Neuroscience & Medicine
Vol.2 No.2(2011), Article ID:5531,3 pages DOI:10.4236/nm.2011.22017
Ile587Val Polymorphism of the eIF2B5 Gene as Susceptibility Factor for Multiple Sclerosis
![]()
1Institute of Neurological Sciences, National Research Council, Cosenza, Italy; 2Department of Neurological and Psychiatric Sciences, “Aldo Moro” University, Bari, Italy; 3Institute of Neurology, University “Magna Graecia”, Catanzaro, Italy.
Email: r.mazzei@isn.cnr.it
Received October 8th, 2010; revised February 19th, 2010; accepted March 1st, 2010.
Keywords: Multiple Sclerosis, Vanishing White Matter Disease, eIF2B Gene, Stress-Sensitive Neurological Disorders
ABSTRACT
Mutations in the eIF2B gene cause the VWM disease. Genetic and biochemical data of MS patient and MRI data showing VWM images similar to MS lesions, encouraged the present study in which we analyzed the eIF2B5 gene in 225 unrelated MS patients to evaluate an overlapping between MS and VWM. A common variation Ile587Val was found very frequent in the MS patients respect normal controls, thus suggesting that Ile587Val should be considered as susceptibility factor in the development of MS. In conclusion, our data strongly highlight a possible involvement of the eIF2B5 in the development of MS.
1. Introduction
Multiple sclerosis (MS) is an inflammatory chronic disease characterized by a demyelinating process, which is followed by neurodegeneration. Although the true etiology remains elusive, MS seems to be a multifactor pathology in which a predisposing genetic background added to an infection plays an important role [1]. A lot of evidence have provided that genetic factors contribute to disease susceptibility; in fact epidemiological studies estimated that the absolute risk of MS in a first-degree relative of a patient is 20 - 40 times the risk in the general population, with the most common case being an affected sibling [2], thus suggesting an autosomal recessive model of inheritance of MS candidate genes.
Despite several linkage and association studies, the strongest and most consistently replicated evidence for a MS susceptibility gene has been localized to the Major Histocompatibility Complex (MHC) superlocus on chromosome 6p21.3 [3]. This data, together with the observed clinical and histopathological heterogeneity of the disease, further supported the hypothesis that the MSprone genotype results from multiple independent or interacting polymorphic genes, each exerting a small or, at most, moderate effect to the overall risk [4]. Then it is likely that genetic heterogeneity exists, meaning that specific genes influence susceptibility and pathogenesis in some individual but not in others. Basing on these grounds, even other genetic elements located within the Human Leukocyte Antigen (HLA) Complex, i.e., Heat Shock Proteins (HSP) that have been shown to be strikingly elevated in MS lesions, could have a role in the pathophysiology of MS [5].
HSPs are similar in function to the eukaryotic translation initiation factor eIF2B. It is an important regulatory pathway for the prevention of synthesis of denaturated proteins during cellular stress, and it functions in parallel with the production of heat shock proteins [6].
The eukaryotic translation initiation factor (eIF2B) consists of five subunits, each encoded by different genes (eIF2B1-5). The eIF2B gene is an important factor in the regulatory pathway that prevents the synthesis of denaturated proteins during cellular stress and whose functions seem to work in parallel with the production of HSPs [6]. Mutations in this gene inherited as autosomal recessive trait are able to cause a stress-sensitive leukoencephalopathy known as Vanishing White Matter disease (VWM), also called Childhood Ataxia with Central Hypomyelination (CACH) (OMIM 603896) [7]. This is a leukoencephalopathy with autosomal recessive inheritance. Magnetic Resonance Images of patients with VWM show a diffuse leukoencephalopathy with evidence of progressive disappearance of the cerebral white matter, which is seen on MRI by signals approaching those of cerebrospinal fluid on all pulse sequences including protondensity and FLAIR sequences. Cavitating leukodistrophy has also been described, especially at the later stages of the disease, but this finding must not be considered a crucial exclusion diagnostic criteria [8]. Previous studies have reported cases of VWM in which MRI was strikingly similar to that observed in MS [9].
