampler) and an Agilent 6410 Triple Quadrupole mass spectrometer equipped with an electrospray ionization source (Agilent Technologies, USA) were used for mass detection. The chromategraphic separation was achieved on a Diamond C18 (150 mm × 4.6 mm, 5 μm) column. All data were acquired employing Agilent 6410 Quantitative Analysis version analyst data processing software.
2.3. LC-MS/MS Conditions
The chromatographic separation was achieved on a Diamond C18 column (150 mm × 4.6 mm, 5 μm) at 30˚C with a thermostated column oven. The mobile phase was methanol mixed with 10 mM ammonium acetate solution, with gradient flow rates and varied ratio of mobile phase (Table 1). The injection volume was 20 μL.
Mass spectrometric analysis was performed on a Series 6410 Triple Quad LC-MS/MS in electrospray positive ionization using multiple reaction monitoring (MRM) mode. The mass transition was m/z 409.2→238.1, 294.1 for amlodipine and m/z 324.2→127.3 for IS, respecttively. The other working parameters of the mass spectrometer were as follows: dwell time (200 ms); protective air of nitrogen gas (9 L/min); gas temperature (350˚C); spray gas pressure (350 Pa); capillary voltage (4000 V); nebulizer pressure (45 psi); the fragment electric voltage (120 V for amlodipine and 110 V for IS); the collision energy
Table 1. The program of amlodipine plasma concentrations determination.
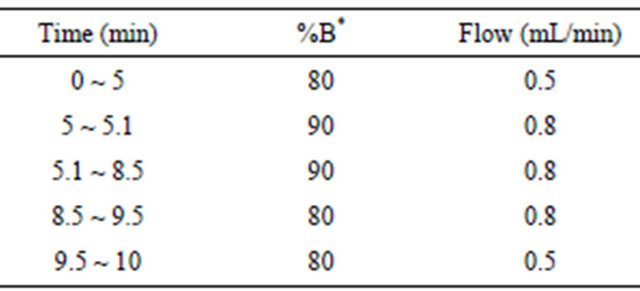
%B*: the proportion of the organic phase.
(12 eV for amlodipine and 17 eV for IS) and the EMV (200 V).
The column was washed with a 95:5 water-methanol (v/v) mobile phase for 40 min and then with methanol for 40 min between batches.
2.4. Preparation of Standard Solutions, Calibration Standard and Quality Control Samples
Because of amlodipine’s photo labile property, the whole process of the experiment must be operated in the dark place.
The accurately weighed amlodipine besylate and gliclazide (IS) were dissolved in methanol to result in 1 mg/mL mother liquor of amlodipine (equivalent to approximately 1 mg/mL of the free base) and IS, respectively. Working standard solutions of amlodipine in the concentration range of 1.25 - 300 ng/mL were prepared by independent dilution of 1 mg/mL stock solution with 10 mM ammonium acetate-methanol (20:80, v/v). The IS was dissolved with 10 mM ammonium acetate-methanol (20:80, v/v) to get an 80 ng/mL standard working solution. All solutions were stored at 4˚C and dark places with no evidence of decomposition, and equilibrated to room temperature before use (approximately 15 min).
The calibration curve standard and quality control (QC) samples were freshly prepared by spiking 20 µL of the appropriate standard solutions with drug-free human plasma. The final concentrations of calibration standard in plasma consist of eight nonzero concentrations (0.05, 0.1, 0.2, 0.5, 1.0, 3.0, 6.0, 12.0 ng/mL), and QC samples, which were used in the validation and during the bioequivalence study, were 0.1 (LQC), 1.0 (MQC) and 10.0 ng/mL (HQC) for amlodipine. The human plasma volume for calibration and QC samples was 0.5 mL.
2.5. Disposition of Samples
A liquid-liquid extraction method was used for the extraction of amlodipine and IS from plasma. 0.5 mL plasma sample was mixed with 20 μL of the IS working solution (80 ng/mL gliclazide in mobile phase) and 4 mL of ethyl acetate, vortex-mixed for 3 min, and centrifuged at 8000 rpm for 5 min. Then, 3.2 mL organic phase was transferred to a clean tube and evaporated to dryness under gentle stream of nitrogen gas at 37˚C. The residue was reconstituted with 200 μL mobile phase, and 20 μL was injected onto the LC-MS/MS for analysis.
2.6. Method Validation
The assays of amlodipine in human plasma were validated for assay specificity, matrix effect, recovery, sensitivity, linearity, precision, accuracy, and stability according to the US Food and Drug Administration (FDA) [26] and Chinese State Food and Drug Administration (SFDA) guidelines for the validation of bioanalytical methods.
