Neuroscience & Medicine
Vol.2 No.3(2011), Article ID:7408,6 pages DOI:10.4236/nm.2011.23031
PKR and PP1C Polymorphisms in Alzheimer’s Disease Risk
![]()
1Laboratory of Molecular Physiology and Channelopathies, Department of Experimental and Health Sciences, Pompeu Fabra University (UPF), Barcelona, Spain; 2NeoCodex, Sevilla, Spain; 3Alzheimer Laboratory, Neurology Department, Santa Creu and Sant Pau Hospital, Biomedical Research Center on Neurodegenerative Diseases (CIBERNED), Barcelona, Spain; 4Fundación Alzheimur, Dementia Unit, Virgen de la Arrixaca Hospital, Murcia, Spain; 5Memory Clinic of ACE Foundation, Catalan Institute of Applied Neurosciences, Barcelona, Spain; 6Neurology Department, Vall d’Hebron Universitary Hospital, Barcelona, Spain.
Email: *paco.munoz@upf.edu
Received June 30th, 2011; revised July 30th, 2011; accepted August 26th, 2011.
Keywords: Alzheimer’s Disease, BACE1, PKR, PP1, eIF2α
ABSTRACT
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by senile plaques and neurofibrillary tangles. Senile plaques are deposits of amyloid ß-peptide (Aß) produced by the cleavage of a transmembrane protein termed Amyloid Precursor Protein (APP). The amyloidogenic cleavage of APP is performed by γ-secretase complex and ß-site APP cleaving enzyme 1 (BACE1), a key enzyme in AD that can be activated by different noxious stimuli. Interestingly, some viruses could activate double-stranded RNA-activated protein kinase (PKR), which phosphorylates Eukaryotic Initiation Factor 2 alpha (eIF2α). This phosphorylation stops global translation to avoid any synthesis of viral infective proteins, but paradoxically up-regulates BACE1 translation. One of the viral mechanisms to circumvent eIF2α phosphorylation is the recruitment of protein phosphatase 1 (PP1), to fully dephosphorylate eIF2α and allow viral protein synthesis. Due to the functional relationship between BACE1, PKR, PP1 and AD we have performed a large (1122 cases and 1191 control individuals) case-control genetic analysis using two biallelic polymorphisms rs2254958 and rs7480390, located within the genes coding for PKR and the catalytic unit A of PP1, respectively. Although a trend to association of the rs2254958 TT genotype with AD risk was found, our results show that neither rs7480390 nor rs2254958 are associated with AD susceptibility.
1. Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease characterized by intracellular neurofibrillary tangles [1] and extracellular amyloid ß-peptide (Aß) aggregates [2-4]. Aß is released from the amyloid precursor protein (APP) when it is processed by the beta-site APP cleaving enzyme 1 (BACE1) and gamma-secretase [5-7]. Mutations in APP or presenilins (PS; part of the gamma secretase complex) trigger an inherited form of the disease, known as familiar AD (FAD) at early ages [8-10]. However, FAD accounts for less than 3% of AD cases [11], whereas sporadic AD accounts for most AD cases [12,13]. Its aetiology is unclear but the APOE-ε4 allele is considered the major risk factor for sporadic AD [14] and some noxious agents such as the neurotropic Herpes simplex Virus Type-1 (HSV1) have been also involved in sporadic AD onset [15-17].
Double-stranded RNA-activated protein kinase (PKR) activation has been recently associated with AD [17-20]. PKR, encoded by the EIF2AK2 gene, is an anti-viral mechanism used by the cell to prevent viral protein synthesis. When a virus infects eukaryotic cells, dsRNA originated from the viral genome activates PKR, which in turn phosphorylates Eukaryotic Initiation Factor 2 alpha (eIF2α) shutting down global protein synthesis [21], and increasing BACE1 protein synthesis as a side effect [17,22]. A mechanism used by HSV1 to avoid the PKRdefensive shutdown of protein synthesis is the de-phosphorylation of eIF2α, which is carried out by the catalytic subunit of protein phosphatase 1 (encoded by the PPP1CA gene) [23].