In view of the fact that eIF2B is essential for regulation of protein synthesis in all cells of the body, especially under different stress conditions, it is not surprising that mutations in the gene subsequently have been reported in other variants including acute fatal infantile forms with multiorgan involvement and Cree leukoencephalopathy; slowly progressive juvenile conditions; and adult-onset disorders associated with ovarian dysgenesis [10].
Genetic and biochemical data of MS patient and MRI data showing VWM images similar to MS lesions, encouraged the present study in which we analyzed the coding region of eukaryotic translation initiation factor 5 (eIF2B5) to evaluate an overlapping between MS and Vanishing White Matter disease (VWM).
2. Patients and Methods
2.1. Patients
We previously analyzed 11 pairs of siblings (22 patients) followed at the Institute of Neurology—University “Magna Graecia” of Catanzaro—Italy, all with definite MS [11]. The group of siblings consisted of 14 females and 8 males, ages ranging 27 - 46 years, with a mean disease onset of 23.7 + 8.3 years, all derived from the Southern Italy. As a control group we selected 10 ageand sex-matched healthy subjects from the same geographic area. In these patients we performed a mutational screening of the eIF2B5 gene in order to verify a possible link between MS and VWM and we initially performed this study because our affected patients were sib pairs, making hypothetical an autosomal recessive model of inheritance. Subsequently, the analysis has been extended, analyzing other 214 unrelated MS patients, all of them diagnosed as clinically established, definite MS, according to Poser criteria and derived from the Southern Italy too. More, 197 ageand sex-matched healthy subjects from the same geographic area were included.
2.2. Molecular Investigation
After informed consent was obtained, DNA was extracted from whole blood samples using standard protocols. A mutational screening of the 16 exons of the eIF2B5 gene and their exon-intron boundaries was performed by PCR using newly designed primer pairs. PCR reactions were carried out in 50 μl reaction volumes containing 130 ng genomic DNA, 2.5U Transgenomic Optimase Polymerase in its 1X Buffer, MgSO4 1.5 mM, dNTPs 200 μM each, primers 20 pmoles each. The amplicons were analyzed by Denaturing Performance Liquid Chromatography (DHPLC) on a WAVE Nucleic Acid Fragment Systems (Transgenomic Inc., Mountain Crewe, CA). Representative samples with an abnormal DHPLC profile were sequenced on an ABI Prism 3130 × l genetic analyzer, using the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems).
2.3. Statistical Analysis
The Hardy-Weinberg equilibrium was tested using Pearson’s chi-square goodness of fit test. The differences in allelic and genotypic distributions between cases and controls were assessed using chi-square test. Odd ratios with 95 percent confidence intervals (95% CI) was estimated by contingency table.
3. Results
In the first group of analyzed subjects, no mutation was found in any of the coding regions in either the patients or the controls, except for a common variation in exon 13 (Ile587Val; rs843358) in the heterozygous state in 6 MS patients and in the homozygous state in only 1 MS patient. This variation was also found in the heterozygous state in 3 out of 10 healthy controls.
Furthermore, mutations were not found in any of the coding regions in either the remaining patients or the controls, except the variation in exon 13 (Ile587Val), detected in the heterozygous state in 83 patients, and in homozygous state in 12 patients. The variation was also found in the heterozygous state in 48 out of 207 control subjects and in homozygous state only in 2 out of control subjects.
4. Discussion
Although MS etiology still remains unclear, the genetic influence on both susceptibility and clinical phenotypes is unequivocal. Studies on candidate genes have focused the interest on different pathways (i.e., the autoimmune cascade, the axonal or myelin targets) but, despite very few results have been reached, each one of them is crucial for adding another wedge of the puzzle.
In our study, we investigated the gene coding the ε subunit of eIF2B because it is the gene in which was found the most of mutations causing the VWM disease and the only one in which were reported mutations causing VWM with milder phenotype [12].