2.6.1. Specificity
Chromatograms of six different lots of blank human plasma were identified for inspecting analytes from the potential interference of endogenous substances at the peak region (amlodipine and IS).
The specificity of the method was evaluated by comparing chromatograms of blank plasma, amlodipine and IS standard, blank plasma spiked with amlodipine and IS, plasma sample from a subject after oral administration of compound amlodipine tablets (10 mg/100mg, amlodipine/losartan).
2.6.2. Matrix Effect
Blank plasma samples were extracted and then spiked with amlodipine at three concentration levels (0.25 ng/mL as low, 2.5 ng/mL as medium and 25 ng/mL as high) and IS in five replicates at each concentration level, respectively, to evaluate the matrix effects of plasma. The corresponding peak areas were compared with those of standard solutions, and peak area ratio was defined as the matrix effect. The matrix suppression was assessed by post-column infusion of an analyte with a syringe pump post column into the MS detector. The extracted blank matrix was injected by an autosampler onto the analytical column. The MS monitored the signal level from post-column infused analyte solution when injecting blank plasma sample.
2.6.3. Recovery
The extraction recoveries of amlodipine were evaluated by comparing peak areas of amlodipine extracted from plasma with those of samples to which the analytes had been added after extraction at equivalent concentrations. Five replicates for each QC level were performed with the established extraction procedure. The extraction recovery of IS was evaluated in a similar way at 3.2 ng/mL.
2.6.4. Calibration Curves
The calibration curve was prepared by analyzing spiked calibration samples at eight different concentration levels on every batch of analysis. Every calibration standard was injected in five replicates. Calibration curves were typically described by the equation y = ax + b, where y corresponds to the peak-area ratio of amlodipine to IS and x represents the plasma concentration of amlodipine. The linearity of calibration curve was assessed by linear regression with a weighting factor of the reciprocal of the concentration squared (1/x2). The low limit of quantification (LLOQ) was evaluated by analyzing five replicates of spiked samples at the concentration of 0.05 ng/mL with acceptable precision.
2.6.5. Precision and Accuracy
The intraor inter-assay precision and accuracy were assessed by analyzing spiked samples at three concentration levels in human plasma (0.1 ng/mL, 1 ng/mL and 10 ng/mL) on the same day or on three consecutive days, and five replicates at each concentration level were applied. The mean value of RE for the accuracy should be within 15% of the actual value except at LLOQ, where it should not deviate by more than 20%. The relative standard deviation (RSD) or CV%, for the precision, determined at each concentration level should not exceed 15% except at LLOQ, where it should not exceed 20%.
2.6.6. Stability
The stability of amlodipine in plasma was evaluated by analyzing samples with five replicates at each concentration level under different study conditions, including the post-extracted samples in the HPLC auto-samples for 0 h (fresh samples) and 7 h at ambient temperature (24˚C ± 2˚C), the plasma samples of three QC levels after storage at −20˚C for 1, 7 and 106 days respectively, and after one or two freeze-thaw cycles (−20˚C to 25˚C). The samples were processed as described above and the criterion for acceptability of the data is the same with that for the precision and accuracy.
2.7. Pharmacokinetic Study
This randomized, open-label, single-dose, two-crossover bioequivalence study was conducted at the Institute of Clinical Pharmacology, Qilu Hospital of Shandong University. The protocol was approved by the Ethics Committee of Qilu Hospital, Shandong University, Jinan, China, and the study was conformed to the principle of the Declaration of Helsinki.
Twenty four healthy Chinese male subjects, aged 21 to 29 years, were involved for the study. All subjects included were within the permitted range for their height and frame, in good health, and had no major organ dysfunction as determined by medical history, physical examination, electrocardiogram (ECG) findings, and routine laboratory testing. All subjects agreed to participate in the trial and signed the approved informed-consent form voluntarily.
Subjects arrived at the study center in the evening before dosing and fasted for at least 10 hours. Each Subject enrolled in the study was uniquely assigned, according to a predetermined randomization schedule which provided the order of treatments. Subjects received two tablets of compound amlodipine tablets (10 mg/100mg, amlodipine/losartan) or two tablets of amlodipine besylate tablets (10 mg) and losartan potassium tablets (100 mg). Subjects ingested the tablets with 250 mL of water without chewing or crushing, no water was allowed 1 h before and 2 h after the administration under supervision of study personnel. A washout period of 14 days separated the two periods. Tolerability was assessed by physician examination and laboratory testing. Each subject received a physical monitoring and laboratory safety tests prior to leaving the institute in the morning of the 17th day.