We hypothesize that the genetic variability within the PKR/eIF2α pathway could have a role in AD susceptibility. Therefore, in the present work we study two genetic markers, rs2254958 and rs7480390 within the EIF2AK2 and PPP1CA genes, to assess if they could be associated with AD.
2. Materials and Methods
2.1. AD and Control Individual Selection
Blood samples were collected from consecutive AD patients in medical centers in Barcelona (Fundació ACEInstitut Català de Neurociències Aplicades), Madrid (Hospital Universitario La Paz-Cantoblanco) and Murcia (Unidad de Demencias, Hospital Virgen de la Arrixaca and Fundación Alzheimur). The referral centers’ ethics committees and Neocodex have approved this research protocol, which was in compliance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association. Written informed consents were obtained from all individuals included in this study.
This analysis comprised 1122 unrelated sporadic AD patients (mean age at diagnosis ± SD: 77.55 ± 7.73 years old) and 1191 unrelated controls (52.14 ± 11.95). Control subjects were recruited from the general population. All AD patients fulfilled DSM-IV criteria for dementia and were diagnosed according to the NINCDS-ADRDA criteria for possible and probable AD [24]. All patients received a thorough clinical and neurological examination and a comprehensive neuropsychological evaluation including tests for general cognition, memory, language, perceptual and constructional abilities and executive functions. Complete blood analysis and neuroimaging studies were performed in all subjects to exclude other potential causes of dementia. Following the guidelines for the diagnosis of AD from the Study group on Behavioral Neurology and Dementia of the Spanish Neurological Society, AD patients were consecutively recruited at the four participating centers. To avoid population stratification problems, all individuals enrolled in this study were white Mediterranean with registered Spanish ancestors (two generations), as recorded by clinical researchers.
2.2. DNA Extraction Procedures
We obtained 5 mL of peripheral blood from all individuals to isolate genomic DNA from leukocytes. DNA extraction was performed in a MagNa Pure LC Instrument (Roche, Basel, Switzerland), using MagNa Pure LC DNA Isolation kit (Roche) in accordance with the manufacturer’s instructions.
2.3. Genetic Association Study
Genotypes for EIF2AK2 rs2254958 polymorphism, located at the 5’-UTR region within a putative exonic splicing enhancer, were obtained using Real-time PCR coupled to Fluorescense Resonance Energy Transfer (FRET) probes. Primers and probes employed for this genotyping protocol are summarized in Table 1. The technique was performed in the LightCycler® 480 System (Roche Diagnostics). Briefly, PCR was carried out in a final volume of 20 µL using 10 ng of genomic DNA, 0.1 µM of forward amplification primer, 0.25 µM of reverse amplification primer, 0.1 µM each detection probe, and 4 µL of LC480 Genotyping Master (5X, Roche Diagnostics, Germany). We used an initial denaturation step of 95˚C for 5 min, followed by 50 cycles of 95˚C for 30 sec, 62˚C for 30 sec, and 72˚C for 30 sec. For melting curve analyses, after an initial denaturation at 95˚C for 2 minutes at a ramp rate of 4.4˚C/second, the samples were incubated at 64˚C for 30 sec and 40˚C for 30 sec at a ramp rate of 2.2˚C/sec, and finally taken to 80˚C with one acquisition per ˚C. Genotypes for PPP1CA rs7480390 polymorphism, located in a putative promoter region, were obtained using Real-time PCR coupled to FRET probes. Briefly, PCR was carried out in a final volume of 20 µL using 10 ng of genomic DNA, 0.1 µM of forward amplification primer, 0.25 µM of reverse amplification primer, 0.1 µM each detection probe, and 4 µL of LC480 Genotyping Master (5X, Roche Diagnostics, Germany). We used an initial denaturation step of 95˚C for 5 min, followed by 50 cycles of 95˚C for 30 sec, 60˚C for 30 sec, and 72˚C for 30 sec. For melting curve analyses, after an initial denaturation at 95˚C for 2 minutes at a ramp rate of 4.4˚C/second, the samples were incubated at 40˚C for 30 sec at a ramp rate of 2.2˚C/sec, and finally taken to 80˚C with one acquisition per ˚C.

