Advances in Microbiology
Vol.3 No.6A(2013), Article ID:38611,11 pages DOI:10.4236/aim.2013.36A009
Substitution of Nitrite Reductase of Thermosynechococcus elongatus BP-1 by the Homologous Gene of Phormidium laminosum
Enzyme and Cell Technology Group, Department of Biochemistry and Molecular Biology, Faculty of Science and Technology, University of the Basque Country (UPV/EHU), Bilbao, Spain
Email: *juanl.serra@ehu.es
Copyright © 2013 Maite Buxens et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 27, 2013; revised September 17, 2013; accepted September 23, 2013
Keywords: Thermophilic Cyanobacteria; Long Flanking Homology Polymerase Chain Reaction; Nitrite Reductase; Gene Transference
ABSTRACT
Even though the nitrate assimilation operon has been extensively studied in Phormidium laminosum, some aspects still remain unclear. The genetic manipulation of this cyanobacterium is problematic that hinders the elucidation of further aspects of nitrogen metabolism. To circumvent this, Thermosynechococcus elongatus BP-1 was selected as a surrogate host and its nirA gene was substituted by the homologous gene of P. laminosum. This process, based on Long Flanking Homology Polymerase Chain Reaction and the natural competence of T. elongatus BP-1, required an intermediate T. elongatus BP-1 ΔnirA::kat mutant, which carries a gene encoding a thermostable kanamycin nucleotidyl transferase in place of nirA_Te. In the presence of nirA_Pl, nirA defective mutants of T. elongatus BP-1 recovered the ability to grow with nitrate as the sole nitrogen source, and showed a phenotype similar to that observed in wild-type cells. The procedure could be useful to substitute other genes from T. elongatus BP-1 with the homologues from P. laminosum in order to study this particular operon. Furthermore, it may be used as a general tool to explore phenotypic changes due to the exchange of a single gene between cyanobacteria.
1. Introduction
Nitrate assimilation in cyanobacteria involves three main steps. Firstly, the anion entry inside the cell mediated by Nrt, the active ABC (ATP-Binding-Cassette) transporter [1-3]. Then, the intracellular nitrate is reduced to ammonium by two consecutive reactions catalyzed by nitrate reductase (NarB) and nitrite reductase (NirA). Finally, the resulting ammonium is incorporated into L-glutamate through the glutamine synthetase-glutamate synthase cycle (for reviews see: [4-6]).
In previous papers, we reported the purification and characterization of the proteins responsible of the uptake and reduction of nitrate and nitrite in the thermophilic cyanobacterium P. laminosum [7-10]. As in other cyanobacteria, in P. laminosum these proteins (NrtA, NrtB, NrtC, NrtD, NarB and NirA) are encoded by genes that are clustered together into an operon that presents the sequence nirA-nrtA-nrtB-nrtC-nrtD-narB [11]. Even though nitrogen assimilation has been studied in a large number of cyanobacteria, the structure and function of the proteins codified by the genes of this operon have been scarcely studied and poorly understood. The phylogenetic relationship among NirA from some cyanobacteria is shown in supplementary Figure S1.
In P. laminosum, although some advances have been made [7-11], the complete understanding of the functionality of this operon seems difficult without using genetic manipulation tools. These methods, frequently applied to analyze genomes and found out the particular roles of some proteins, could come in handy to continue investigating the roles and interactions of these proteins in P. laminosum. However, when considering the genetic manipulation of cyanobacteria, some strains present disadvantages over others. For instance, the genetic modification of P. laminosum is difficult mainly due to its filamentous character and because its genome has not been sequenced so far. Several transformation strategies have been developed and applied in some cyanobacteria, especially in those whose genomes have been sequenced [12-17], as is the case of T. elongatus BP-1. This unicellular, rod-shaped, non-N2-fixing cyanobacterium, originally isolated from a hot spring in Beppu, Japan [18], seems to have branched very close to the evolution of cyanobacteria. One of the most distinctive characteristics of T. elongatus BP-1 is its thermophilic character (showing an optimum growth temperature of approximately 55˚C). Furthermore, the genome of T. elongatus BP-1 has been sequenced [19] making this cyanobacterium a suitable candidate for genetic studies [20,21]. Moreover, T. elongatus BP-1 presents a remarkable natural competence. This property, found in some prokaryotes [22-25], allows the incorporation of naked DNA into their genome without resorting to techniques such as electroporation or conjugation (for reviews see: [26-30]). Formerly, the DNA fragments engineered for gene substitution were constructed by using PCR, restriction enzymes and ligases. As PCR has become a more common technique, methods to synthesize these fragments by using exclusively PCR and thus avoiding restriction enzymes are being established [31-34]. Such is the case of Long Flanking Homology Polymerase Chain Reaction (LFHPCR). This technique, initially applied in Saccharomyces cerevisiae (denoted as Short Flanking Homology-Polymerase Chain Reaction (SFH-PCR) [35-38], was afterwards adapted to be used in many other microorganisms [13,31-34,39]. The synthesized DNA fragment, called “conversion cassette”, consists of a marker gene flanked by the upstream and downstream regions of the target gene to be substituted. The capacity of the “conversion cassette” to originate the desired modifications, avoiding the alteration of any other region of the genome, relies on the process of homologous recombination [40-42].
With the goal of integrating the previous knowledge and experience acquired in nitrogen metabolism in P. laminosum and to be able to perform genetic transformation strategies, we have established a method that allows the substitution of a target gene of T. elongatus BP-1 with a homologous from P. laminosum. In this paper, we apply this process to obtain recombinant T. elongatus BP-1 cells carrying a substitution of nitrite reductase (nirA_Te) with the homologous from P. laminosum (nirA_Pl) and characterize their phenotype at different temperatures.
2. Materials and Methods
2.1. Materials
Oligonucleotides (Table 1) were obtained from Bonsaitech (Madrid, Spain) and dNTPmix from GE Healthcare (Uppsala, Sweden). Pfu polymerase and BioTaq polymerase were obtained from Biotools B&M Labs (Madrid, Spain) and from Bioline (London, United Kingdom), respectively. All other chemicals were of analytical or molecular biology grade.
2.2. Strains and Growth Conditions
The thermophilic, unicellular, non-diazotrophic cyanobacterium Thermosynechococcus elongatus BP-1 (NIES- 2133) was obtained from Prof. M. Rögner (University of Bochum, Germany). Cells were grown photoautotrophically in 250 ml Erlenmeyer flasks containing 100 ml of sterile mineral medium BG11 [43]. Flasks were continu-

Table 1. Oligonucleotides used for LFH-PCR and for checking transformation (TC).
Boldface and underlined text indicate homology to kat, or to nirA from P. laminosum, respectively. F, forward; R, reverse; TC, transformation check.
ously shaked in orbital incubators (Kuhner Shaker X, Basel, Switzerland) at 140 - 180 rpm. Light intensity (10 - 50 µE∙m−2∙s−1) and temperature (45˚C or 55˚C) were fixed depending on the type of assay. The air atmosphere inside the incubator was enriched with 1% (v/v) CO2. Nitrogen-free BG11 culture medium (denoted here as BG110) was used supplemented with the indicated nitrogen source in each case. Thus, when T. elongatus BP-1 cells were cultured with nitrate as the sole nitrogen source, BG110 medium was supplemented with 17 mM NaNO3 (denoted as BG11 medium) before autoclaving. When cells were cultured with ammonium, the sterile BG110 medium was supplemented with 2 mM (NH4)2SO4 using a concentrated stock solution of this salt previously sterilized by filtration (0.2 μm Millex®- FG filter, Millipore Co., Bedford, MA, USA). When agar plates of solid medium were required, BG110 medium and 1.5% (w/v) agar in ultrapure water (MilliQ, Millipore Co., Bedford, MA, USA) were autoclaved separately and mixed. In this moment, kanamycin (when indicated) and the necessary nitrogen source were added. The nitrogen source was added at the same final concentration used in liquid culture. Due to the high temperature used to grow cells of T. elongatus BP-1, in order to prevent desiccation, a thick layer of BG11-agar was solidified in the plates. Moreover, plates were incubated in a humid atmosphere.