Blood samples were drawn at pre-dose (baseline) and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96, 144 and 168 hours post-dose during each study period. Samples were immediately collected into sodium-heparinized tubes and centrifuged at 5000 rpm for 5 minutes at 4˚C within 30 minutes of collection. Plasma was separated, transported into EP tubes, and stored at −20˚C for pending analysis.
2.8. Statistical Analysis
The bioequivalence of the test and reference formulations was assessed by calculating individual test/reference ratios for the peak of amlodipine concentration (Cmax), area under the plasma concentration-time curve from 0 to last measurable time (AUC0-t) amd from 0 to infinity (AUC0-∞). The pharmacokinetic parameters including Cmax, time to maximum plasma concentration (Tmax) were calculated directly from the measured data. The terminal elimination half-life (t1/2), AUC0-t and AUC0-∞ were calculated by using Drug and Statistics Software-Version 2.0 (DAS, version 2.0.1, by Sun Ruiyuan et al., China).
All data are presented as mean ± standard deviation. Statistical significance was assessed by two-way factorial analysis of variance test followed by Duncan’s multiple comparison between groups. Based on guidance from the Food and Drug Administration (FDA) definition, a 90% confidence interval (CI) of the In-transformed ratios should fall between 80% and 125%. A probability level of p < 0.05 was considered to be statistically significant.
3. Results
3.1. Method Validation
3.1.1. Specificity
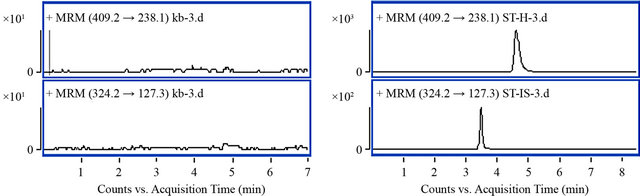
Typical MRM chromatograms of blank plasma, amlodipine and IS standard, blank plasma spiked with amlodipine and IS, plasma from a subject after administration of amlodipine test tablets spiked with IS were shown in Figure 2. At least six blank plasma samples from different individuals were examined for the specificity of the method. Retention times of amlodipine and IS were 4.8 and 3.7 min, respectively. No significant interference were observed in the endogenous blank human plasma at the retention times of amlodipine and IS during analysis.
3.1.2. Matrix Effect and Extraction Recovery
Matrix effect and extraction recovery data from plasma of individual subjects who did not receive amlodipine were summarized in Table 2. The extraction recoveries of amlodipine at three concentrations in plasma were all over 60%, and IS over 80%. No matrix suppression from blank plasma was observed during analysis, indicated that the analytical method could be kept free endogenous substances in human plasma.
3.1.3. Calibration Curve and LLOQ
Correlation coefficient (r2), which indicates linearity over the calibration range of amlodipine, was calculated by using the 1/x2 model. The calibration curve was linear over the amlodipine concentration ranges of 0.05 - 12 ng/mL in human plasma with a coefficient of correlated (r2) > 0.99. A typical equation of the calibration curve was y = 0.6630x − 0.00065 (r2 = 0.9918).
The LLOQ (0.05 ng/mL) was defined with acceptable accuracy and precision. The analyte response at the LLOQ should be at least five times the response of blank baseline. A good signal-to-noise ratio (S/N > 10) was observed at the LLOQ. Intra-assay precision and accuracy were satisfactory for the LLOQ, and RSD values were less than 5% (Table 3).
3.1.4. Precision and Accuracy
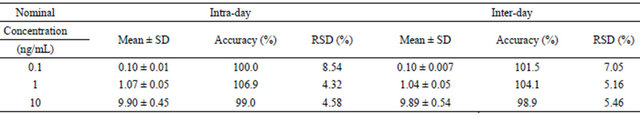
Precision and accuracy data for intraand inter-day plasma samples of amlodipine are shown in Table 4. The intra-day and inter-day precision values (RSD) were less than 8.6%, and the accuracies were ranged from 98.9% to 106.9%. The results demonstrated that the precision and accuracy of this assay were within the acceptable range and the method was accurate and precise.
 (a)
(a)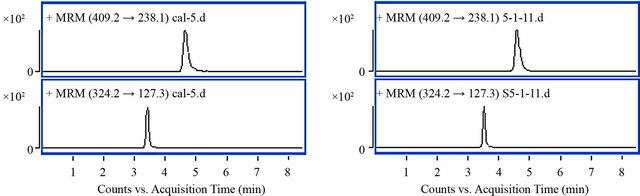 (b)
(b)
Figure 2. Typical MRM chromatograms of amlodipine and IS. ((a) Blank human plasma; (b) Standard of amlodipine (2.5 ng/mL) and IS (8 ng/mL); (c) Blank human plasma spiked with amldoipine (1 ng/mL) and IS (3.2 ng/mL); (d) Plasma sample of subject NO. 5 after 8 h of oral administration in period 1).