Table 1. Amplification primers and FRET probes employed in the genotyping protocols.
APOE-ε4 genotyping protocol has been previously described [25].
2.4. Statistics
We explored the polymorphisms association with AD phenotype using different tests adapted from Sasieni based on chi-squared test (http://ihg2.helmholtz-muenchen. de/). SPSS statistical package was used to develop binary logistic regression models adjusting for age, gender and APOE dosage.
3. Results
3.1. EIF2AK2 rs2254958 Polymorphism and AD Susceptibility
A bi-allelic polymorphism (rs2254958) located in a highly conserved SRp55 binding enhancer element of the EIF2AK2 gene (which encodes PKR protein) has been previously associated with age of AD onset and susceptibility to AD [20]. We carried out an independent casecontrol genetic association study, and we found that rs2254958 allele and genotype frequencies were not statistically different between AD patients and controls (Table 2). In order to address whether the APOE-ε4 allele could be a confounding factor in our analyses, we used the Odds ratio homogeneity test but, in contrast to Bullido et al. 2008 [20], we did not detect any interaction between APOE-ε4 and rs2254958 (p = 0.580). However, we observed a trend towards association of the rs2254958 TT genotype (recessive model) with AD susceptibility when we carried out a binary logistic regression analysis adjusting for gender and APOE-ε4 allele (Table 3). Finally, we performed a Cox regression analysis to test the effect of the rs2254958 TT genotype on the age of AD onset, adjusting for APOE-ε4 allele and gender, but no statistically significant results were obtained (data not shown).
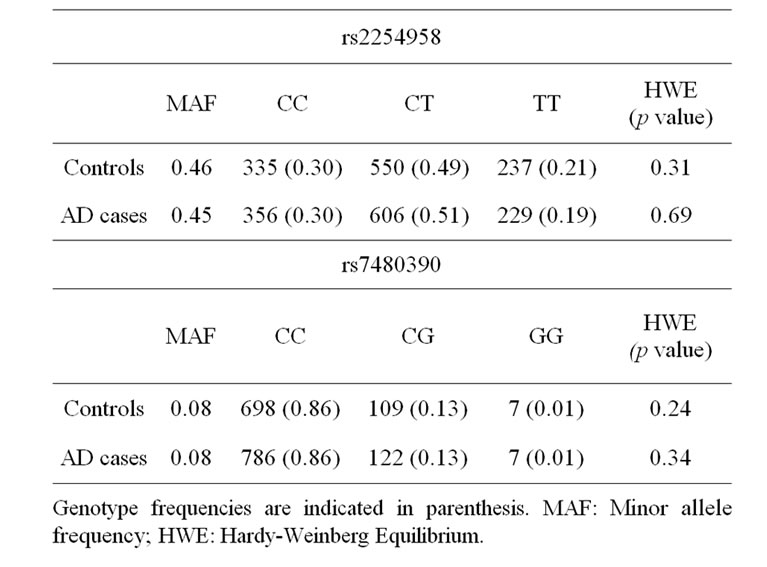
Table 2. Genotype frequency distribution.
