American Journal of Analytical Chemistry
Vol.5 No.3(2014), Article ID:43227,4 pages DOI:10.4236/ajac.2014.53026
Optimization of Quantitative Analysis of Naproxin Sodium Using UV Spectrophotometery in Different Solvent Mediums
1Department of Chemistry, University of Karachi, Karachi, Pakistan
2Department of Chemistry, Federal Urdu University for Arts, Science and Technology, Karachi, Pakistan
3Department of Environmental Science, Sindh Madressatul Islam University, Karachi, Pakistan
Email: *mhzuberi1@gmail.com
Copyright © 2014 Muhammad Hashim Zuberi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Muhammad Hashim Zuberi et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received November 14, 2013; revised December 19, 2013; accepted December 27, 2013
KEYWORDS
Naproxen Sodium; UV Spectrophotometry; Pharmaceutical Analysis
ABSTRACT
A new rapid, simple and reproducible UV spectrophotometric method was developed and validated for the estimation of Naproxen Sodium (NpSd) in bulk and pharmaceutical formulation. The quantification of NpSd was done at 230 nm in methanol and in buffer of pH 6.8 and 9. Beer’s law was obeyed in the concentration range of 4 - 36 (r2 = 0.999) in methanol and 5 - 25 μgmL−1 in buffer of pH 6.8 and 9 (r2 = 0.988 and 0.997) respectively. The apparent molar absorptivity values were also calculated in all mediums. All parameters according to ICH guideline were tested and validated. The detection and quantitation limits were found to be 0.054, 0.083, 0.073 and 0.181, 0.251, 0.211 μgmL−1 respectively. These methods were applied directly to the analysis of the pharmaceutical tablet preparations (Anex® tablet 250 mg). The results demonstrated that the procedure is accurate, precise and reproducible (relative standard deviation < 3%), while being simple, cheap and less time consuming and hence can be suitably applied for the estimation of NpSd in dosage forms and dissolution studies.
1. Introduction
Naproxen (NpSd), a non-steroidal anti-inflammatory drug, is chemically [(+)-2-(6-methoxy-2-naphthyl) propionic acid with anti-inflammatory, analgesic and antipyretic properties, commonly used in the treatment of rheumatoid arthritis and other rheumatic or musculoskeletal disorders, dysmenorrhea and acute gout (Figure 1). It is often preferred to acetylsalicylic acid (aspirin) because of its better absorption following oral administration and poses fewer adverse effects. It works by inhibition of both the COX-1 and COX-2 enzymes with consequent decrease in prostaglandin concentrations in various fluids and tissues. Anti-inflammatory effects of NpSd are generally thought to be related to its inhibition of cyclooxygenase and formulated in tablets or suppositories, it

Figure 1. Naproxen sodium.
is used in the treatment of rheumatoid arthritis and other rheumatic or musculoskeletal disorders, dysmenorrhea and acute gout [1].
Naproxen in commercial formulations has been determined by coulometry [2], UV spectrophotometry [3-6], heavy atom-induced room temperature phosphorescence [7], UPLC [8] and high-performance liquid chromatography (HPLC) [9-14]. Present paper was undertaken with the aim to develop an accurate, simple and reliable UV method for estimation of naproxen API and applied to commercial formulations. These studies were performed in different pH environments: simulated gastric juice (0.1 N HCl) and simulated intestinal environment (pH 6.8 and 9). A detailed survey of analytical literature reveals that there is no UV spectrophotometric study on NpSd in different pH mediums simulating human body compartments. The results obtained were validated as the ICH guidelines i.e. Lambert and Beer’s law validation, accuracy, precision, limits of detection and quantification. The proposed method is successfully applied for the analysis of the drug in bulk form and its pharmaceutical formulation. The results were in good agreement with those obtained by the official and reported methods.
2. Experimental
2.1. Instruments
Shimadzu 1801 double beam UV-visible spectrophotometer possessing a fixed slit width (2 nm) with quartz cells of 10-mm (1.0 cm) cell path length connected to a P-IV computer loaded with Shimadzu UVPC version 3.9 software and a HP Desk Jet 1200 printer were used to record the absorption spectra.