Table 2. Matrix effect and extraction recovery of amlodipine and IS in human plasma (n = 5).
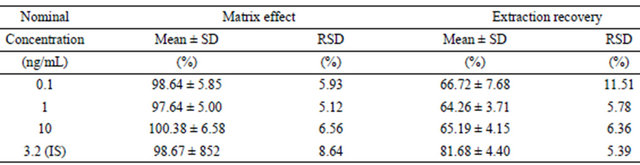
Table 3. The analyte response at the LLOQ in human plasma (n = 5).
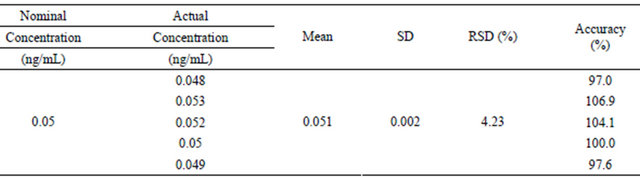
Table 4. Intraday and interday precision and accuracy for analysis of amlodipine in human plasma (n = 5).
3.1.5. Stability
The stability results of amlodipine in plasma were summarized in Table 5. The results indicated that amlodipine were stable in plasma after storage at −20˚C for 106 days, and for two freeze-thaw cycles. Processed samples were allowed to stand at room temperature in reconstituted solution for 7 h prior to analysis, with no observed effect on results of quantification. The data conform to the acceptance criteria.
3.2. Pharmacokinetics
The whole assay was conducted strictly in accordance with the current Good Clinical Practices. In total, 24 healthy male subjects completed the study, and underwent PK and safety evaluations. The demographic profiles did not differ compared with the previous, all oral doses were well tolerated by the subjects, as no adverse events reported. Mean PK parameters derived from 24 male subjects after the administration of 10 mg amlodipine tablets and the 90% CI for the treatment ratio (test: reference) of AUC0-t, AUC0-∞, Cmax were all listed in Table 6. The mean plasma concentration-time curve after a single oral dose of test and reference tablets formulation of amlodipine was illustrated in Figure 3. In addition, the calculated 90% CI for mean Cmax, AUC0-t and AUC0-∞ were within the 80% - 125% bioequivalence limit defined by the US Food and Drug Administration. Supported by these data, pharmacokinetic parameters (Tmax, Cmax, AUC0-t, AUC0-∞ and t1/2) were similar between the reference and test products.
4. Discussion
4.1. Method Development
4.1.1. Selection of IS
A stable analyte has to be used as IS to deal with sample matrix effects. It was difficult to find a compound that could ideally mirror the analyte to serve as a suitable IS. The chosen of IS should match the chromatographic properties, recovery and ionization properties of the analyte. In the initia phase, several compounds were investigated, such as lorazepam, matrine, ammothamnine and theocin, but the former three had long retention time and theocin had double humps. Finally gliclazide, a readily available compound, was selected as the IS in positive ion mode. Its chromatographic behavior and extraction
Table 5. Stability data of amlodipine in human plasma at various conditions (n = 5).
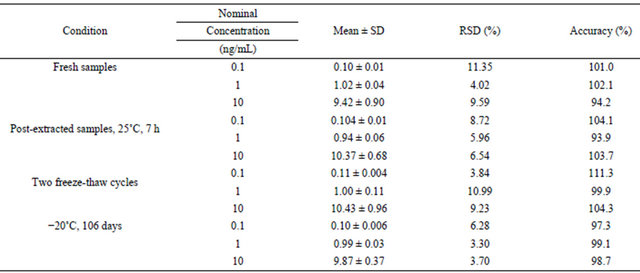
Table 6. PK parameters of amlodipine after a single dose of compound amlodipine tablets (10 mg/100mg, amlodipine/losartan) and amlodipine tablets (10 mg)/losartan tablets (100 mg) after oral administration to 24 healthy male subjects.