The thermophilic, filamentous, non-diazotrophic cyanobacterium Phormidium laminosum strain OH-1-p.Cl1 (renamed as Geitlerinema sp., PCC 8501) was grown axenically at 45˚C in 250 ml Erlenmeyer flasks containing 100 ml of sterile mineral medium D [44] supplemented with 0.5 g∙l−1 NaHCO3 as an additional carbon source. Cultures were grown with continuous illumination at a light intensity of 100 E∙m−2∙s−1.
Escherichia coli NovaBlue SingleTM cells were used to propagate the different pT7Blue vectors used. Cells were grown in LB medium with IPTG, X-gal, carbenicillin and tetracycline according to the instructions indicated in the Perfectly Blunt Cloning Kit (Novagen, Darmstadt, Germany).
2.3. Genomic DNA Purification
The genomic DNA of cyanobacterial cells was extracted and purified using Nucleo Spin Tissue kit (Machery Nagel, Düren, Germany). A previous step was included, in which cells were incubated at 37˚C for 1 h with 50 mM Tris-HCl, pH 8.0, supplemented with 50 mM EDTA, 1% (v/v) Triton X-100, 20 mg∙ml−1 lysozyme and 0.3 mg∙ml−1 RNase [45]. The obtained DNA was stored at −20˚C.
2.4. Long Flanking Homology-Polymerase Chain Reaction
Oligonucleotides used to amplify genomic DNA of T. elongatus BP-1 (Table 1) were designed considering sequences from Cyanobase (http://genome.kazusa.or.jp/ cyanobase). The sequence of P. laminosum nitrite reductase gene (nirA_Pl) was obtained from GenBank ID: Z19598.1. In all reactions 1 U of Pfu was used, and the amplification mixture (50 µl) contained 0.2 mM of each dNTP, 5 µl of (10X) PCR buffer, 2 mM MgCl2, 1 µM of each oligonucleotide and the corresponding template.
To obtain the conversion cassette for nirA_Te replacement with kat (C1), four different PCRs were carried out (Figure 1). In the first, primers FU1 and RU1 were used to amplify the upstream fragment U1. In the

Figure 1. Schematic diagram of the LFH-PCR used. Small arrows indicate primers (see Table 1); kat, encodes a thermostable kanamycin resistance gene; nirA_Pl encodes nitrite reductase from P. laminosum. U, upstream region; M, marker gene; D, downstream region. Straight and curved lines indicate linear and genomic DNA, respectively. Circled numbers 1, 2, 3 and 4 indicate the four different PCR performed.
second, the downstream fragment D1 was obtained, using primers FD1 and RD1. In both PCRs 10 ng of T. elongatus BP-1 genomic DNA was used as a template under the same conditions: heating to 94˚C for 2 min; 30 cycles of denaturation at 94˚C for 30 s, annealing at 58˚C for 1 min, extension at 72˚C for 2 min, and finally an additional extension at 72˚C for 10 min. A third PCR allowed the amplification of M1 using primers FM1 and RM1. In this reaction, plasmid pKT1 was used as a template for kat with the following conditions: 2 min denaturation at 94˚C followed by 30 cycles of denaturation at 94˚C for 30 s, annealing at 62˚C for 1 min, extension at 72˚C for 2 min, and finally an additional extension at 72˚C for 10 min.
To prepare the conversion cassette to replace kat with nirA_Pl (C2), four different PCRs were carried out. In the first, primers FU2 and RU2 were used to amplify the upstream fragment U2. In the second, downstream fragment D2 was obtained, using primers FD2 and RD2. In both PCRs, 10 µl of T. elongatus BP-1 nirA/kat+ genomic DNA was used as a template, under the same conditions used to amplify U1 and D1. A third PCR allowed the amplification of M2 using FM2 and RM2 oligonucleotides. In this reaction 10 ng of P. laminosum genomic DNA was used as a template, under the following conditions: 2 min denaturation at 94˚C followed by 30 cycles of denaturation at 94˚C for 30 s, annealing at 63˚C for 1 min, extension at 72˚C for 4 min, and finally an additional extension at 72˚C for 10 min. After gel extraction, purification and concentration of fragments U2, M2 and D2 from agarose gels, a fourth PCR was performed using approximately 200 ng U2, 400 ng M2 and 200 ng D2. PCR conditions were as follows: heating to 94˚C for 2 min; 30 cycles of 94˚C for 30 s, 60˚C for 1 min and 72˚C for 8 min; followed by a final extension at 72˚C for 10 min.
2.5. DNA Cloning and Plasmid Purification
When circular DNA was used, the fragments corresponding to the conversion cassettes were extracted from agarose gels using DNA Gel Extraction Kit (Millipore Co, Mildford, MA, USA) and cloned into pT7Blue vector using Perfectly Blunt Cloning Kit. Finally, plasmid DNA was purified with Qiaprep® Spin Miniprep Kit (Qiagen, Hilden, Germany) when used for sequencing, or with NucleoBond®Xtra Midi Plus Kit (Macherey Nagel, Düren, Germany) when used to perform T. elongatus BP-1 transformation assays.
2.6. T. elongatus BP-1 Transformation Assays
Conversion cassette C1, pT7Blue-C1 and pT7Blue-C2 were concentrated (to 1.5 µg∙µl−1) and used for transformation of exponentially growing, T. elongatus BP-1. Transformation assays were performed according to (Iwai et al., 2004), with some modifications. To obtain T. elongatus BP-1 nirA−/kat+ cells, 6 µg of C1 or different amounts (from 6 µg to 50 µg) of pT7Blue-C1 were used. In this case, selection was carried out on solid BG110 medium supplemented with ammonium sulphate (BG110AS) and with 40 µg∙ml−1 kanamycin. To insert nirA from P. laminosum in substitution of the T. elongatus BP-1 gene, different amounts (from 6 µg to 50 µg) of pT7Blue-C2 were used, and selection was performed in solid BG11 medium.
2.7. Analytical Methods
Growth of T. elongatus BP-1 was determined by measuring the turbidity of the cultures at 730 nm in a DU®800 UV/Vis spectrophotometer (Beckman Coulter Inc., Fullerton, CA, USA). DNA concentration and purity was determined by measuring its absorbance at 260 nm and 280 nm, or using agarose gels by comparison to a known concentration of DNA markers. To perform agarose gel electrophoresis of DNA, 1% (w/v) agarose in Tris-acetate-EDTA (TAE) was used. Gels contained 5 µl of SYBR® Safe DNA Stain (Invitrogen, Carlsbad, CA, USA). DNA was supplemented with 0.25% (w/v) bromophenol blue and 30% (v/v) glycerol loading buffer. In all gels, 5 µl of DNA HyperLadder I (Bioline, Randolph, MA, USA) were also loaded. Electrophoresis was carried out using a horizontal Mini-Sub Cell GT (BioRad, Hercules, PA, USA). An image analyzer GenoSmart (VWR International, Darmstadt, Germany) was used to visualize the bands. Recovery of DNA fragments from agarose gels was carried out with DNA Gel Extraction Kit (Millipore Co., Milford, MA, USA).
To screen transformants, colonies were picked from the corresponding plates, and colony PCR was performed. To lyse cells, 50 µl of buffer containing 1% (v/v) Triton X-100, 20 mM Tris-HCl, pH 8.0 and 500 mM EDTA, was added to each colony.