As eIF2B5 gene mutations were not found in the eleven MS sib pairs, it was believable that VWM and familial MS are distinct diseases. On the other hand, a
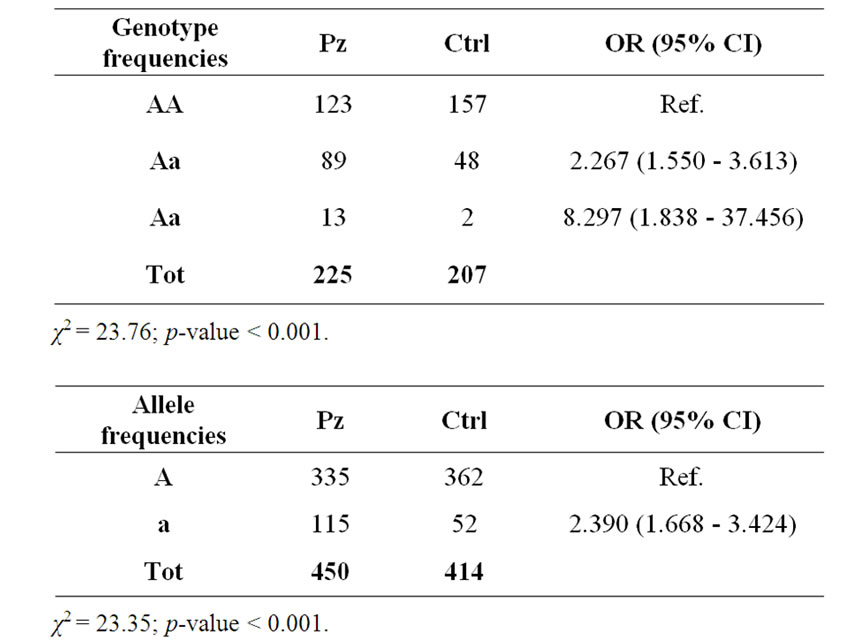
Table 1. Allele and genotype frequency of the eIF2B5 gene polymorphism (Ile587Val) in MS patients and in normal subjects. Hardy-Weinberg equilibrium: Pz: χ2 = 0.352; p-value = 0.553; Ctrl: χ2 = 0.642; p-value = 0.423.
common variation Ile587Val was found in the exon 13 in 7 out of them, and in 3 control subject only. This finding had a very little relevance because of the slenderness of sample group, but encouraged us to enlarge our cohort despite other authors did not find any association between this polymorphism and MS [13].
In fact, analyzing other 214 unrelated MS patients and 197 controls, we found that the polymorphism Ile587Val was very frequent in the MS patients respect normal controls. A statistical analysis (χ2-test) suggests that polymorphism Ile587Val in exon 13 of the EIF2B5 gene should be considered as susceptibility factor in the development of MS (χ2 = 23.76; p < 0.001) (Table 1).
In conclusion, our data strongly suggest a possible involvement of the eIF2B5 in the development of MS and that variations in the MS patient strongly depend on geographic and ethnic factors, thus suggesting the importance of knowledge the epigenetic contribution in the pathogenesis of MS.