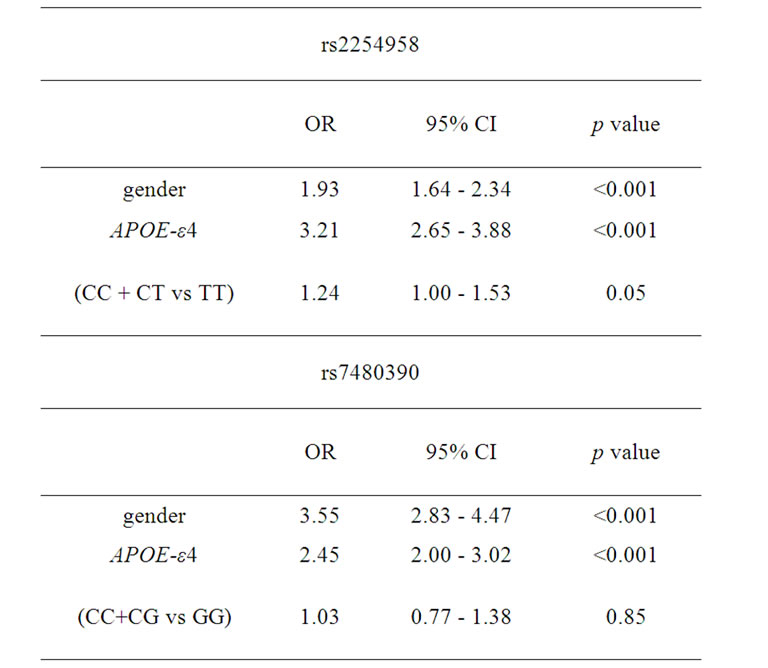
Table 3. Binary logistic regression analyses adjusting for gender and APOE-ε4 allele.
3.2. PPP1CA rs7480390 Polymorphism and AD Susceptibility
We found that the allele and genotype frequencies of rs7480390 were not statistically different between AD patients and controls (Table 2). In order to address whether the APOE-ε4 allele could be a confusing factor in our analyses, we used the Odds ratio homogeneity test. We did not detect any interaction between APOE-ε4 and rs7480390 (p = 0.785) even when a binary logistic regression analysis adjusting by gender and APOE-ε4 allele was calculated (Table 3).
4. Discussion
The causes of sporadic AD are still unknown. Several polymorphism in different genes have been reported to increase AD risk such as APOE-ε4 allele [26-28] or neprilysin [29], a protein that degrades Aß. However the relationship of these proteins with the onset of the disease is unclear. In the present work we focused in the genetic study of two intracellular proteins (PKR and PP1) that can influence the expression levels of BACE1, the rate-limiting enzyme in the production of Aß. PKR is activated by double stranded RNA from viral origin and phosphorylates eIF2α in serine 51. The phosphorylation of this factor allows BACE1 translation from its basally dormant mRNA [17]. On the other hand, PP1 is recruited by the HSV1 protein ICP34.5 to de-phosphorylate eIF2α in an attempt to circumvent the PKR/eIF2α defensive mechanism and allow global protein synthesis (together with viral protein synthesis) to go on in the host cell. Paradoxically, under these circumstances BACE1 translation from its mRNA remains repressed as in basal conditions.