Samples were heated at 95˚C for 10 min and then centrifuged (3000 × g, 10 min). Finally, 4 µl of the supernatant was used as a template to perform PCR verifications with the corresponding oligonucleotides.
3. Results
The process followed to obtain T. elongatus BP-1 cells carrying nirA_Pl consisted of two differentiated steps. In a first stage, nirA_Te was eliminated from T. elongatus BP-1 and substituted by kat, a gene coding for a thermostable kanamycin nucleotidyl transferase variant from Staphylococcus aureus [46]. In the second step, the resulting cells (ΔnirA::kat mutant) were transformed with nirA_Pl, which was inserted in the same locus. To obtain both types of cells a constructed DNA fragment (conversion cassette) is used to target a particular gene or sequence without promoting any other modification within of T. elongatus BP-1 genome. Construction of such conversion cassettes was achieved using LFH-PCR (Figure 1) and was followed by transformation of T. elongatus by natural competence. Results obtained during each step were as follows.
3.1. Construction of T. elongatus BP-1 ΔnirA::kat Mutant
To obtain this first cell type, the conversion cassette C1, targeted to replace nirA by kat without promoting any other modification within the genome of T. elongatus BP-1, was constructed. For this purpose, fragments U1, M1 and D1 were amplified in three different PCRs (Figure 2(a)) and assembled by a fourth PCR. The length of U1 and D1 is critical for transformation efficiency and it was determined from similar studies carried out in Synechocystis [15-17]. Thus, C1 consisted of the kat selection marker, flanked by the upstream and downstream regions nirA, U1 (508 bp) and R1 (536 bp). The correct assembly of the C1 conversion cassette was confirmed by agarose electrophoresis (Figure 2(b)) and sequencing.
Taking into account that there is only a single copy of nirA in the genome of T. elongatus BP-1, and that the expression of this gene is completely required by cells to grow in media with nitrate or nitrite as the sole nitrogen source, BG110 medium supplemented with ammonium (BG110AS) was used to grow ΔnirA mutants.
Initially, T. elongatus BP-1 cells were transformed with linear C1, the resulting mutants were selected in BG110AS plates supplemented with kanamycin and, finally, colony PCR was performed to verify the genotype of the transformants. To force the complete segregation of the genome, colonies were re-streaked in BG110AS
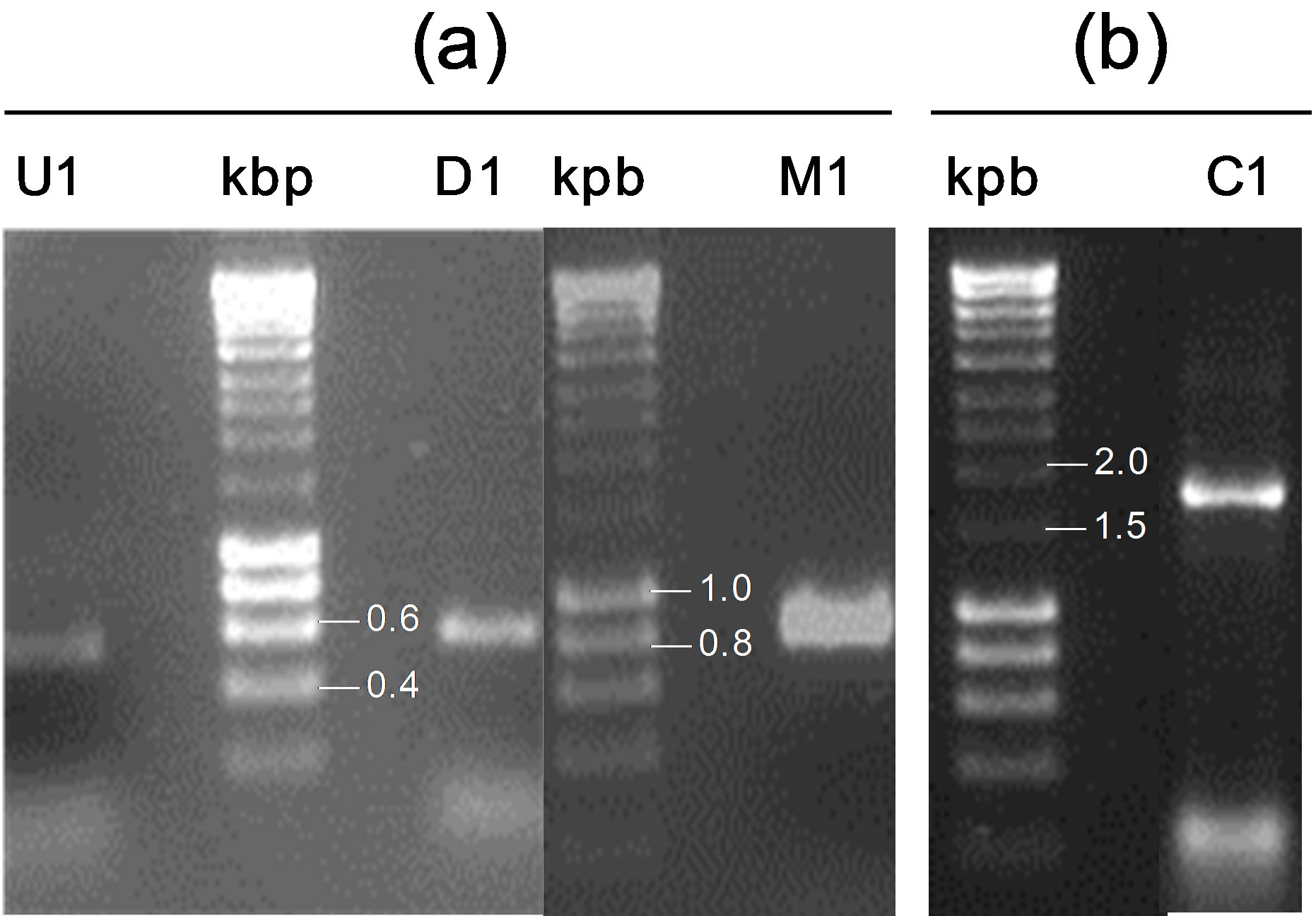
Figure 2. Analysis in agarose gels of (a) the PCR synthesized fragments to construct C1, and (b) the resulting C1 conversion cassette. U1, upstream fragment; D1, downstream fragment; M1, fragment containing the marker gene (kat).
plates with increasing kanamycin concentrations and analyzed by PCR. After 10 days, several colonies were able to grow in the presence of kanamycin, indicating that cells had acquired kat. PCR analysis, using FU1 and RD1 oligonucleotides, was done to ascertain the correct location of the insert and to guarantee the absence of nirA in the genome. These primers annealed at both ends of C1, and amplification by PCR indicated whether nirA (1536 bp) had been or not replaced by kat (approx. 800 bp). The PCR analysis by agarose electrophoresis shown in Figure 3(A.a) compares wild-type (lane WT) and transformed cells (lane T). In the case of transformants, two bands can be detected. This result suggested that transformation was incomplete and merodiploid cells, containing both nirA in some copies of their genome and kat in others, were obtained. Since T. elongatus BP-1 cells contain various copies of its whole genome [19], care has to be taken to guarantee that all copies of the target gene were replaced by gene conversion. Merodiploidity evidenced by PCR was confirmed phenotypically as cells were able to grow both in BG110AS medium supplemented with kanamycin and in medium with nitrate (BG11 medium) without kanamycin (Figure 3B.c).