REFERENCES
- R. G. Urdinguio, J. V. Sanchez-Mut and M. Esteller, “Epigenetic mechanisms in neurological diseases: genes, sindrome, and terapie,” The Lancet Neurology, Vol. 8, No. 11, 2009, pp. 1056-1072. doi:10.1016/S1474-4422(09)70262-5
- A. D. Sadovnick, P. A. Baird and R. H. Ward, “Multiple sclerosis: updated risk for relatives,” American Journal of Medical Genetics, Vol. 29, No. 3, 1988, pp. 533-541. doi:10.1002/ajmg.1320290310
- O. Olerup and J. Hillert, “HLA class II-associated genetic susceptibility in multiple sclerosis: a critical evaluation,” Tissue Antigens, Vol. 38, No. 2, 1991, pp. 1-15. doi:10.1111/j.1399-0039.1991.tb02029.x
- J. R. Oksenberg, L. F. Barcellos, B. A. Cree, S. E. Baranzini, T. L Bugawan., O. Khan, R. R. Lincoln, A. Swerdlin, E. Mignot, L. Lin, D. Goodin, H. A. Erlich, S. Schmidt, G. Thomson, D. E. Reich, M. A. Pericak-Vance, J. L. Haines and S. L. Hauser, “Mapping Multiple Sclerosis susceptibility to the HLA-DR Locus in African Americans,” The American Journal of Human Genetics, Vol. 74, No. 1, 2004, pp. 160-167. doi:10.1086/380997
- C. F Brosnan, L. Battistini, Y. L Gao, C. S. Raine and D. A. Aquino, “Heat Shock Proteins and Multiple Sclerosis: A Review,” Journal of Neuropathology Experimental Neurology, Vol. 55, No. 4, 1996, pp. 389-402. doi:10.1097/00005072-199604000-00001
- C. G. Proud, “Regulation of Eukaryotic Initiation Factor eIFB,” Progress in Molecular Subcellular Biology, Vol. 26, 2001, pp. 95-114.
- P. A. Leegwater, G. Vermeulen, A. A. Könst, S. Naidu, J. Mulders, A. Visser, P. Kersbergen, D. Mobach, D. Fonds, C. G. van Berkel, R. J. Lemmers, R. R. Frants, C. B. Oudejans, R. B. Schutgens, J. C. Pronk and M. S. van der Knaap, “Subunits of the Translation Initiation Factor eIF2B are Mutant in Leukoencephalopathy with Vanishing White Matter,” Nature Genetics, Vol. 29, 2001, pp. 383-388. doi:10.1038/ng764
- A. Fogli, R. Schiffmann, E. Bertini, S. Ughetto, P. Combes, E. Eymard-Pierre, C. R. Kaneski, M. Pineda, M. Troncoso, G. Uziel, R. Surtees, D. Pugin, M. P. Chaunu, D. Rodriguez and O. Boespflug-Tanguy, “The effect of genotype on the natural history of elF2B-Related leukodistrophies,” Neurology, Vol. 62, No. 9, 2004, pp. 1509-1517. doi:0.1212/01.WNL.0000123259
- M. S. van der Knaap, W. Kamphorst, P. G. Barth, C. L. Kraaijeveld, E. Gut and J. Valk, “Phenotypic Variation in Leukoencephalopathy with Vanishing White Matter,” Neurology, Vol. 51, No. 2, 1998, pp. 540-547.
- M. J. Noetzel, “Diagnosing ‘undiagnosed’ leukodistrophies: The role of molecular genetics,” Neurology, Vol. 62, 2004, pp. 847-848.
- C. M. Poser, D. W. Paty, L. Scheinberg, W. I. McDonald, F. A. Davis, G. C. Ebers, K. P. Johnson, W. A. Sibley, D. H. Silberberg and W. W. Tourtellotte, “New diagnostic criteria for multiple sclerosis: guidelines for research protocols,” Annals of Neurology, Vol. 13, No. 3, 1983, pp. 227-231. doi:10.1002/ana.410130302
- M. S. van der Knaap, P. A. Leegwater, C. G. van Berkel, C. Brenner, E. Storey, M. Di Rocco, F. Salvi and J. C. Pronk, “Arg113His mutation in eIF2Bepsilon as cause of leukoencephalopathy in adults,” Neurology, Vol. 62, No. 9, 2004, pp. 1464-1465.
- J. Pronk, G. Scheper, R. van Andel, C. van Berkel, C. Polman, B. Uitdehaag and M. van der Knaap, “No evidence that polymorphisms of the vanishing white matter disease genes are risk factors in multiple sclerosis,” Multiple Sclerosis, Vol. 14, No. 8, 2008, pp. 1123-1126.