Most humans are infected by HSV1 [30,31]. eIF2α phosphorylation after HSV1 infection might be an important event in sporadic AD aetiology [32,33], as phosphorylated eIF2α enables BACE1 translation promoting amyloidogenic pathway [22] (Figure 1). Interestingly PKR could be also activated by other stimuli than dsRNA viruses such as siRNA (reviewed in [34]) or oxidative stress [35]. It would be also possible the existence of a leak in eIF2α phosphorylation due to a dysregulation of PKR or PP1 activities. Therefore, uncovering the genetic variability underlying eIF2α phosphorylation after PKR activation and eIF2α de-phosphorylation after PP1 activation is critical to understand the role of this pathway in the aetiology of sporadic AD pathology. In fact, a genetic association between rs2254958 polymorphism within EIF2AK2 gene with the risk of AD has been found [20]. In our study we found a trend to association of the same marker with AD susceptibility in a recessive model. However our results show that the at-risk allele (T allele) described herein is the opposite (C allele) to that reported by Bullido and coworkers, and according to the original study, the effect of the rs2254958 occurs even in the ab-
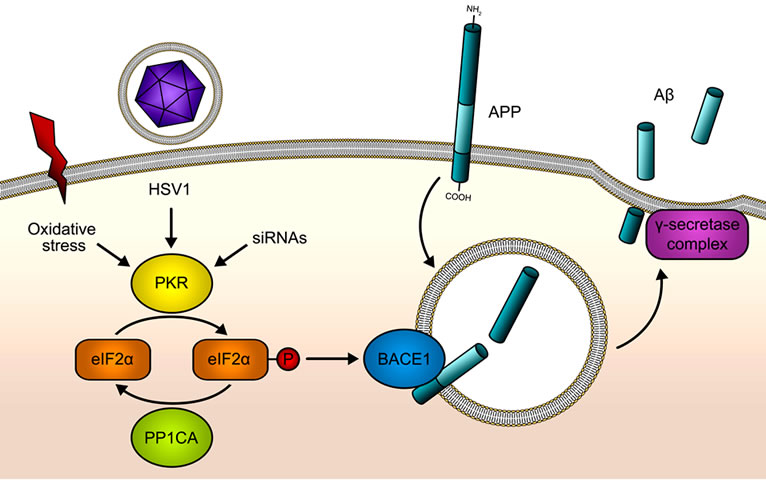
Figure 1. PKR/eIF2α pathway in Aß production. Double stranded RNA (dsRNA) from HSV1 or other viral origins is sensed by PKR, which becomes activated after the binding of the dsRNA, by oxidative stress action or by the binding of siRNAs. As a consequence eIF2α is phosphorylated at Serine 51 by PKR. This phosphorylation will shut down global protein synthesis at the level of translation initiation in a defensive mechanism when PKR is activated by viruses. HSV1 is able to evade this anti-viral PKR pathway through the recruitment of PP1CA, an eIF2α phosphatase. BACE1 and other genes with specific features in the 5’UTR of its mature mRNAs respond inversely to the phosphorylation of eIF2α: their translation is activated when eIF2α is phosphorylated at Serine 51. Therefore, the activation of the PKR/eIF2α pathway will lead to BACE1 up-regulation, promoting the ß-cleavage of APP, which followed by the γ- cleavage will increase Aß production.
sence of APOE-ε4 allele whereas we did not detect any interaction among these variants. Contrary to the reported results, we did not observe any influence of the rs2254958 variant over the age of AD onset. These discrepancies between the two studies could be due to differences in sample size, mean age of AD onset in patients and characteristics of the control group.
We did not detect any association of the PPP1CA rs7480390 polymorphism with AD, therefore we discarded a relevant role of this polymorphism in AD pathology.
In spite of these results, we cannot discard that other polymorphisms within EIF2AK2, PPP1CA or other genes involved in the PKR/eIF2α pathway could be related to AD ethiology. Further studies would be necessary to elucidate the real relevance of this pathway and its interaction with other environmental factors in AD susceptibility.
5. Acknowledgements
This work was supported by the Spanish Ministerio de Ciencia y Tecnología (FIS: PI07/0593 and PI10/00587; ISCIII-RETIC RED HERACLES RD06/0009/002- FEDER). The authors wish to thank to all the individuals and technicians who participated in this work. The authors thank I. Hernández, A. Mauleon, M. Rosende-Roca, P. Martínez-Lage, M. Alegret, S. Ruíz and L. Tárraga (Memory Clinic of Fundació ACE & Institut Català de Neurociències Aplicades; Barcelona, Spain) for their technical support; to the personnel of Unidad de Demencias, Hospital Universitario Virgen de la Arrixaca (Murcia, Spain); and Memory Unit of University Hospital La Paz-Cantoblanco (Madrid, Spain) for their continued effort to support this project.