The obtained result was not completely satisfactory because nirA should be removed from all genome copies of T. elongatus BP-1 before complementing cells with nirA of P. laminosum. To improve transformation efficiency, cells were transformed with circular DNA. In order to perform the transformation assay with circular DNA, C1 was cloned into pT7Blue, and after transformation and selection, the mutants were genotypically analyzed. Wild-type (Figure 3(A.b), lane WT) and transformed cells (Figure 3(A.b), lanes T1, T2 and T3) were analyzed. In this case, the absence of a 2500 bp product in T1, T2 and T3 (Figure 3(A.b)) confirmed the total replacement of all copies of nirA_Te in the transformants. Moreover, transformation was verified phenotypically by assessing cell growth in medium with nitrate as the sole nitrogen source. In this case, mutants grew in medium BG110AS supplemented with kanamycin, but were unable to grow with nitrate as the sole nitrogen source (BG11 medium) (Figure 3(B.d)).
3.2. Incorporation of P. laminosum nirA Gene into the Chromosome of T. elongatus BP-1 ΔnirA::kat Mutant
To construct this cell type, previously obtained T. elongatus BP-1 ΔnirA::kat mutants were transformed with pT7Blue-C2 circular DNA. To construct the conversion cassette C2, fragments U2, M2 and D2 were amplified in three different PCRs (Figure 4(a)) and assembled by a fourth PCR. U2 and D2 were the same regions amplified when designing C1. In this occasion, C2 consisted of
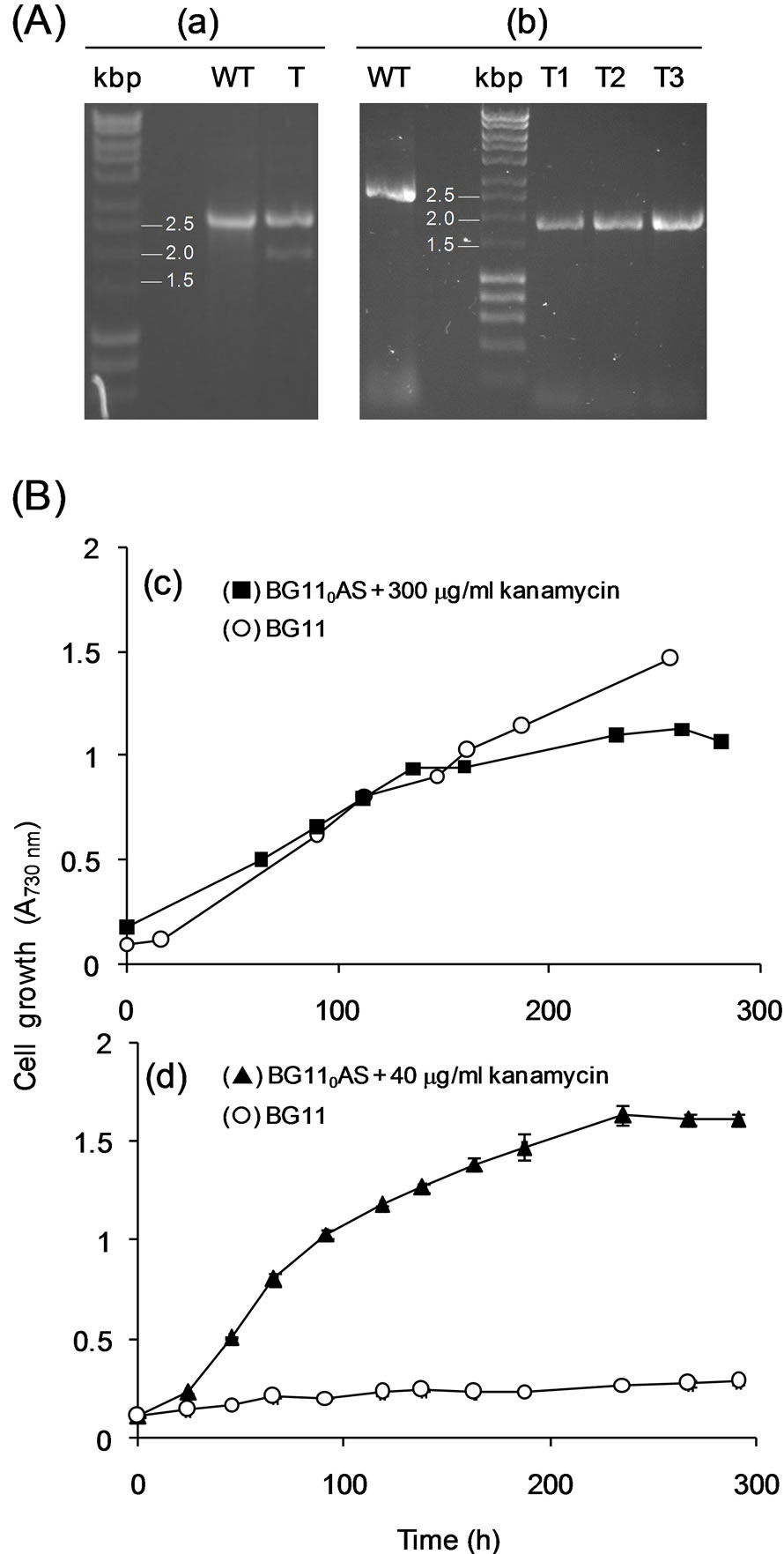
Figure 3. Mutant analysis of T. elongatus BP-1. (A) Genotypic assessment of cells transformed with the conversion cassettes C1 (a) and pt7Blue-C1 (b). Oligonucleotides used were FD1 and RD1, and the templates for PCR were genomic DNA extracted from wild type (WT) or transformed (T) colonies. In panel (b), T1, T2 and T3 correspond to transformants grown in BG110AS medium supplemented with 40, 100 and 300 µg∙ml−1 kanamycin, respectively. (B) Phenotypic assessment of cells transformed with the conversion cassettes C1 (c) and pt7Blue-C1 (d). Cell growth was assessed in BG11 and BG110AS media supplemented with 40 or 300 µg∙ml−1 kanamycin. Each point in curves represents mean values (±standard deviations) of data obtained from two independent cultures.
nirA_Pl as the marker gene, flanked by the upstream and downstream regions of kat in T. elongatus BP-1 ΔnirA::kat mutants. The C2 construct was confirmed by agarose electrophoresis (Figure 4(b)), sequenced and

Figure 4. Analysis in agarose gels of (a) the PCR synthesized fragments to construct C2, and (b) the resulting C2 conversion cassette. U2, upstream fragment; M2, fragment containing nirA_Pl; D2, downstream fragment.
finally cloned to create pT7Blue-C2. This construct was used to transform T. elongatus BP-1 ΔnirA::kat mutants. Once transformed, selection was carried out in BG11 plates. Colonies were picked and analysis by PCR was performed using FU2 and RD2.
The presence of 2500 bp products confirmed that cells had incorporated nirA from P. laminosum (Figure 5(A.a)).
Furthermore, genotypic verification was also done with primers annealing at both ends of nirA from P. laminosum (FnirA_Pl and RnirA_Pl) to guarantee the identity of the insertion. Transformation was checked (Figure 5(A.b)) confirming that cells had incorporated nirA instead of kat. However, when transferred to liquid medium after several attempts, mutants were unable to grow optimally in BG11 medium. In order to improve the genotype of the transgenic cells, the assay was repeated lowering the temperature used for transformation and cell growth. After repeating transformation at 48˚C, a higher number of transformants was obtained, whose genotype was confirmed with FU2, RD2 (Figure 5(B.a)) and FnirA_Pl, RnirA_Pl (Figure 5(B.b)). After an initial incubation in BG110AS medium to facilitate growth, cells were transferred to BG11 medium where optimum growth was observed (Figures 6(a) and (b)).