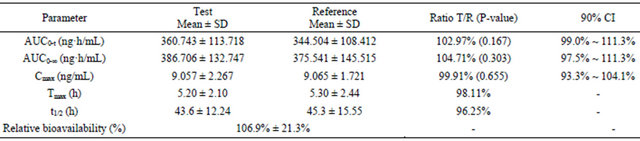
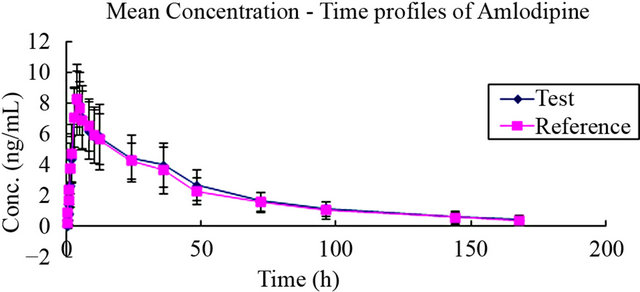
Figure 3. Mean plasma concentration-time profile of amlodipine after administration of the compound amlodipine tablets (10 mg/100mg, amlodipine/losartan) and amlo-dipine tablets (10 mg)/losartan tablets (100 mg). (Mean ± SD, n = 24.)
efficiency were similar to amlodipine, stable in plasma and reproducible in the LC/MS/MS system. In addition, there were no interferences from amlodipine and endogenous substances.
4.1.2. Sample Pre-Treatment
A sample pre-treatment method should remove interferences from the biological sample and also be reproducible with a high recovery involving a minimum number of working steps. Protein precipitation (PPT), liquid-liquid extraction (LLE) and solid-phase extraction (SPE) were tested to obtain a simple and excellent plasma preparation procedure. PPT was easy to dilute the sample, but failed to sufficiently remove endogenous interference. SPE had too many disturbances, which contributed to poor reproducibility and poor recovery. What is more, the column for SPE is relatively expensive for a large quantity of samples. LLE was advantageous for producing chromatographia clean samples with sufficient efficiency and specificity, and it also minimized cost. In addition, LLE was essential for minimizing ion suppression and matrix effects in HPLC-MS/MS analysis. LLE with various extraction solvents, including ethyl acetate, diethyl ether-dichloromethane (2:1, v/v) and diethyl ether, was investigated and evaluated for acceptable extraction recoveries and matrix effect. Diethyl ether, extraction recoveries were about 20.12% and unacceptable, chloroform was easy to emulsify when the sample was made vortex and mixed, and its recover was about 31.20%. Ethyl acetate was adopted with the weakest matrix effect, no-concentration-dependent extraction recovery and acceptable matrix effect. Furthermore, sodium hydroxide (1 mol/L) added to extraction solvents was also tested, and there were not significant difference for peak area and retention time of amlodipine.
Solvents for reconstituting residues were also investigated to optimize the chromatographic behaviors. Acetonitrile, methanol, 10 mM ammonium acetate and mobile phase were compared and evaluated for optimizing peak shape and maximum peak response. Finally, mobile phase with good peak shape and maximum response was adopted.
4.1.3. Liquid Chromatography
A simple chromatographic separation was developed for amlodipine with acquisition of good separation. Compositions of mobile phase were investigated to optimize the chromatographic behaviors of amlodipine. The feasibility of various mixtures of solvents such as acetonitrile and methanol using different buffers, such as ammonium acetate, ammonium formate and formic acid, along with altered flow-rates (in the range of 0.5 - 1 mL/min), was tested to identify the optimal mobile phase that produced the best sensitivity, efficiency and peak shape. Finally, chromatographic separation was operated by gradient flow rates and varied ratio of mobile phase system consisting of methanol −10 mM ammonium acetate to suppress the matrix effect from blank plasma in positive ionization mode. As the temperature increased, the peak area of amlodipine became smaller and retention time appeared earlier, therefore, the column temperature was maintained at 30˚C.
4.1.4. Mass Spectrometry Optimization
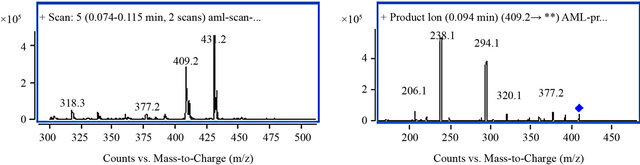
MS parameters were tuned in both positive and negative ionization modes for amlodipine and IS. However, a good response was found in positive ionization mode. The solutions containing amlodipine and IS were injected directly into the mass spectrometer. Under these conditions, the analytes yielded major ions at [M + H]+ m/z 409.2 and [M + Na]+ m/z 431.2 for amlodipine and [M + H]+ m/z 324.2 for IS. Each of precursor ions was subjected to collision-induced dissociation to determine the resulting product ions. The product ion mass spectra of amlodipine and IS are shown in Figure 4. Compared with [M + H]+ mode, a poor linearity for [M + Na]+ (data not shown) and a good response was found in [M + H]+ mode. Finally, the most sensitive and abundant mass transitions was monitored from m/z 409.2→238.1 (quantitation), 294.1 (qualitation) for amlodipine, and 324.2→ 127.3 for IS.
