Advances in Alzheimer's Disease
Vol.1 No.3(2012), Article ID:26142,6 pages DOI:10.4236/aad.2012.13011
Neuronal protection by a variant of GAPDH pseudogene P44 in AD
![]()
Department of Biochemistry, Kansas City University of Medicine and Biosciences, Kansas City, USA; *Corresponding Author: nseidler@kcumb.edu
Received 24 October 2012; revised 26 November 2012; accepted 3 December 2012
Keywords: GAPDH, Alzheimer’s Disease; Pseudogene; GAPDHP44; SNP; Apoptotic Nuclear Translocation; Siah; GOSPEL
ABSTRACT
GAPDH is a conserved enzyme that binds diverse proteins, such as Siah during apoptotic nuclear translocation. There is one somatic GAPDH gene, but over 60 pseudogenes, the expression of which is nebulous. A single nucleotide polymorphism (SNP) in the GAPDHP44 pseudogene exhibits a beneficial allele in AD. The objective of this study was to examine the P44 gene and to propose a mechanism for the putative protein and its impact on AD. We examined the sequences in the putative coding region of the human GAPDHP44 gene and the upstream genetic elements using a bioinformatics approach. We compared the amino acid sequences of the putative gene product with that of the parent GAPDH protein. There is a TATA box 24 nt upstream from, and a Kozak sequence at, putative transcription and translation start sites, respecttively. The upstream region also has sequences (7 - 16 nt) paralogous to those in parent gene introns; one shows homology to a known enhancer element. The resulting protein would contain 139 aa due to a stop codon, roughly the same size as the dinucleotide domain (151 aa) of the parent protein. The SNP is in a region (residues 80 - 120) that binds to the protein GOSPEL. We propose that the beneficial SNP may cause a glutamine to glutamate substitution. NMDA-stmulated neurons undergo GAPDH nitrosylation, Siah translocation, but can be rescued by GOSPEL binding to GAPDH. Our model suggests that the putative P44 protein may regulate GAPDH-GO-SPEL interaction and the beneficial SNP may ameliorate AD.
1. INTRODUCTION
Alzheimer’s disease (AD) is an age-related disorder characterized by gradual loss in cognitive function. It is the leading cause of dementia, which affects an estimated 35.6 million people worldwide, according to alz.org. In the United States alone, there are 20,000 “baby boomers” that turn 65 years of age every day, thus entering the age of risk. Data from at-risk individuals (i.e. both geneticand age-associated) suggest that the pathophysiological processes of AD begin more than a decade prior to the emerging indications of dementia. In preclinical AD, there are many neuritic and diffuse amyloid b (Ab)-peptide-containing plaques in the neocortex. Additionally, there are many neurofibrillary tau protein-containing tangles in the entorhinal cortex and in the CA1 region of the hippocampus with little to no cell loss in these areas of the brain during the preclinical phase of AD. Changes in the CSF levels of these proteins correspond to the pathological landmarks that are thought to occur well before clinical manifestations. In mild AD, one begins to notice cognitive impairment and very mild dementia, and attendant to this, one sees a substantial amount of cell loss in the entorhinal cortex and hippocampus/CA1. Despite the suggestion of Ab-peptide as a critical early factor in the pathogenesis of AD, anti-Ab-peptide therapies have failed to achieve a significant level of success. Our lab is interested in an alternate approach to viewing AD, namely the role of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the pathogenesis of AD.
GAPDH is a highly conserved glycolytic oxido-reductase enzyme that has many non-glycoly tic “moonlighting” functions [1], one of which includes regulating neuronal apoptosis [2]. The diversity of function exhibited by the GAPDH protein remains a mystery. In humans, there are two parent GAPDH genes (i.e. somatic and spermatogenic) as well as many processed pseudogenes that were derived from the parent somatic gene [3], representing the paralogous GAPDH genes.