2.2. Preparation of Solutions
Three different stock solutions of NpSd reference standard (100 μg∙mL−1) were prepared by dissolving 10 mg of NpSd in 100 mL of methanol, phosphate buffer of pH 6.8 and 9. Aliquot was diluted to a concentration range 4 - 36 μg∙mL−1 in methanol and 5 - 25 μg∙mL−1 all above mediums. Anex® tablets (PharmEvo (Pvt) Ltd.) were purchase from local market each containing 250 mg of NpSd. Twenty tablets were weighed to obtain mean tablet weight, then were crushed to powder and preceded in the same way. The NpSd solutions were scanned in the UV region and were quantified at 230 nm in methanol and buffer of pH 6.8 and 9, respectively.
3. Method Validation
3.1. Linearity, Accuracy and Precision
The method was validated according to International Conference on Harmonization [15] for validation of analytical procedures in order to determine the linearity, sensitivity, precision and accuracy in all mediums (methanol, buffer of pH 6.8 and 9). Table 1 summarizes the statistical results evaluated from the above observations. Precision of the method was determined by adding known amounts of pure drug (50%, 100%, and 150%) in triplicate to the solution of exciepients (10% each) (Table 2). For the accuracy of the developed method, standard addition method was done. Different concentrations of pure drug (8, 10 and 12 μg∙mL−1) were added to a known preanalysed formulation sample and the total concentration
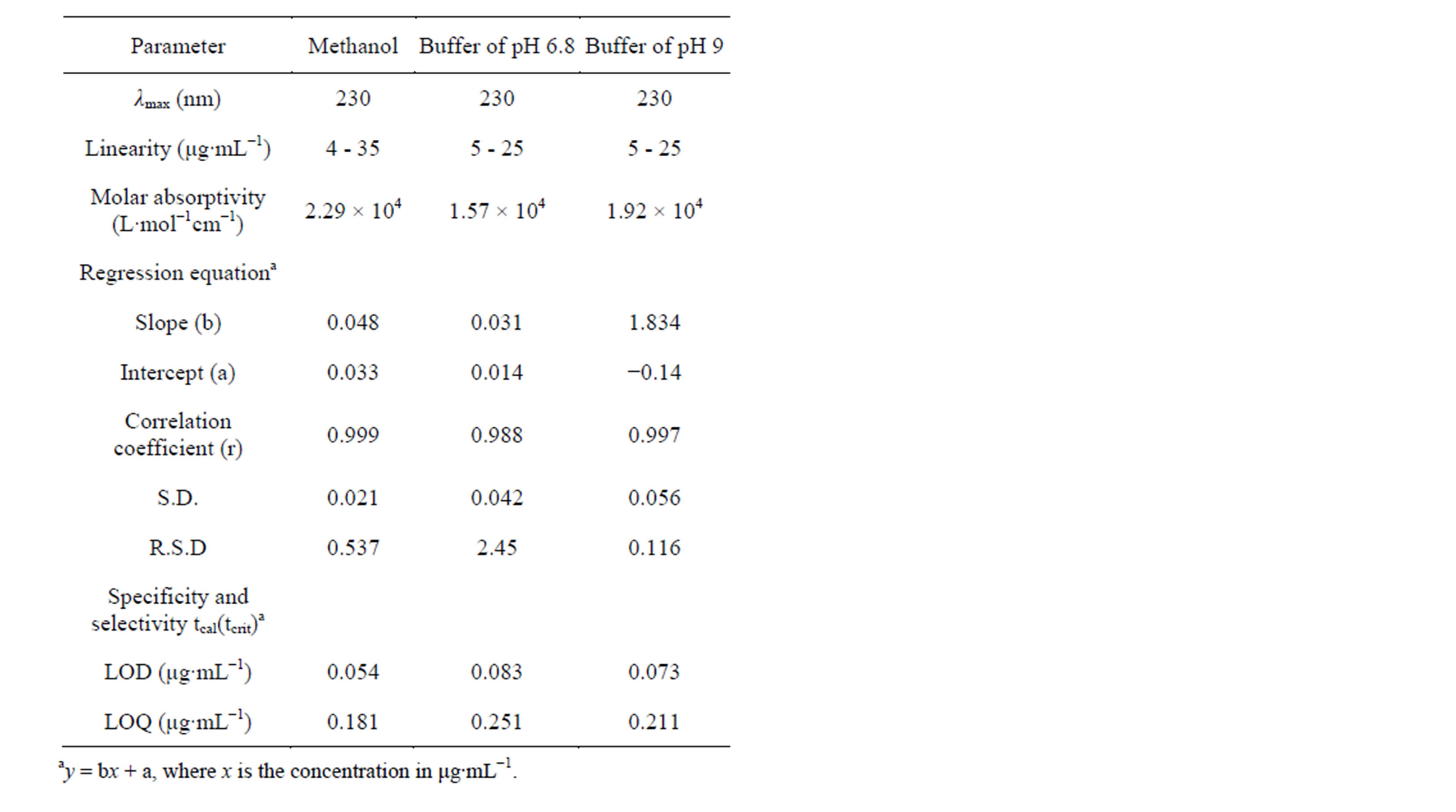
Table 1. Optical characteristics and precision data.