REFERENCES
- J. Lewis, D. W. Dickson, W. L. Lin, L. Chisholm, A. Corral, G. Jones, S. H. Yen, N. Sahara, L. Skipper, D. Yager, C. Eckman, J. Hardy, M. Hutton and E. McGowan, “Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP,” Science, Vol. 293, No. 5534, 2001, pp. 1487-1491. doi:10.1126/science.1058189
- G. G. Glenner and C. W. Wong, “Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein,” Biochemical and Biophysical Research Communications, Vol. 120, No. 3, 1984, pp. 885-890.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, “Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 82, No. 12, 1985, pp. 4245-4249.
- D. J. Selkoe, C. R. Abraham, M. B. Podlisny and L. K. Duffy, “Isolation of Low-Molecular-Weight Proteins from Amyloid Plaque Fibers in Alzheimer’s Disease,” Journal of Neurochemistry, Vol. 46, No. 6, 1986, pp. 1820-1834.
- S. B. De, P. Saftig, K. Craessaerts, H. Vanderstichele, G. Guhde, W. Annaert, F. K. Von and L. F. Van, “Deficiency of Presenilin-1 Inhibits the Normal Cleavage of Amyloid Precursor Protein,” Nature, Vol. 391, No. 6665, 1998, pp. 387-390. doi:10.1038/34910.
- R. Vassar, B. D. Bennett, S. Babu-Khan, S. Kahn, E. A. Mendiaz, P. Denis, D. B. Teplow, S. Ross, P. Amarante, R. Loeloff, Y. Luo, S. Fisher, J. Fuller, S. Edenson, J. Lile, M. A. Jarosinski, A. L. Biere, E. Curran, T. Burgess, J. C. Louis, F. Collins, J. Treanor, G. Rogers and M. Citron, “Beta-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE,” Science, Vol. 286, No. 5440, 1999, pp. 735-741. doi:10.1126/science.286.5440.735.
- D. Del Toro, M. Coma, I. Uribesalgo, F. X. Guix and F. J. Munoz, “The Amyloid Betaprotein Precursor and AlzHeimer’s Disease. Therapeutic Approaches,” Current Medicinal Chemistry - Central Nervous System Agents, Vol. 5, 2005, pp. 271-281.
- D. Scheuner, C. Eckman, M. Jensen, X. Song, M. Citron, N. Suzuki, T. D. Bird, J. Hardy, M. Hutton, W. Kukull, E. Larson, E. Levy-Lahad, M. Viitanen, E. Peskind, P. Poorkaj, G. Schellenberg, R. Tanzi, W. Wasco, L. Lannfelt, D. Selkoe and S. Younkin, “Secreted Amyloid BetaProtein Similar to That in the Senile Plaques of Alzheimer’s Disease Is Increased in Vivo by The Presenilin 1 and 2 and APP Mutations Linked to Familial Alzheimer’s Disease,” Nature Medicine, Vol. 2, No. 8, 1996, pp. 864- 870.
- M. Bentahir, O. Nyabi, J. Verhamme, A. Tolia, K. Horre, J. Wiltfang, H. Esselmann and S. B. De, “Presenilin clinical Mutations Can Affect Gamma-Secretase Activity by Different Mechanisms,” Journal of Neurochemistry, Vol. 96, No. 3, 2006, pp. 732-742. doi:10.1111/j.1471-4159.2005.03578.x
- T. Wakabayashi and S. B. De, “Presenilins: Members of the Gamma-Secretase Quartets, but Part-Time Soloists too,” Physiology (Bethesda), Vol. 23, 2008, pp. 194-204. doi:10.?1152/?physiol.00009.2008
- K. S. Vetrivel, Y. W. Zhang, H. Xu and G. Thinakaran, “Pathological and Physiological Functions of Presenilins,” Molecular Neurodegeneration, Vol. 1, 2006, p. 4
- J. C. Lambert and P. Amouyel, “Genetic Heterogeneity of Alzheimer’s Disease: Complexity and Advances,” Psychoneuroendocrinology, Vol. 32, Supplement 1, 2007, pp. S62-S70.