There is increased interest in the role of GAPDH in neurodegenerative diseases, particularly in light of the pervasive multifunctionality of GAPDH [4]. Curiously, senile plaques in autopsy samples from AD patients were observed to contain GAPDH [5]. Furthermore, overexpression of GAPDH in neuronal cultures contributes to apoptotic cell death. Another study used a proteomic ap proach in studying AD [6]. These authors looked at thehippocampal proteome in patients with AD by two-dimensional electrophoresis and mass spectrometry. They identified two protein spots as being GAPDH and showed that they were at significantly higher than control levels, consistent with an overexpression of GAPDH in AD. This suggests that either multiple transcripts of GAPDH were presented or that a post-transcriptional modification occurred. In addition to S-nitrosylation of critical residues, GAPDH undergoes many other types of chemical modification [7]. Interestingly, Mazzola and Sirover [8] observed that individuals with AD have reduced levels of GAPDH oxido-reductase activity compared to control subjects. However, gene expression remains unchanged in the individuals afflicted with AD, as determined by levels of GAPDH protein. The authors propose that the loss of GAPDH activity associated with AD is due to post-translational modifications. Both nuclear and nonnuclear fractions of GAPDH exhibited decreased oxidoreductase activity in AD. Chemical modification may precede GAPDH’s involvement in neuronal apoptosis. Additionally, several other studies implicate GAPDH in neuronal apoptosis [9-11], supporting the linkage between AD and GAPDH.
The amyloid hypothesis, as it stands in isolation of other environmental/genetic triggers, appears to be incomplete. Current therapies have fallen short of their expected outcomes. The role of GAPDH may provide an insight to the molecular mechanism of the disease, and therefore, knowledge in this area may offer a new route for effective AD therapies.
2. GENETIC LINKAGE BETWEEN AD AND GAPDH
Several studies have linked the various GAPDH paralogous genes with AD [12-15]. A large-scale single nucleotide polymorphism (SNP) association study [12] observed that a beneficial SNP occurs in the region of a GAPDH pseudogene. Individuals with the AA or AG alleles at rs2029721 had a lower risk of late-onset AD than those individuals with the GG alleles [14], confirming earlier work on this SNP [12]. The G allele is the ancestral form. The SNP, which is designated rs2029721, is located on chromosome 12 (specifically, 12q14.2). The official name of the pseudogene is GAPDHP44. This gene represents a processed pseudogene that was retrotransposed from the single somatic GAPDH gene, involving reverse transcription of the processed mRNA and stable insertion of the DNA copy—a curiously non-viral process. There are over 60 GAPDH pseudogenes in the human genome. The fact that a significant odds ratio was observed for a SNP in a GAPDH pseudogene suggests that the pseudo-gene has biologic function.
GAPDHP44 gene is located on the forward strand. There are no intervening sequences in this gene. The anti-sense strand of this pseudogene sits in an intron of the protein phosphatase 1H gene (gene name, PPM1H), which is transcribed on the reverse strand.
We were interested in exploring possible biological functions for this pseudogene, particularly given its association with late-onset AD. It is suggested [12,14] that the A and G alleles may represent a missense mutation, implying that a protein product may be directing the beneficial aspect of this SNP.
3. BIOLOGICAL ROLES OF GAPDHP44
There are several possible biological roles that one can propose for the SNP at rs2029721. The effects of this SNP may act through either the GAPDHP44 or the PPM- 1H gene. Both genes, which exist on opposite strands, appear to be expressed in brain.
We think that the SNP acts through GAPDHP44. There are two possible mechanisms by which this can occur. First, the gene may be producing a translatable transcript, which produces a functional protein. The putative protein product may exhibit biological properties that contribute to the functional diversity seen with GAPDH [16]. Second, the gene itself may act in an anti-sense manner, regulating parent GAPDH mRNA stability and expression.
An alternate mechanism may involve the SNP acting through PPM1H. The anti-sense of the GAPDHP44 pseudogene is located in the intron of PPM1H. Interestingly, the PPM1H gene is overexpressed in brains of patients with late-onset AD [14]. This alternate mechanism involves two possible scenarios. Since a PPM1H intron contains sequences that are complementary to parent GAPDH mRNA, one can envision the intronic processing may result in RNA interference products, such as microRNA, regulating parent GAPDH mRNA translation. Additionally, the SNP may affect processing of the PPM1H intron, playing a role in PPM1H expression.