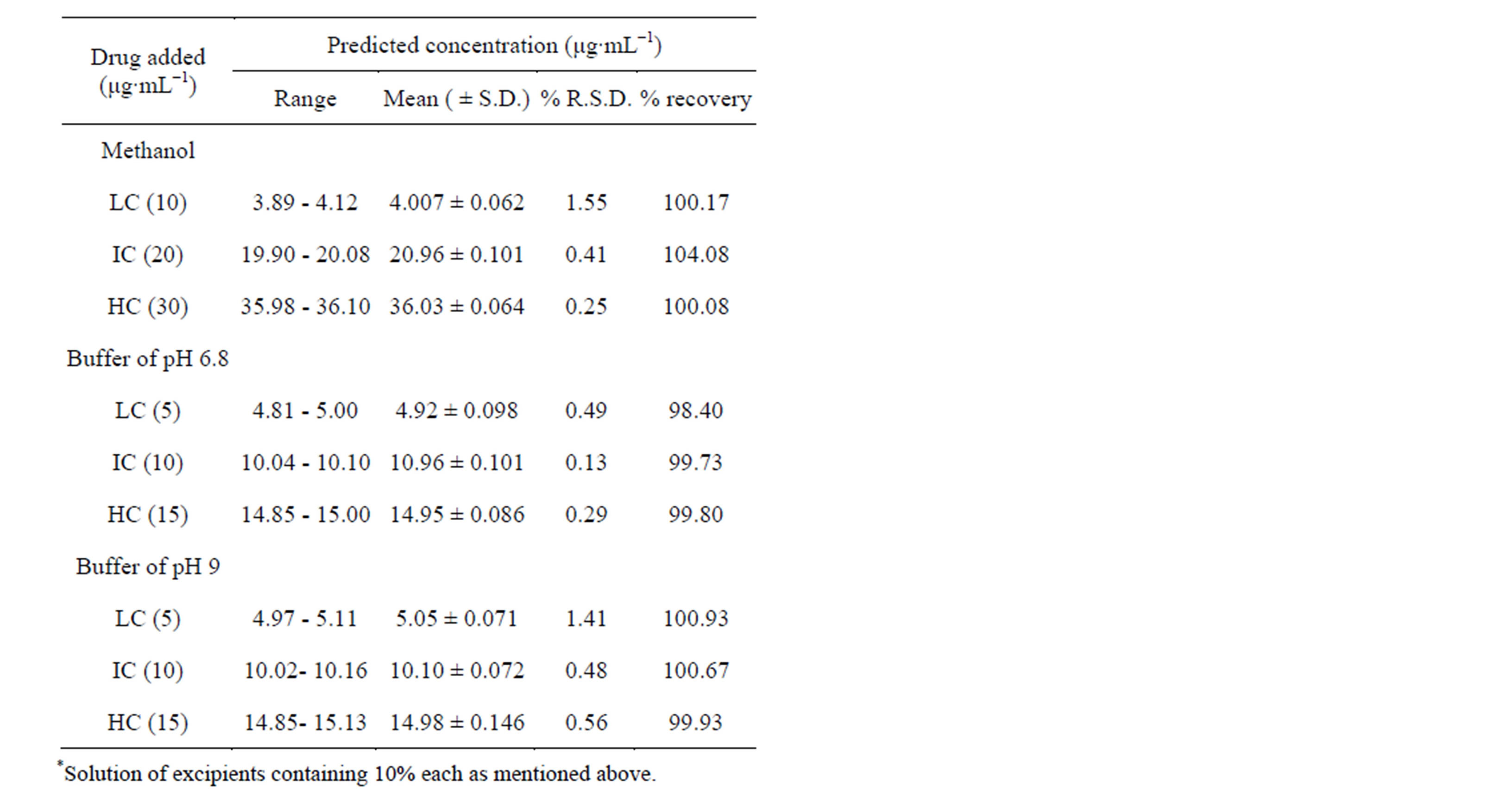
Table 2. Precision and accuracy of the proposed method in presence of excipients*.