- L. Bertram and R. E. Tanzi, “Alzheimer’s Disease: One Disorder, Too Many Genes?” Human Molecular Genetics, Vol. 13, Spec No. 1, 2004, pp. R135-R141.
- D. S. Borgaonkar, L. C. Schmidt, S. E. Martin, M. D. Kanzer, L. Edelsohn, J. Growdon and L. A. Farrer, “Linkage of Late-Onset Alzheimer’s Disease with Apolipo-Protein E Type 4 on Chromosome 19,” Lancet, Vol. 342, No. 8871, 1993, pp. 625.
- R. F. Itzhaki and M. A. Wozniak, “Herpes Simplex Virus Type 1 in Alzheimer’s Disease: The Enemy within,” Journal of Alzheimer’s Disease, Vol. 13, No. 4, 2008, pp. 393-405.
- M. A. Wozniak and R. F. Itzhaki, “Antiviral Agents in Alzheimer’s Disease: Hope for the Future?” Therapeutic Advances in Neurological Disorders, Vol. 3, No. 3, 2010, pp. 141-152.
- G. ILL-Raga, E. Palomer, M. Wozniak, E. RamosFernández, MònicaBosch-Morato, M. Tajes, F. Guix, J. Galán, J. Clarimón, C. Antúnez, L. M.Real, C. Fandos, M. Boada, R. Itzhaki and F. J. Munoz, “Activation of PKR Causes Amyloid ß-Peptide Accumulation via De-Repression of BACE1 Expression,” PlosOne, Vol. 6, No. 6, 2011, p. e21456.doi:10.1371/journal.pone.0021456.g001
- R. C. Chang, A. K. Wong, H. K. Ng and J. Hugon, “Phosphorylation of Eukaryotic Initiation Factor-2alpha (eIF2alpha) is Associated with Neuronal Degeneration in Alzheimer’s Disease,” Neuroreport, Vol. 13, No. 18, 2002, pp. 2429-2432.
- A. L. Peel and D. E. Bredesen, “Activation of the Cell Stress Kinase PKR in Alzheimer’s Disease and Human Amyloid Precursor Protein Transgenic Mice,” Neurobiology of Disease, Vol. 14, No. 1, 2003, pp. 52-62.
- M. J. Bullido, A. Martinez-Garcia, R. Tenorio, I. Sastre, D. G. Munoz, A. Frank and F. Valdivieso, “Double Stranded RNA Activated EIF2 Alpha Kinase (EIF2AK2; PKR) is Associated with Alzheimer’s Disease,” Neurobiology of Aging, Vol. 29, No. 8, 2008, pp. 1160-1166. doi:10.1016/j.neurobiolaging.2007.02.023
- B. R. Williams, “PKR; A Sentinel Kinase for Cellular Stress,” Oncogene, Vol. 18, No. 45, 1999, pp. 6112-6120.
- T. O’Connor, K. R. Sadleir, E. Maus, R. A. Velliquette, J. Zhao, S. L. Cole, W. A. Eimer, B. Hitt, L. A. Bembinster, S. Lammich, S. F. Lichtenthaler, S. S. Hebert, S. B. De, C. Haass, D. A. Bennett and R. Vassar, “Phosphorylation of the Translation Initiation Factor eIF2alpha Increases BACE1 Levels and Promotes Amyloidogenesis,” Neuron, Vol. 60, No. 6, 26-12-2008, pp. 988-1009. doi:10.1016/j.neuron.2008.10.047.
- B. He, M. Gross and B. Roizman, “The Gamma(1)34.5 Protein of Herpes Simplex Virus 1 Complexes with Protein Phosphatase 1alpha to Dephosphorylate the Alpha Subunit of the Eukaryotic Translation Initiation Factor 2 and Preclude the Shutoff of Protein Synthesis by Double-Stranded RNA-Activated Protein Kinase,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 94, No. 3, 1997, pp. 843-848.