We think that only one of these several mechanisms predominates. We propose that the protein product, which is generated from the GAPDHP44 gene, plays a direct role in the beneficial effects associated with the SNP. By careful analysis of this gene, we developed a working model describing how a variant of the GAPDHP44 gene can provide neuronal protection.
4. EXPRESSION OF GAPDHP44
There is evidence that the intronless GAPDHP44 gene is expressed in various regions of the brain, including the hippocampus, cerebellum, temporal cortex and prefrontal cortex [12]. This is consistent with the anatomical progression of AD. The hippocampal region, which is involved in memory, is an area particularly affected in AD. Another less compelling evidence of GAPDHP44 expression is indicated by the documentation of a transcript ID (i.e. ENST00000513513) that can be found at the Ensembl website, although no annotation is given.
We know that the PPM1H gene is actively expressed in brain as indicated by electronic databases. Curiously, the database at bioGPS.org shows that expression of this gene is highest in neuronal tissue. With active transcripttion of the PPM1H gene, it is reasonable to suggest that the complementary strand, which contains the GAPDHP- 44 gene, would be in an open configuration accessible to transcriptional machinery, enhancing the likelihood of GAPDHP44 expression.
The upstream sequence of GAPDHP44 pseudogene was obtained from the RefSeqNC_000012.11 numbered from 63,148,800 - 63,149,007. We visually inspected the sequence upstream of the translated region and identified viable consensus sequences (Figure 1).
The putative TATA box sequence is highlighted in bold and shaded. The transcription and translation start sites are noted with arrows. The transcribed but un-translated region is shown in brackets. The Kozak consensus sequence [17,18] is underlined. Spaces are included for ease of viewing.
A putative TATA box sequence is found 24 nucleotides upstream of the putative start site that is identified as the beginning of the GAPDHP44 pseudogene. This observation is consistent with the expected location of the RNA polymerase II binding site [19]. The translational start site in the GAPDHP44 pseudogene does not begin with the typical AUG codon; the start codon observed in the GAPDHP44 pseudogene is ACG. While the universal initiation codon for eukaryotes is AUG, other codons (namely, ACG and CUG) were observed as initiation codons in a few mammalian mRNAs. While the ACG normally codes for threonine, it is possible that the initiator tRNA may incorporate the start amino acid methionine [20] using ACG; thus acting as a translational initiation codon in the pseudogene GAPDHP44.
We observed another curious phenomenon. The 5’untranslated region (i.e. shown in brackets in Figure 1) contained sequences identical to those found in the introns of the parent GAPDH. Sequences from introns A, B, C, E and G of the parent somatic GAPDH gene are conserved in the noncoding region of the GAPDHP44 gene (Figure 2).
The first 71 nucleotides of GAPDHP44, which did not show any similarity to the coding region of the somatic GAPDH, interestingly, showed multiple comparisons with elements located in the introns of the somatic GAPDH gene (Figure 2). While this finding is curious, the

Figure 1. Genetic elements in pseudogene GAPDHP44.
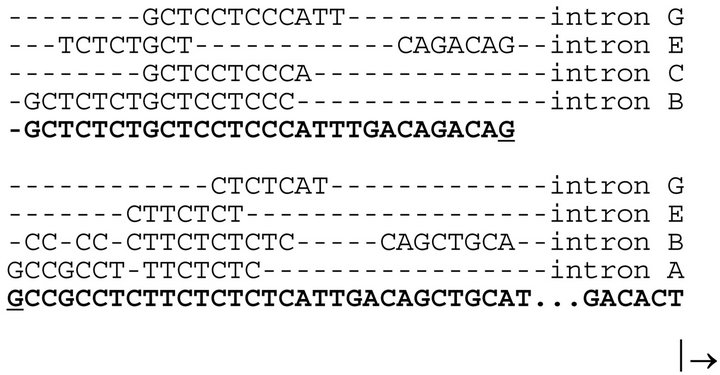
Figure 2. Conserved sequences in the 5’untranslated region of the GAPDHP44 gene.
significance is not yet known. It is unlikely that this phenomenon is random and may provide clues as to the biological role of pseudogenes. These elements may represent conserved enhancer sequences that may play a role both in the somatic GAPDH as well as in promoting expression of the GAPDHP44 gene. This would be consistent with the proposed hypothesis of GAPDHP44 being expressed.