was determined (Table 3). The percent recovery of the added pure drug was calculated as follows:
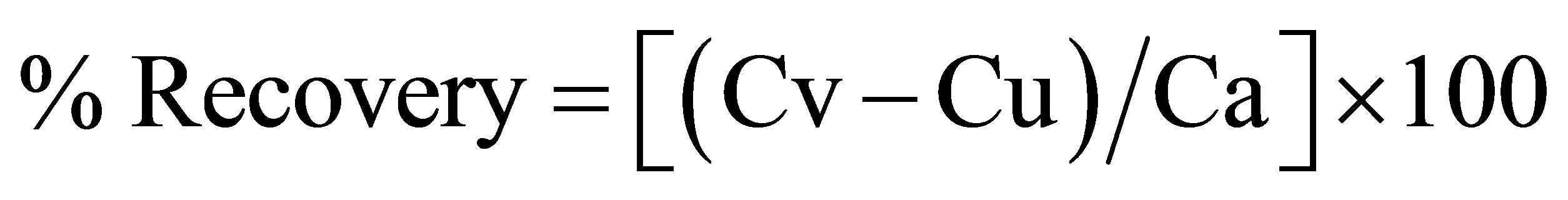
where Cv was the total drug concentration measured after standard addition, Cu, drug concentration in the for-

Table 3. Accuracy of the proposed method (standard addition technique)*.
mulation and Ca, drug concentration added to formulation. Under the experimental conditions described above, a linear regression equation (intercept and slope) for NpSd was established. The high values of the correlation coefficient and the values of Y-intercepts close to zero indicate good linearity of the calibrations.
3.2. Specificity and Selectivity
NpSd solution (8, 10 and 12 μg∙mL−1) were prepared in all selected medias along with and without common excipients (magnesium stearate, purified talc, lactose, maiz starch, povidone K 30, hydroxypropylmethyl cellulose, twin 80) all the solutions were scanned from 400 to 200 nm at a speed of 400 nm∙min−1.
In a separate study, drug concentration of 10 µg∙mL−1 was prepared independently from pure drug stock solution in selective media and analyzed (n = 9). Paired t-test at 95% level of significance was performed to compare the means of absorbance (Table 3).
3.3. Detection and Quantification Limits or Sensitivity
The limit of detection (LOD) is the lowest amount of analyte in a sample that can be detected, but not necessarily quantitated under the stated experimental conditions. The lower limit of detection of NpSd was shown in Table 1. Limit of quantitation (LOQ) is the lowest amount of analyte in a sample that can be determined
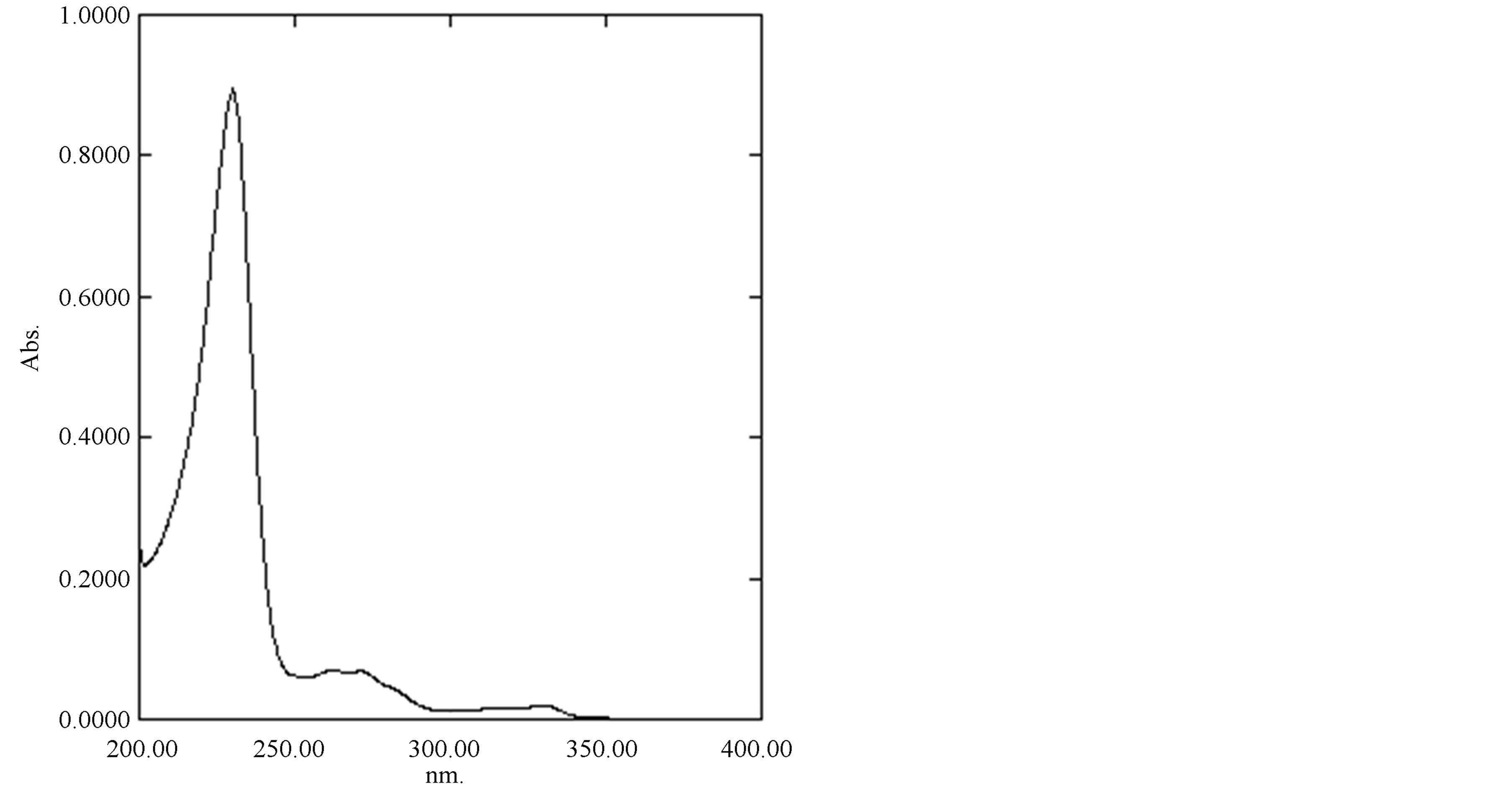