3.3. Nitrate Assimilation of T. elongatus BP-1 ΔnirA::kat Mutants Complemented with P. laminosum nirA
Finally, to determine whether nirA_Pl could completely restore the capacity of complemented cells to grow with nitrate and also to determine whether growth was affected by the presence of nirA (from T. elongatus BP-1 or from P. laminosum), the growth of P. laminosum cells and T. elongatus BP-1, both wild-type and recombinant cells carrying nirA from P. laminosum, was assessed at

Figure 5. Agarose gel analysis of PCR performed with DNA extracted from several colonies of T. elongatus BP-1 transformed at (A) 52˚C and (B) 48˚C. Transformation was checked with primers (a) FU2 and RD2, and (b) FnirA_Pl and RnirA_Pl.
45˚C (Figure 6(a)) and 55˚C (Figure 6(b)). These temperatures were determined as optimum to grow cells of P. laminosum and T. elongatus BP-1, respectively. As expected, P. laminosum was unable to grow at 55˚C and grew optimally at 45˚C. In case of T. elongatus BP-1, although slight differences were observed between wildtype and chimeric cells carrying P. laminosum nirA, this effect was more noticeable at 55˚C than at 45˚C.
4. Discussion
Globally, studies related to bacterial transformation are targeted to eliminate, disrupt or insert a particular gene or group of genes within their genome or plasmids. In all cases, marker genes are needed for this purpose. These markers, which allow selection of transformed cells, usually code for proteins that provide a selective advantage to its host. In this study, the process followed to obtain transgenic cells (i.e., T. elongatus BP-1 cells bearing the nirA gene from P. laminosum) is a combination of all the above-mentioned strategies. Although direct substitution of nirA_Te with nirA_Pl may have been feasible, it might have been difficult to distinguish both cell types phenotypically due to the fact that the proteins codified by both genes have the same catalytic activity.
One of the requirements to carry out the transforma-
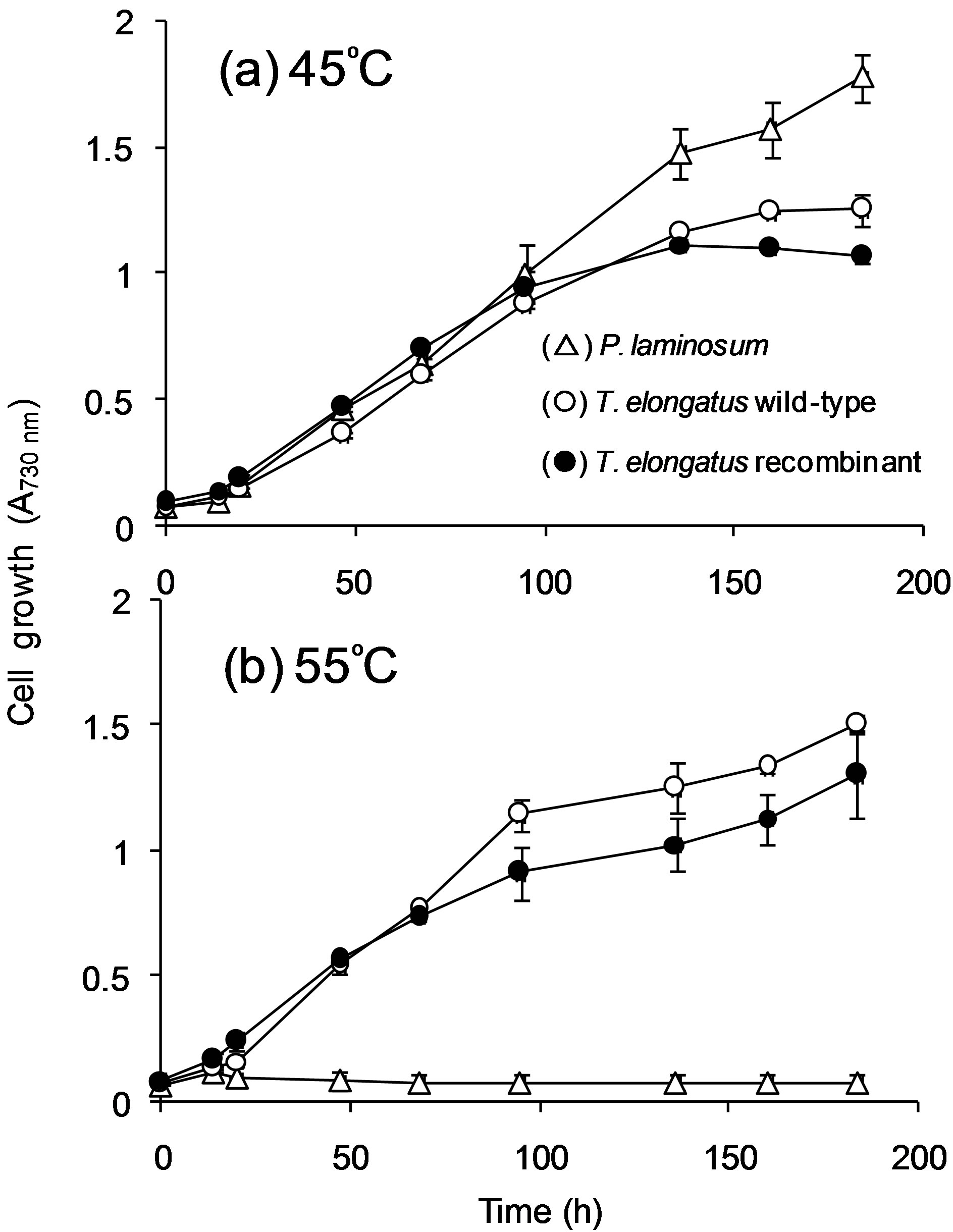
Figure 6. Cell growth in BG11 medium at (a) 45˚C, and (b) 55˚C of P. laminosum and wild-type and recombinant T. elongatus BP-1. Each point in curves represents mean values (±standard deviations) of data obtained from two independent cultures.
tion is to have a marker gene that allows selecting transformants successfully. Therefore, the construction of the first cell type (i.e., T. elongatus BP-1 ΔnirA::kat) required a marker gene stable enough under the culture conditions of T. elongatus BP-1 (55˚C under constant illumination). Although many selection markers are available for the transformation of mesophilic microorganisms, few are suitable for the transformation of thermophilic cyanobacteria. The use of a gene coding for a thermostable kanamycin nucleotidyl transferase (kat) allowed the selection of mutants at high temperature in BG110AS plates supplemented with kanamycin.
Verification of mutants transformed with linear C1 (Figure 3(A.a)) indicated that this initial attempt failed, giving place to merodiploid cells bearing kat in some copies of their genome and nirA in others. Incomplete gene deletion or substitution usually stems from the deleted gene (in this case nirA) being essential. Nevertheless, this is not the case because cells cultured using ammonium as nitrogen source do not need to express nitrite reductase to grow. When working with merodiploid microorganisms, re-streaking colonies in medium containing increasing concentration of antibiotic usually promotes the genome segregation and complete substitution of the target gene by the antibiotic resistance marker [13]. However, re-streaking of colonies in the presence of increasing concentrations of kanamycin did not improve genome segregation. Other authors have reported the same problem when transforming cells with linear DNA [32]. One explanation could be based on some DNA uptake sequences found in other bacteria. Some authors have reported the importance of DNA uptake sequences (DUS) [29,47] for efficient transformation. The fact that the lack of these sequences would have limited the entrance of C1 to the inside of T. elongatus BP-1 seems improbable, as other researchers have reported transformation in this cyanobacterium independently of the DNA sequence used [20,21]. In the present work, the best explanation for this result might be the lower transformation efficiency of T. elongatus BP-1 cells compared to some other bacteria and cyanobacteria. The quantity of C1 used may not have been enough for the complete substitution of nirA by kat gene, and/or this linear DNA might have been digested by nucleases, resulting in reduced transformation efficiency [48].