Intronic sequences (i.e. seven to sixteen nucleotides in length and found in the parent somatic GAPDH gene) are also found clustered in the noncoding region of the GAPDHP44 gene (bolded). The arrow pointed to the right indicates the translation start site.
As mentioned above, the complementary strand to the GAPDHP44 pseudogene is located in the PPM1H gene. Active expression therefore of the PPM1H mRNA would maintain this region of the chromosome in a decondensed state, increasing the likelihood of expressing the GAPDHP44 gene. Interestingly, the PPM1H gene is overexpressed in the brains of patients with late-onset AD [14], suggesting a co-expression of the GAPDHP44 gene.
We recently looked at a computational assessment of GAPDH pseudogenes with the assumption that they are expressed and that protein products would potentially exhibit viable biological activity [21]. The biological property that was assessed was membrane-binding, which may contribute to the multi-functionality of GAPDH [1, 22]. The first alpha helix in the dinucleotide-binding domain is thought to play a role in membrane interaction, in part due to its amphipathic property. In the event that this region was significantly modified, we postulated [21] that it would suggest a disablement of the pseudogene. While most of the pseudogenes that were investigated did not show disablement, the GAPDHP44 failed to show a stable amphiphathic helix, suggesting that the protein would likely not interact with membranes. This observation is consistent with our current model described below.
In examining the GAPDHP44 protein, we found that there are eight stop codons. In the event that this gene is translated, the initial 139 amino acid residues would be intact prior to the first stop codon. We propose that this approximately 15 kDa truncated GAPDHP44 protein may have biological function. Curiously, the somatic GAPDH dinucleotide-binding site is exactly comprised of the first 151 amino acid residues, suggesting that the GAPDHP44 protein would contain a nearly complete dinucleotide-binding site. Furthermore, this truncated GAPDHP44 protein also contains an intact GOSPEL-binding site. GOSPEL is a protein that regulates the GAPDH-induced cell death cascade.
5. PROPOSED MECHANISM FOR GAPDHP44
The biological implications of the expression of a truncated GAPDH isoform are intriguing. Interestingly, when the gene products of pseudogene GAPDHP44 and parent GAPDH are compared (Figure 3), we observed an 83% identity and an 86% similarity using the Blosum 62 (i.e. values greater than zero were designated as similar substitutions). One question would be to determine the integrity of the phosphatidylserine binding site, which is located at amino acid residues 70 - 94 [23]. In comparing this region we observed a greater than 10% difference in sequence identity, suggesting that this region may be affected in some small way. The N-terminal alpha helix defined by amino acid residues 12 - 23 is thought to also interact with biological membranes [24]. In comparing this region, only 75% identity is seen and considered a significant disablement of the protein functionality [21]. These two observations suggest that the membrane association of this truncated protein may be negatively affected, resulting in this protein being largely soluble and not membrane bound.
Additionally, of the 23 amino acid substitutions, there are 4 cysteine residues that are found in this stretch of protein. The high number of cysteine residues would make it quite susceptible to chemical modification by S-nitrosylation involving nitric oxide. Cysteine residues are not very abundant and are often found in functionally important areas [25].
The truncated gene product of GAPDHP44 (given as “P”) is compared with the same region from somatic GAPDH (given as “G”). Sequence differences are shown in grey and bolded. The dashes represent the continued structure of the parent GAPDH protein.
We explored the biological consequence of the SNP,

Figure 3. Comparison of GAPDH isoforms.
which results in a codon change from CAG to CAA. Both of these codons code for glutamine, however, the literature suggests that the fidelity of this incorporation is not absolute [26,27]. Therefore, we propose that a glutamate residue may be inserted in place of a glutamine residue, particularly in times nutritional compromise. This slight change in amino acid sequence may be responsible for the biological consequences. This SNP alteration would result in an amino acid substitution at position 113 (Figure 4).