- G. McKhann, D. Drachman, M. Folstein, R. Katzman, D. Price and E. M. Stadlan, “Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease,” Neurology, Vol. 34, No. 7, 1984, pp. 939-944.
- R. Ramirez-Lorca, M. Boada, M. E. Saez, I. Hernandez, A. Mauleon, M. Rosende-Roca, P. Martinez-Lage, M. Gutierrez, L. M. Real, J. Lopez-Arrieta, J. Gayan, C. Antunez, A. Gonzalez-Perez, L. Tarraga and A. Ruiz, “GAB2 Gene Does Not Modify the Risk of Alzheimer’s Disease in Spanish APOE 4 Carriers,” Journal of Nutrition Health and Aging, Vol. 13, No. 3, 2009, pp. 214-219.
- A. Varez-Arcaya, O. Combarros, J. Llorca, M. SanchezGuerra, J. Berciano and J. L. Fernandez-Luna, “The - 491 TT Apolipoprotein E Promoter Polymorphism Is Associated with Reduced Risk for Sporadic Alzheimer’s Disease,” Neuroscience Letters, Vol. 304, No. 3, 2001, pp. 204-208. doi:10.1016/S0304-3940(01)01790-6
- J. C. Lambert, F. Pasquier, D. Cottel, B. Frigard, P. Amouyel and M. C. Chartier-Harlin, “A New PolymerPhism in the APOE Promoter Associated with Risk of Developing Alzheimer’s Disease,” Human Molecular Genetics, Vol. 7, No. 3, 1998, pp. 533-540. doi:10.1093/hmg/7.3.533
- E. H. Corder, A. M. Saunders, W. J. Strittmatter, D. E. Schmechel, P. C. Gaskell, G. W. Small, A. D. Roses, J. L. Haines and M. A. Pericak-Vance, “Gene Dose of ApoliPoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families,” Science, Vol. 261, No. 5123, 1993, pp. 921-923.
- J. Clarimon, F. J. Munoz, M. Boada, L. Tarraga, J. Sunyer, J. Bertranpetit and D. Comas, “Possible Increased Risk for Alzheimer’s Disease Associated with Neprilysin Gene,” Journal of Neural Transmission, Vol. 110, No. 6, 2003, pp. 651-657.
- G. A. Jamieson, N. J. Maitland, G. K. Wilcock, J. Craske and R. F. Itzhaki, “Latent Herpes Simplex Virus Type 1 in Normal and Alzheimer’s Disease Brains,” Journal of Medical Virology, Vol. 33, No. 4, 1991, pp. 224-227.
- M. A. Wozniak, S. J. Shipley, M. Combrinck, G. K. Wilcock and R. F. Itzhaki, “Productive Herpes Simplex Virus in Brain of Elderly Normal Subjects and Alzheimer’s DisEase Patients,” Journal of Medical Virology, Vol. 75, No. 2, 2005, pp. 300-306.
- M. J. Ball, “Limbic Predilection in Alzheimer Dementia: Is Reactivated Herpesvirus Involved?” Canadian Journal Neurological Sciences, Vol. 9, No. 3, 1982, pp. 303-306.
- R. F. Itzhaki, W. R. Lin, G. K. Wilcock and B. Faragher, “HSV-1 and Risk of Alzheimer’s Disease,” Lancet, Vol. 352, No. 9123, 1998, p. 238.
- S. R. Nallagatla, R. Toroney and P. C. Bevilacqua, “Regulation of Innate Immunity through RNA Structure and the Protein Kinase PKR,” Current Opinion in Structural Biology, Vol. 21, No. 1, 2011, pp. 119-127. doi10.1016/j.sbi.2010.11.003
- G. Li, C. Scull, L. Ozcan and I. Tabas, “NADPH Oxidase Links Endoplasmic Reticulum Stress, Oxidative Stress, and PKR Activation to Induce Apoptosis,” Journal of Cell Biology, Vol. 191, No. 6, 2010, pp. 1113-1125. doi:10.1083/jcb.201006121.