In the case of the cyanobacterium Synechocystis sp. PCC6803, where transformation has been extensively evaluated, some authors [15-17] reported that efficiency is higher when circular DNA was used in the conversion cassette. In the case of T. elongatus BP-1, colony PCR (Figure 3(A.b)) and growth assessment (Figure 3(B.d)) confirmed that gene substitution was complete, strongly suggesting that transformation with circular DNA is more efficient than with its linear counterpart.
T. elongatus BP-1 ΔnirA::kat mutant grown in BG110AS medium was transformed with pt7Blue-C2. Due to the low integration efficiency experienced with linear cassette C1, cells were transformed with pT7BlueC2 and not with linear C2. As reported in the Results section, the first attempts to complement T. elongatus BP-1 ΔnirA::kat mutants were unsuccessful. Even though mutant analysis by PCR indicated that cells had incorporated nirA effectively replacing kat (Figure 5(A.a)), amplification with FnirA_Pl and RnirA_Pl was only detected in two colonies (Figure 5(A.b)). Furthermore, the obtained colonies were few and yellowish, presenting a color similar to that observed when cells were subjected to nitrogen starvation or under other situations related to metabolic stress. It is important to highlight that these cells have been transferred from an ammonium to a nitrate containing medium. Ammonium is the easiest assimilable source of inorganic nitrogen because it does not require further reduction and can be taken up by cells through either active transport or diffusion mechanisms [49-51]. However, the uptake of nitrate and/or nitrite from the external medium by cells requires the expression of a number of proteins involved in their transport to the cytoplasm and further reduction to ammonium. Consequently, when a nitrogen source is shifted to a less favorable one, cells need some time to synthesize or balance the levels of the required newly synthesized proteins. However, even after allowing extra acclimatization, transformants showed a phenotype typical of cells unable to grow adequately with nitrate.
Although data are insufficient to confirm it, the results point towards the differences in nirA thermostability between P. laminosum and T. elongatus BP-1. Considering that P. laminosum (which shows optimal growth at 45˚C) presents a lower thermophilicity compared to T. elongatus BP-1, the temperature of 52˚C used to select and cultivate mutants may have been too high for the expression or correct function of P. laminosum nirA. This explanation could be reinforced by previous data obtained in our laboratory of the nitrite reductase activity from wild-type P. laminosum [7]. These results indicated that the purified nirA was completely thermostable in vitro at temperatures up to 40˚C, but its activity decreased rapidly at higher temperatures, although 45˚C was estimated as the optimum temperature for cell growth under laboratory conditions.
To discard temperature as a factor compromising the correct expression of nirA in P. laminosum, the transformation assay was repeated lowering the temperature to 48˚C. After selecting some colonies from BG11 plates and growing them at moderate light intensity (initially at 48˚C in liquid BG110AS to increase cell density, and then in BG11 medium), some colonies were able to grow in BG11 medium (i.e., showing a normal phenotype), and unable to grow in medium supplemented with kanamycin.
Finally, when comparing wild-type and transgenic T. elongatus BP-1 growth curves (Figure 6), even though there were no significant differences, these were greater at 55˚C (Figure 6(b)) than at 45˚C (Figure 6(a)), probably due to differences in thermostability between both nitrite reductases.
5. Conclusion
In conclusion, we have constructed transgenic cells of T. elongatus BP-1 in which nirA was substituted by the homologous gene of P. laminosum. These recombinant cells can grow in BG11 medium, indicating that the P. laminosum nirA gene was expressed in an active form in cells of T. elongatus BP-1. Slight differences between the growth of wild-type and transgenic cells of T. elongatus BP-1 were observed, probably due to the different thermostability of NirA of both cyanobacteria. The followed process to construct transgenic cells of T. elongatus BP-1 was mainly based on the LFH-PCR technique and the remarkable natural competence showed by this cyanobacterium. Moreover, the thermostable kanamycin nucleotidyl transferase-encoding kat gene has also been very helpful in the intermediate stage of the process. This procedure can be applied in the future to transfer other genes present in nirA-permease-narB operon from P. laminosum to T. elongatus BP-1 and would be a useful tool to explore changes in phenotype due to the exchange of a single gene between cyanobacteria.
5. Acknowledgements
This work was supported by the University of the Basque Country (UPV/EHU) (Grants GIU07/55 and GIU11/25). We are grateful to Prof. J. Berenguer and Dr. A. Hidalgo (Autonomous University of Madrid, Spain) for providing us with the pKT1 plasmid carrying the kat gene. A strain of T. elongatus BP-1 cells was kindly provided by Prof. M. Rögner (University of Bochum, Germany). MB was the recipient of a scholarship from the UPV/EHU.
REFERENCES
- T. Omata, X. Andriesse and A. Hirano, “Identification and Characterization of a Gene Cluster Involved in Nitrate Transport in the Cyanobacterium Synechococcus sp. PCC 7942,” Molecular and General Genetics, Vol. 236, No. 2-3, 1993, pp. 193-202. http://dx.doi.org/10.1007/BF00277112
- I. Luque, E. Flores and A. Herrero, “Molecular Mechanism for the Operation of Nitrogen Control in Cyanobacteria,” EMBO Journal, Vol. 13, No. 12, 1994, pp. 2862- 2869.
- S. Maeda and T. Omata, “Nitrite Transport Activity of the ABC-Type Cyanate Transporter of the Cyanobacterium Synechococcus elongatus,” Journal of Bacteriology, Vol. 191, No. 10, 2009, pp. 3265-3272. http://dx.doi.org/10.1128/JB.00013-09
- E. Flores and A. Herrero, “Nitrogen Assimilation and Nitrogen Control in Cyanobacteria,” Biochemical Society Transactions, Vol. 33, 2005, pp. 164-167. http://dx.doi.org/10.1042/BST0330164
- E. Flores, J. E. Frías, L. M. Rubio and A. Herrero, “Photosynthetic Nitrate Assimilation in Cyanobacteria,” Photosynthesis Research, Vol. 83, No. 2, 2005, pp. 117-133.