The sequences of the GOSPEL binding domain are shown. The GAPDHP44 variants (given as “P-E” and “P-Q” for the missense Gln113Glu mutation) are compared and juxtaposed to the same region from the somatic GAPDH (given as “G”). The differences with the parent GAPDH are shown in grey and bolded. The variant difference is shown blackened with white letters.
The Siah protein (i.e. seven in absentia homolog) is a known apoptotic-inducing protein. GAPDH, which can be modified by nitrosylation agents such as nitric oxide, binds the Siah protein, which translocates GAPDH into the nucleus. Once in the nucleus, Siah initiates the cell death cascade [28]. Additionally, nuclear-translocated GAPDH also activates p300/CBA, contributing to cell death [29]. The GOSPEL (i.e. GAPDH’s competitor Of Siah Protein Enhances Life) protein has a protective cell function, as the binding of GAPDH to GOSPEL prevents the Siah protein from being translocated into the nucleus of the cell, as well as preventing nitrosylated GAPDH from entering the nucleus. The location of the GOSPEL binding site is at the periphery of domain 1 of the GAP-DH protein.
We propose that the truncated protein product of the

Figure 4. Comparison of the GOSPEL-binding domain.
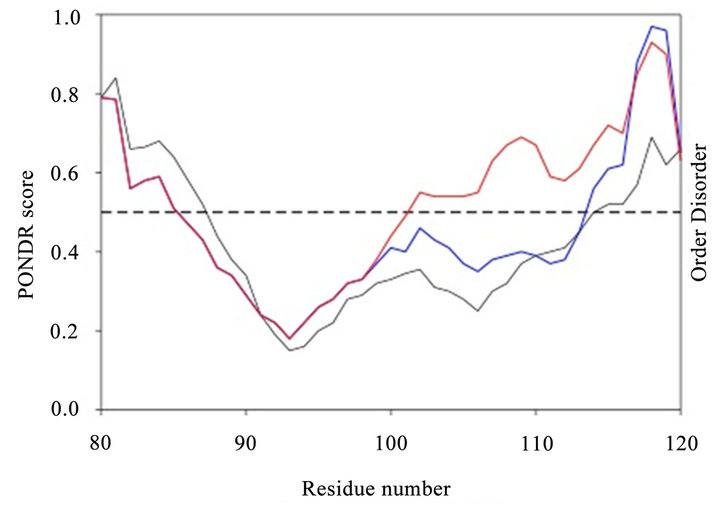
Figure 5. Analysis using PONDR program.
GAPDHP44 pseudogene may play a role in this process. This protein may be involved in regulating GOSPELGAPDH interaction by competing for interaction with residues on GOSPEL. It is known that residues 80 - 120 of GAPDH bind with GOSPEL. This region, which is found in the GAPDHP44 protein, can presumably interact with GOSPEL, freeing somatic GAPDH and allowing it to participate in the cell-death cascade. With the presence of the SNP, which may contribute to a missense mutation, the resulting GAPDHP44 protein may interact less with GOSPEL, allowing it bind GAPDH and promote cell viability.
We used the PONDR algorithm to compare the regions that bind GOSPEL (Figure 5). The parent somatic GAPDH contains largely an ordered domain in the center part of the region. The sequence associated with the GAPDHP44 protein exhibited properties similar to that of the parent protein. When the SNP variant was examined, the region showed an increased amount of disorder. This observation suggests that the disorder associated with the SNP variant of the P44 putative protein may contribute to biological properties that differ such as diminished GOSPEL binding.
The GOSPEL binding domains of parent GAPDH (black line), Figure 5. GAPDHP44 variant P-Q (blue line) and GAPD-HP44 variant P-E (red line) were analyzed by PONDR (Predictor of Natural Disordered Regions) from Molecular Kinetics, Inc. We used the VL-XT predictor as described by the vendor. The dashed line represents the se paration from predicted ordered and disordered regions.
The GOSPEL binding region of parent GAPDH is 34.2% disordered. The GAPDHP44 variant that contains the ancestral allele with a glutamine residue at position 113 is not that different from the parent GAPDH. It is 31.7% disordered. Interestingly, the GAPDHP44 variant with the missense mutation (i.e. Gln113Glu) is considerably more disordered, reaching 61% overall disorder. This finding supports the contention that the P-E variant may not bind as effectively to GOSPEL allowing it to interact with native GAPDH preserving cell viability and conferring neuronal protection.