Above all, the sensitive and specific LC-MS/MS method in this study allowed a better characterization of the PK profiles of amlodipine.
 (a)
(a)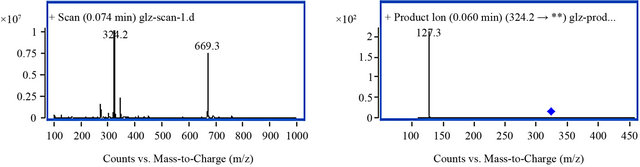 (b)
(b)
Figure 4. MS spectrum of amlodipine and IS with full-scan positive product ion mass spectra. ((a) Precursor ion spectrum of amlodipine; (b) Product ion spectrum of amlodipine; (c) Precursor ion spectrum of IS; (d) Product ion spectrum of IS).
4.2. Bioequivalence Evaluate
As reported in the literatures, amlodipine is extensively metabolized in the liver and then redistributed into the systemic circulation, resulting in a long time to Cmax (6 - 12 hours) and a long elimination half-life t1/2 (30 - 50 hours) in healthy subjects [27]. Amlodipine has large volume of distribution and it exhibits a high degree of protein binding (98%). Bioavailability is 60% - 65% due to first pass metabolism [18,28]. Compared with single preparations of amlodipine, our results show that there is no significant difference in PK parameters of amlodipine.
After two-one side t-test following ANOVA to Cmax, AUC0-t and AUC0-∞, no significant difference was found among compound amlodipine tablets (10 mg/100mg, amlodipine/losaran) and 10 mg amlodipine tablets in 24 healthy male subjects. The 90% CI of test and reference mean ratio for Cmax, AUC0-t and AUC0-∞ were all ranged from 80% to 125%. Therefore, it was concluded that both formulations (test and reference) achieved equivalent bioavailability.
5. Conclusions
In summary, a highly sensitive, specific, reproducible and high-throughput LC-MS/MS method was developed and validated based on the procedure of LLE for quantification of amlodipine pharmacokinetic and bioequivalence studies. The extraction procedure and LC-MS/MS conditions were optimized inorder to improve the sensitivity and robustness of the method. Finally, the achieved limit of quantification (0.05 ng/mL) was sensitive for plasma amlodipine determination and for its the pharmacokinetic parameters calculation, and the procedure was fully validated to meet the requirements for sensitivity, accuracy and precision defined by State Food and Drug Administration and GLP Guidelines for Industry where more than 2200 samples were analyzed.
The amlodipine relative bioavailability between 10 mg amlodipine/100mg losartan compound amlodipine tablets as test and 10 mg amlodipine tablet as reference was assessed by calculating individual test/reference ratios for AUC0-t and pharmacokinetic profiles indicated bioequivalence since all ratios were as proposed by FDA. According to the validation parameters, it concluded that the developed method could be useful for amldoipine bioequivalent studies and routine therapeutic drug monitoring with desired precision and accuracy, and this study may be extended to determine the pharmacokinetics of amlodipine in patients and also to examine the drug-food or drug-drug interaction in combination therapy.
6. Acknowledgements
This study was conducted as a part of the projects (2012ZX09303-016-003) supported by the Major National Science and Technology Project, People Republic of China.
REFERENCES
- W. C. Cushman, “Are There Benefits to Specific Antihypertensive Drug Therapy?” American Journal of Hypertension, Vol. 16, No. 11, 2003, pp. 31S-35S.