- M. I. Muro-Pastor, J. C. Reyes and F. J. Florencio, “Ammonium Assimilation in Cyanobacteria,” Photosynthesis Research, Vol. 83, No. 2, 2005, pp. 135-150. http://dx.doi.org/10.1007/s11120-004-2082-7
- J. M. Arizmendi and J. L. Serra, “Purification and Some Properties of the Nitrite Reductase from the Cyanobacterium Phormidium laminosum,” Biochimica et Biophysica Acta, Vol. 1040, No. 2, 1990, pp. 237-244. http://dx.doi.org/10.1016/0167-4838(90)90082-Q
- D. Nagore, M. Llarena, M. J. Llama and J. L. Serra, “Characterization of the N-Terminal Domain of NrtC, the ATP-Binding Subunit of ABC-Type Nitrate Transporter of the Cyanobacterium Phormidium laminosum,” Biochimica et Biophysica Acta, Vol. 1623, No. 2-3, 2003, pp. 143-153. http://dx.doi.org/10.1016/j.bbagen.2003.09.001
- M. Llarena, M. J. Llama and J. L. Serra, “Purification and Properties of NrtC and NrtD, the ATP-Binding Subunits of the ABC Nitrate/Nitrite Transporter of Phormidium laminosum,” Biochimica et Biophysica Acta, Vol. 1760, No. 12, 2006, pp. 1819-1826. http://dx.doi.org/10.1016/j.bbagen.2006.08.006
- D. Nagore, B. Sanz, J. Soria, M. Llarena, M. J. Llama, J. J. Calvete and J. L. Serra, “The Nitrate/Nitrite ABC Transporter of Phormidium laminosum: Phosphorylation State of NrtA Is Not Involved in Its Substrate Binding Activity,” Biochimica et Biophysica Acta, Vol. 176, No. 2, 2006, pp. 172-181. http://dx.doi.org/10.1016/j.bbagen.2005.12.011
- F. Merchán, K. L Kindle, M. J. Llama, J. L. Serra and E. Fernández, “Cloning and Sequencing of the Nitrate Transport System from the Thermophilic Filamentous Cyanobacterium Phormidium laminosum: Comparative Analysis with the Homologous System from Synechococcus sp. PCC 7942,” Plant Molecular Biology, Vol. 28, No. 4, 1995, pp. 759-766. http://dx.doi.org/10.1007/BF00021199
- S. Naithani, J. M. Hou and P. R. Chitnis, “Targeted Inactivation of the psaK1, psaK2 and psaK3 Genes Encoding Subunits of Photosystem I in the Cyanobacterium Synechocystis sp. PCC 6803,” Photosynthesis Research, Vol. 63, No. 3, 2000, pp. 225-236. http://dx.doi.org/10.1023/A:1006463932538
- G. Taroncher-Oldenburg and G. Stephanopoulos, “Targeted, PCR-Based Gene Disruption in Cyanobacteria: Inactivation of the Polyhydroxyalkanoic Acid Synthase Genes in Synechocystis sp. PCC6803,” Applied and Microbial Biotechnology, Vol. 54, No. 5, 2000, pp. 677-680. http://dx.doi.org/10.1007/s002530000450
- H. Katoh, S. Itoh, J. R. Shen and M. Ikeuchi, “Functional Analysis of psbV and a Novel c-Type Cytochrome Gene psbV2 of the Thermophilic Cyanobacterium Thermosynechococcus elongatus strain BP-1,” Plant and Cell Physiology, Vol. 46, No. 6, 2001, pp. 599-607. http://dx.doi.org/10.1093/pcp/pce074
- G. I. Kufryk, M. Sachet, G. Schmetterer and F. J. Vermaas Wim, “Transformation of the Cyanobacterium Synechocystis sp. PCC 6803 as a Tool for Genetic Mapping: Optimization of Efficiency,” FEMS Microbiology Letters, Vol. 26, No. 2, 2002, pp. 215-219. http://dx.doi.org/10.1111/j.1574-6968.2002.tb11012.x
- S. V. Shestahov, “Gene-Targeted and Site-Directed Mutagenesis of Photosynthesis Genes in Cyanobacteria,” Photosynthesis Research, Vol. 73, No. 1-3, 2002, pp. 279-284. http://dx.doi.org/10.1023/A:1020432004692
- X. Zang, B. Liu, S. Liu, K. Arunakamura and X. Zhang, “Optimal Conditions for Transformation of Synechocystis sp. PCC 6803,” Journal of Microbiology, Vol. 45, No. 3, 2007, pp. 241-245.
- T. Yamaoka, K. Satoh and S. Katoh, “Photosynthetic Activities of a Thermophilic Blue-Green Alga,” Plant and Cell Physiology, Vol. 19, No. 6, 1978, pp. 943-954.
- Y. Nakamura, T. Kaneko, S. Sato, M. Ikeuchi, H. Katoh, S. Sasamoto, A. Watanabe, M. Iriguchi, K. Kawashima, T. Kimura, Y. Kishida, C. Kiyokawa, M. Kohara, A. Matsumoto, N. Nakazaki, S. Shimpo, M. Sugimoto, C. Takeuchi, M. Tamada and S. Tabata, “Complete Genome Structure of the Thermophilic Cyanobacterium Thermosynechococcus elongatus BP-1,” DNA Research, Vol. 9, No. 4, 2002, pp. 123-130. http://dx.doi.org/10.1093/dnares/9.4.123
- K. Onai, M. Morishita, Y. Kaneko, S. Tabata and M. Ishiura, “Natural Transformation of the Thermophilic Cyanobacterium Thermosynechococcus elongatus BP-1: a Simple and Efficient Method for Gene Transfer,” Molecular Genetics and Genomics, Vol. 271, No. 1, 2004, pp. 50-59. http://dx.doi.org/10.1007/s00438-003-0953-9
- M. Iwai, H. Katoh, M. Katayama and M. Ikeuchi, “Improved Genetic Transformation of the Thermophilic Cyanobacterium Thermosynechococcus elongatus BP-1,” Plant and Cell Physiology, Vol. 45, No. 2, 2004, pp. 171- 175. http://dx.doi.org/10.1093/pcp/pch015
- M. G. Lorenz and W. Wackernagel, “Bacterial Gene Transfer by Natural Genetic Transformation in the Environment,” Microbiology Reviews, Vol. 58, No. 3, 1994, pp. 563-602.
- R. D. Lunsford, “Streptococcal Transformation: Essential Features and Applications of a Natural Gene Exchange System,” Plasmid, Vol. 39, No. 1, 1998, pp. 10-20. http://dx.doi.org/10.1006/plas.1997.1323
- C. M Thomas and K. M. Nielsen, “Mechanisms of, and Barriers to, Horizontal Gene Transfer between Bacteria,” Nature Reviews in Microbiology, Vol. 3, No. 9, 2005, pp. 711-721. http://dx.doi.org/10.1038/nrmicro1234
- O. Johnsborg and L. S. Håvarstein, “Regulation of Natural Genetic Transformation and Acquisition of Transforming DNA in Streptococcus pneumonia,” FEMS Microbiology Reviews, Vol. 33, No. 3, 2009, pp. 627-642. http://dx.doi.org/10.1111/j.1574-6976.2009.00167.x
- B. Averhoff and A. Friedrich, “Type IV Pili-Related Natural Transformation System: DNA Transport in Mesophilic and Thermophilic Bacteria,” Archives in Microbiology, Vol. 180, No. 6, 2003, pp. 385-393. http://dx.doi.org/10.1007/s00203-003-0616-6
- B. Averhoff, “DNA Transport and Natural Transformation in Mesophilic and Thermophilic Bacteria,” Journal of Bioenergetics and Biomembranes, Vol. 36, No. 1, 2004, pp. 25-33. http://dx.doi.org/10.1023/B:JOBB.0000019595.66733.fa
- I. Chen, P. J. Christie and D. Dubnau, “The Ins and Outs of DNA Transfer in Bacteria,” Science, Vol. 310, No. 5753, 2005, pp. 1456-1460. http://dx.doi.org/10.1126/science.1114021