6. CONCLUSIONS
Current AD therapies and treatments have limited efficacy, creating a need for continued research to slow the progression of the disease and further reduce the symptoms. GAPDH and the pseudogene GAPDHP44 is one such area for further research. Somatic GAPDH has a known role in neuronal apoptosis. We propose that GAPDHP44 may be expressed as a truncated protein, which may regulate the interaction of somatic GAPDH with GOSPEL. The proven AD-beneficial SNP in GAPDHP- 44 may result in an amino acid substitution (i.e. Gln- 113Glu). We propose that the putative protein variant with this amino acid substitution affects the GOSPEL binding site, liberating GOSPEL, allowing for somatic GAPDH interaction, preventing neuronal apoptosis.
The clinical significance of this study relative to the treatment of AD is associated with delaying the onset of symptoms which appears correlated to neuronal loss. In preclinical AD, there are many protein deposits in the entorhinal cortex and in the CA1 region of the hippocampus but with little to no cell loss in these areas of the brain [30]. When one begins to notice cognitive impairment and mild dementia, this is when one can identify a substantial cell loss in the entorhinal cortex and hippocampus/CA1. Differential expression of the P44 variant may ameliorate the symptoms of AD.
REFERENCES
- Seidler, N.W. (2012) Compartmentation of GAPDH. Advances in Experimental Medicine and Biology, 985, 61- 101. doi:10.1007/978-94-007-4716-6_3
- Hara, M.R., et al. (2006) Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proceedings of National Academy of Sciences of the USA, 103, 3887-3889. doi:10.1073/pnas.0511321103
- Seidler, N.W. (2012) Basic biology of GAPDH. Advances in Experimental Medicine and Biology, 985, 1-36. doi:10.1007/978-94-007-4716-6_1
- Seidler, N.W. (2012) Multiple binding partners. Advances in Experimental Medicine and Biology, 985, 249-267. doi:10.1007/978-94-007-4716-6_8
- Sunaga, K., Takahashi, H., Chuang, D.M. and Ishitani, R. (1995) Glyceraldehyde-3-phosphate dehydrogenase is overexpressed during apoptotic death of neuronal cultures and is recognized by a monoclonal antibody against amyloid plaques from Alzheimer’s brain. Neuroscience Letters, 200, 133-136. doi:10.1016/0304-3940(95)12098-O
- Sultana, R., Boyd-Kimball, D., Cai, J., et al. (2007) Proteomics analysis of the Alzheimer’s disease hippo-campal proteome. Journal of Alzheimer’s disease, 11, 153-164.
- Seidler, N.W. (2012) Target for diverse chemical modifications. Advances in Experimental Medicine and Biology, 985, 179-206. doi:10.1007/978-94-007-4716-6_6
- Mazzola, J.L. and Sirover, M.A. (2001) Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. Journal of Neurochemistry, 76, 442-449. doi:10.1046/j.1471-4159.2001.00033.x
- Ishitani, R. and Chuang, D.M. (1996) Glyceraldehyde- 3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proceedings of National Academy of Sciences of the USA, 93, 9937-9941. doi:10.1073/pnas.93.18.9937
- Ishitani, R., Sunaga, K., Hirano, A., Saunders, P., Katsube, N. and Chuang, D.M. (1996) Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. Journal of Neurochemistry, 66, 928-935. doi:10.1046/j.1471-4159.1996.66030928.x
- Ishitani, R., Tanaka, M., Sunaga, K., Katsube, N. and Chuang, D.M. (1998) Nuclear localization of overexpressed glyceraldehyde-3-phosphate dehydrogenase in cultured cerebellar neurons undergoing apoptosis. MolPharmacol, 53, 701-707.