- C. Borghi and A. F. Cicero, “Rationale for the Use of a Fixed-Dose Combination in the Management of Hypertension: Efficacy and Tolerability of Lercanidipine/ Enalapril,” Clinical Drug Investigation, Vol. 30, No. 12, 2010, pp. 843-854. doi:10.1007/BF03256912
- T. Philipp, T. R. Smith, R. Glazer, M. Wernsing, J. Yen, J. Jin, et al., “Two Multicenter, 8-Week, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Studies Evaluating the Efficacy and Tolerability of Amlodipine and Valsartan in Combination and as Monotherapy in Adult Patients with Mild to Moderate Essential Hypertension,” Clinical Therapeutics, Vol. 29, No. 4, 2007, pp. 563-580. doi:10.1016/j.clinthera.2007.03.018
- F. Kolocouri, Y. Dotsikas, C. Apostolou, C. Kousoulos and Y. L. Loukas, “Simultaneous Determination of Losartan, EXP-3174 and Hydrochlorothiazide in Plasma via Fully Automated 96-Well-Format-Based Solid-Phase Extraction and Liquid Chromatography-Negative Electrospray Tandem Mass Spectrometry,” Analytical and Bioanalytical Chemistry, Vol. 387, No. 2, 2007, pp. 593-601. doi:10.1007/s00216-006-0990-4
- J. Y. Jia, M. Q. Zhang, Y. M. Liu, Y. Liu, G. Y. Liu, S. J. Li, et al., “Pharmacokinetics and Bioequivalence Evaluation of Two Losartan Potassium 50-mg Tablets: A Single-Dose, Randomized-Sequence, Open-Label, Two-Way Crossover Study in Healthy Chinese Male Volunteers,” Clinical Therapeutics, Vol. 32, No. 7, 2010, pp. 1387- 1395. doi:10.1016/j.clinthera.2010.06.018
- J. Bhatt, S. Singh, G. Subbaiah, B. Shah, S. Kambli and S. Ameta, “A Rapid and Sensitive Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Estimation of Amlodipine in Human Plasma,” Biomedical Chromatography, Vol. 21, No. 2, 2007, pp. 169-175. doi:10.1002/bmc.730
- S. Tatar and S. Atmaca, “Determination of Amlodipine in Human Plasma by High-Performance Liquid Chromatography with Fluorescence Detection,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 758, No. 2, 2001, pp. 305-310. doi:10.1016/S0378-4347(01)00197-9
- D. Murdoch and R. C. Heel, “Amlodipine. A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Use in Cardiovascular Disease,” Drugs, Vol. 41, No. 3, 1991, pp. 478-505. doi:10.2165/00003495-199141030-00009
- F. Scharpf, K. D. Riedel, H. Laufen and M. Leitold, “Enantioselective Gas Chromatographic Assay with Electron-Capture Detection for Amlodipine in Biological Samples,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 655, No. 2, 1994, pp. 225-233. doi:10.1016/0378-4347(94)00068-9
- S. C. Monkman, J. S. Ellis, S. Cholerton, J. M. Thomason, R. A. Seymour and J. R. Idle, “Automated Gas Chromatographic Assay for Amlodipine in Plasma and Gingival Crevicular Fluid,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 678, No. 2, 1996, pp. 360-364. doi:10.1016/0378-4347(95)00526-9
- G. Bahrami and S. Mirzaeei, “Simple and Rapid HPLC Method for Determination of Amlodipine in Human Serum with Fluorescence Detection and Its Use in Pharmacokinetic Studies,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 36, No. 1, 2004, pp. 163-168. doi:10.1016/j.jpba.2004.05.016
- A. Zarghi, S. M. Foroutan, A. Shafaati and A. Khoddam, “Validated HPLC Method for Determination of Amlodipine in Human Plasma and Its Application to Pharmacokinetic Studies,” Farmaco, Vol. 60, No. 9, 2005, pp. 789-792. doi:10.1016/j.farmac.2005.06.012
- K. Shimooka, Y. Sawada and H. Tatematsu, “Analysis of Amlodipine in Serum by a Sensitive High-Performance Liquid Chromatographic Method with Amperometric Detection,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 7, No. 11, 1989, pp. 1267-1272. doi:10.1016/0731-7085(89)80130-X
- M. Josefsson, A. L. Zackrisson and B. Norlander, “Sensitive High-Performance Liquid Chromatographic Analysis of Amlodipine in Human Plasma with Amperometric Detection and a Single-Step Solid-Phase Sample Preparation,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 672, No. 2, 1995, pp. 310-313. doi:10.1016/0378-4347(95)00231-7
- K. K. Pandya, M. satia, T. P. Gandhi, I. A. Modi, R. I. Modi and B. K. Chakravarthy, “Detection and Determination of Total Amlodipine by High-Performance ThinLayer Chromatography: A Useful Technique for Pharmacokinetic Studies,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 667, No. 2, 1995, pp. 315-320. doi:10.1016/0378-4347(95)00016-C
- Y. Feng, L. Zhang, Z. Shen, F. Pan and Z. Zhang, “Analysis of Amlodipine in Human Plasma by Liquid Chromatography-Mass Spectrometry,” Journal of Chromatographic Science, Vol. 40, No. 1, 2002, pp. 49-53.