- H. L. Hamilton and J. P. Dillard, “Natural Trans-Formation of Neisseria gonorrhoeae: From DNA Donation to Homologous Recombination,” Molecular Microbiology, Vol. 59, No. 2, 2006, pp. 376-385. http://dx.doi.org/10.1111/j.1365-2958.2005.04964.x
- O. Johnsborg, E. Vegard and L. S. Håvarstein, “Natural Genetic Transformation: Prevalence, Mechanisms and Function,” Research in Microbiology, Vol. 158, No. 10, 2007, pp. 767-778. http://dx.doi.org/10.1016/j.resmic.2007.09.004
- A. Urban, S. Neukirchen and K.-E. Jaeger, “A Rapid and Efficient Method for Site-Directed Mutagenesis Using One-Step Overlap Extension PCR,” Nucleic Acids Research, Vol. 25, No. 11, 1997, pp. 2227-2228. http://dx.doi.org/10.1093/nar/25.11.2227
- R. Kong and X. Xu, “Tree-Piece-Ligation PCR and Application in Disruption of Chlorophyll Synthesis Genes in Synechocystis sp. PCC 6803,” Current Microbiology, Vol. 44, No. 4, 2002, pp. 241-245. http://dx.doi.org/10.1007/s00284-001-0032-6
- H. Kuwayama, S. Obara, T. Mario, M. Katoh, H. Urushihara and Y. Tanaka, “PCR-Mediated Generation of a Gene Disruption Construct without the Use of DNA Ligase and Plasmid Vectors,” Nucleic Acid Research, Vol. 30, No. 2, 2002, Article ID: e2. http://dx.doi.org/10.1093/nar/30.2.e2
- J. Wendland, “PCR-Based Methods Facilitates Targeted Gene Manipulations and Cloning Procedures,” Current Genetics, Vol. 44, No. 3, 2003, pp. 115-123. http://dx.doi.org/10.1007/s00294-003-0436-x
- A. Baudin, K. O. Ozier, A. Denouel, F. Lacroute and C. Cullin, “A Simple Efficient Method for Direct Gene Deletion in Saccharomyces cerevisiae,” Nucleic Acids Research, Vol. 21, No. 14, 1993, pp. 3329-3330. http://dx.doi.org/10.1093/nar/21.14.3329
- A. Wach, A. Brachat, R. Poehlmann and P. Philippsen, “New Heterologous Modules for Classical or PCR-Based Gene Disruptions in Saccharomyces cerevisiae,” Yeast, Vol. 10, No. 13, 1994, pp. 1793-1808. http://dx.doi.org/10.1002/yea.320101310
- M. C. Lorenz, R. S. Muir, E. Lim, J. McElver, S. C. Weber and J. Heitman, “Gene Disruption with PCR Products in Saccharomyces cerevisiae,” Gene, Vol. 158, No. 1, 1995, pp. 113-117. http://dx.doi.org/10.1016/0378-1119(95)00144-U
- P. Manivasakam, S. C. Weber, J. McElver and R. H. Schiestl, “Micro-Homology Mediated PCR Targeting in Saccharomyces cerevisiae,” Nucleic Acids Research, Vol. 23, No. 14, 1995, pp. 2799-2800. http://dx.doi.org/10.1093/nar/23.14.2799
- A. Wach, “PCR-Synthesis of Marker Cassettes with Long Flanking Homology Regions for Gene Disruptions in S. cerevisiae,” Yeast, Vol. 12, No. 3, 1996, pp. 259-265. http://dx.doi.org/10.1002/(SICI)1097-0061(19960315)12:3<259::AID-YEA901>3.0.CO;2-C
- J. M. Koomey and S. Falkow, “Cloning of the recA Gene of Neisseria gonorrhoeae and Construction of Gonococcal recA Mutants,” Journal of Bacteriology, Vol. 169, No. 2, 1987, pp. 790-795.
- A. Kowalczykowski and A. Eggelston, “Homologous Pairing and DNA Strand-Exchange Proteins,” Annual Review of Biochemistry, Vol. 63, 1994, pp. 991-1043. http://dx.doi.org/10.1146/annurev.bi.63.070194.005015
- D. Kidane and P. L. Graumann, “Intracellular Protein and DNA Dynamics in Competent Bacillus subtilis Cells,” Cell, Vol. 122, No. 1, 2005, pp. 73-84. http://dx.doi.org/10.1016/j.cell.2005.04.036
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. CohenBazire, “Purification and Properties of Unicellular BlueGreen Algae (Order Chroococcales),” Bacteriology Reviews, Vol. 35, No. 2, 1971, pp. 171-205.
- R. W. Castenholz, “Laboratory Culture of Thermophilic Cyanophytes,” Schweizerische Zeitschrift fur Hydrologie, Vol. 32, No. 2, 1970, pp. 538-551.
- J. A. Smoker and S. R. Barum, “Rapid Small-Scale DNA Isolation from Filamentous Cyanobacteria,” FEMS Microbiology Letters, Vol. 56, No. 1, 1988, pp. 119-122. http://dx.doi.org/10.1111/j.1574-6968.1988.tb03161.x
- H. Liao, T. Mckenzie and R. Hageman, “Isolation of a Thermostable Enzyme Variant by Cloning and Selection in a Thermophile,” Proceedings of the National Academy of Sciences USA, Vol. 83, 1986, pp. 576-580. http://dx.doi.org/10.1073/pnas.83.3.576
- H. O. Smith, M. L. Gwinn and S. L. Salzberg, “DNA Uptake Signal Sequences in Naturally Transformable Bacteria,” Research in Microbiology, Vol. 150, No. 9-10, 1999, pp. 603-616. http://dx.doi.org/10.1016/S0923-2508(99)00130-8
- T. Blomqvist, H. Steinmoen and L. S. Håvarstein, “Natural Genetic Transformation: A Novel Tool for Efficient Genetic Engineering of the Dairy Bacterium Streptococcus thermophilus,” Applied and Environmental Microbiology, Vol. 72, No. 10, 2006, pp. 6751-6756. http://dx.doi.org/10.1128/AEM.01156-06
- M. L. Montesinos, A. M. Muro-Pastor, A. Herrero and E. Flores, “Ammonium/Methylammonium Permeases of a Cyanobacterium. Identification and Analysis of Three Nitrogen-Regulated amt Genes in Synechocystis sp. PCC 6803,” Journal of Biological Chemistry, Vol. 273, No. 47, 1998, pp. 31463-31470. http://dx.doi.org/10.1074/jbc.273.47.31463
- M. F. Vázquez-Bermúdez, J. Paz-Yepes, A. Herrero and E. Flores, “The NtcA Activated amt1 Gene Encodes a Permease Required for Uptake of Low Concentrations of Ammonium in the Cyanobacterium Synechococcus sp. PCC 7942,” Microbiology, Vol. 148, No. 3, 2002, pp. 861-869.
- M. I. Muro-Pastor and F. J. Florencio, “Regulation of Ammonium Assimilation in Cyanobacteria,” Plant Physiology and Biochemistry, Vol. 41, No. 6-7, 2003, pp. 595-603. http://dx.doi.org/10.1016/S0981-9428(03)00066-4
Supplement
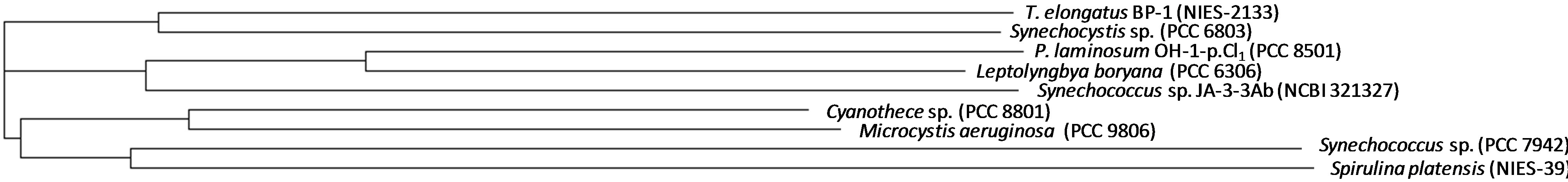
Figure S1. Phylogenetic tree reflecting nitrite reductase protein (NirA) of some cyanobacteria from Chroococcales (T. elongatus BP-1, Synechocystis sp. PCC 6803, Synechococcus sp. PCC 7942, Synechococcus sp. JA-3-3Ab, Cyanothece sp. PCC 8801, Microcystis aeruginosa PCC 9806) and Oscillatoriales (P. laminosum OH-1-p clone1, Leptolyngya boryana, Spirulina platensis NIES-39) order. Sequences were obtained from NCBI and alignment was performed with clustalW2 (EMBL-EBI).
NOTES
*Corresponding author.