- Li, Y., et al. (2004) Association of late-onset Alzheimer’s disease with genetic variation in multiple members of the GAPD gene family. Proceedings of National Academy of Sciences of the USA, 101, 15688-15693. doi:10.1073/pnas.0403535101
- Lee, J.H., et al. (2008) Further examination of the candidate genes in chromosome 12p13 locus for late-onset Alzheimer disease. Neurogenetics, 9, 127-138. doi:10.1007/s10048-008-0122-8
- Lin, P.I., et al. (2006) Exploring the association of glyceraldehyde-3-phosphate dehydrogenase gene and Alzheimer disease. Neurology, 67, 64-68. doi:10.1212/01.wnl.0000223438.90113.4e
- Bertram, L., McQueen, M.B., Mullin, K., Blacker, D. and Tanzi, R.E. (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nature Genetics, 39, 17-23. doi:10.1038/ng1934
- Seidler, N.W. (2012) Functional diversity. Advances in Experimental Medicine and Biology, 985, 103-147. doi:10.1007/978-94-007-4716-6_4
- Kozak, M. (1986) Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell, 44, 283-292. doi:10.1016/0092-8674(86)90762-2
- Kozak, M. (1992) Regulation of translation in eukaryotic systems. Annual Reviews: Cell and Developmental Biology, 8, 197-225. doi:10.1146/annurev.cb.08.110192.001213
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. (2002) Molecular biology of the cell. 4th Edition, Garland Science, New York.
- Drabkin, H.J. and RajBhandary, U.L. (1998) Initiation of protein synthesis in mammalian cells with codons other than AUG and amino acids other than methionine. Molecular and Cellular Biology, 18, 5140-5147.
- Theisen, C.S., Seidler, K.A. and Seidler, N.W. (2012) Computational criteria for the disablement of human GAPDH pseudogenes. In: Arabnia, H.R. and Tran, Q.-N., Eds., Proceedings of the 2012 International Conference on Bioinformatics and Computational Biology, CSREA Press, Las Vegas, 158-165.
- Montalbano, A.J., Theisen, C.S., Fibuch, E.E. and Seidler, N.W. (2012) Isoflurane enhances the moonlighting activity of GAPDH: Implications for GABAA receptor trafficking. ISRN Anesthesiology, 2012, Article ID: 970795.
- Kaneda, M., Takeuchi, K., Inoue, K. and Umeda, M. (1997) Localization of the phosphatidylserine-binding site of glyceraldehyde-3-phosphate dehydrogenase responseble for membrane fusion. Journal of Biochemistry, 122, 1233-1240. doi:10.1093/oxfordjournals.jbchem.a021886
- Alvarez-Dominguez, C., et al. (2008) Characterization of a Listeria monocytogenes protein interfering with Rab5a. Traffic, 9, 325-337. doi:10.1111/j.1600-0854.2007.00683.x
- Marino, S.M. and Gladyshev, V.N. (2010) Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. Journal of Molecular Biology, 404, 902-916. doi:10.1016/j.jmb.2010.09.027
- O’Donoghue, P., Sheppard, K., Nureki, O. and Söll, D. (2011) Rational design of an evolutionary precursor of glutaminyl-tRNAsynthetase. Proceedings of National Academy of Sciences of the USA, 108, 20485-20490. doi:10.1073/pnas.1117294108
- Wilcox, M. and Nirenberg, M. (1968) Transfer RNA as a cofactor coupling amino acid synthesis with that of protein. Proceedings of National Academy of Sciences of the USA, 61, 229-236. doi:10.1073/pnas.61.1.229
- Hara, M.R., et al. (2005) S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nature Cell Biology, 7, 665-674. doi:10.1038/ncb1268
- Sen, N., et al. (2008) Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nature Cell Biology, 10, 866-873. doi:10.1038/ncb1747
- Morris, J.C. and Price, J.L. (2001) Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. Journal of Molecular Neuroscience, 17, 101-118. doi:10.1385/JMN:17:2:101
Abbreviations
AD: Alzheimer’s disease;
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase;
P44: Pseudogene44;
SNP: Single nucleotide polymorphism;
Ab: Amyloidb;
PPM1H: Protein phosphatase 1H;
Siah: Seven in absentia homolog;
GOSPEL: GAPDH’s competitor of Siah Protein Enhances Life.