- B. Streel, C. Laine, C. Zimmer, R. Sibenaler and A. Ceccato, “Enantiomeric Determination of Amlodipine in Human Plasma by Liquid Chromatography Coupled to Tandem Mass Spectrometry,” Journal of Biochemical and Biophysical Methods, Vol. 54, No. 1-3, 2002, pp. 357-368. doi:10.1016/S0165-022X(02)00133-1
- J. Shentu, L. Fu, H. Zhou, X. J. Hu, J. Liu, J. Chen, et al., “Determination of Amlodipine in Human Plasma Using Automated Online Solid-Phase Extraction HPLC-Tandem Mass Spectrometry: Application to a Bioequivalence Study of Chinese Volunteers,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 70, No. 2012, pp. 614-618.
- M. Carvalho, C. H. Oliveira, G. D. Mendes, M. Sucupira, M. E. Moraes and G. De Nucci, “Amlodipine Bioequivalence Study: Quantification by Liquid Chromatography Coupled to Tandem Mass Spectrometry,” Biopharmaceutics & Drug Disposition, Vol. 22, No. 9, 2001, pp. 383- 390. doi:10.1002/bdd.282
- A. K. Sarkar, D. Ghosh, A. Das, P. S. Selvan, K. V. Gowda, U. Mandal, et al., “Simultaneous Determination of Metoprolol Succinate and Amlodipine Besylate in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry Method and Its Application in Bioequivalence Study,” Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, Vol. 873, No. 1, 2008, pp. 77-85. doi:10.1016/j.jchromb.2008.07.040
- A. V. Ramani, P. Sengupta and R. Mullangi, “Development and Validation of a Highly Sensitive and Robust LC-ESI-MS/MS Method for Simultaneous Quantitation of Simvastatin Acid, Amlodipine and Valsartan in Human Plasma: Application to a Clinical Pharmacokinetic Study,” Biomedical Chromatography, Vol. 23, No. 6, 2009, pp. 615-622. doi:10.1002/bmc.1161
- V. B. Ravi, J. K. Inamadugu, N. R. Pilli, V. Sreenivasulu and V. Ponneri, “Simultaneous Determination of Telmisartan and Amlodipine in Human Plasma by LC-MS/MS and Its Application in a Human Pharmacokinetic Study,” Journal of Pharmaceutical Analysis, Vol. 2, No. 5, 2012, pp. 319-326. doi:10.1016/j.jpha.2012.03.008
- X. Wei, G. Yang, L. Qi and Y. Chen, “Determination of Nicardipine and Amlodipine in Human Plasma Using On-Line Solid-Phase Extraction with a Monolithic Weak Cation-Exchange Column,” Talanta, Vol. 77, No. 3, 2009, pp. 1197-1202. doi:10.1016/j.talanta.2008.08.034
- H. Chang, J. Li, X. Guan, F. Sun, Z. Qian, K. Bi, et al., “Simultaneous Determination of Amlodipine and Bisoprolol in Rat Plasma by a Liquid Chromatography/ Tandem Mass Spectrometry Method and Its Application in Pharmacokinetic Study,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 71, No. 2012, pp. 104-110.
- Y. Ma, F. Qin, X. Sun, X. Lu and F. Li, “Determination and Pharmacokinetic Study of Amlodipine in Human Plasma by Ultra Performance Liquid ChromatographyElectrospray Ionization Mass Spectrometry,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 43, No. 4, 2007, pp. 1540-1545. doi:10.1016/j.jpba.2006.11.015
- C. T. Viswanathan, S. Bansal, B. Booth, A. J. DeStefano, M. J. Rose and J. Sailstad, et al., “Quantitative Bioanalytical Methods Validation and Implementation: Best Practices for Chromatographic and Ligand Binding Assays,” Pharmaceutical Research, Vol. 24, No. 10, 2007, pp. 1962-1973. doi:10.1007/s11095-007-9291-7
- Y. Liu, J. Jia, G. Liu, S. Li, C. Lu and C. Yu, “Pharmacokinetics and Bioequivalence Evaluation of Two Formulations of 10-mg Amlodipine Besylate: An Open-Label, Single-Dose, Randomized, Two-Way Crossover Study in Healthy Chinese Male Volunteers,” Clinical Therapeutics, Vol. 31, No. 4, 2009, pp. 777-783. doi:10.1016/j.clinthera.2009.04.013
- F. Abad-Santos, J. Novalbos, M. A. Galvez-Mugica, S. Gallego-Sandin, S. Almeida, F. Vallee, et al., “Assessment of Sex Differences in Pharmacokinetics and Pharmacodynamics of Amlodipine in a Bioequivalence Study,” Pharmaceutical Research, Vol. 51, No. 5, 2005, pp. 445- 452. doi:10.1016/j.phrs.2004.11.006
NOTES
*Project supported by the Major National Science and Technology Project (2012ZX09303-016-003).
#Corresponding author.