Figure 2. Absorption spectrum of Naproxen sodium.
with acceptable precision and accuracy under the stated experimental conditions [15]. The LOQ value for NpSd was shown in Table 1.
4. Results and Discussion
Naproxen sodium (NpSd) yields a characteristic curve in all mediums when scanned in the ultraviolet wavelength range. The scan (Figure 2) shows absorption maxima at 230 nm in all solvent mediums. The molar absorptivity in methanol and in buffer pH 6.8 and 9 was found to be 2.299 × 104 L∙mol−1cm−1, 1.57 × 104 and 1.92 × 104 L∙mol−1cm−1. which was in good agreement and hence 230 nm was chosen as the analytical wavelength. In these methods, Beer’s law was valid in the concentration range of 4 to 36 μg∙mL−1 in methanol and 5 to 25 μg∙mL−1 in buffers (pH 6.8 and 9). The satisfactory correlation coefficient and intercept values were obtained as shown in Table 1. Precision of the method was studied in presence of standard exciepients and different solvents (Table 2) and was applied to commercial pharmaceutical formulations using stantdard addition method whose percent recovery was shown in Table 3. The method was validated for linearity, accuracy, precision, specificity, limit of detection, and limit of quantitation. It was observed that the excipients in the tablets were not interfering in the analysis of the active compounds.
5. Conclusions
This method gave a successful result for the quantitative resolution of the reference standard and pharmaceutical dosage formulation. Using this analytical procedure, a good analytical performance was obtained for the determination of NpSd. The results suggest that this method is a powerful tool with very simple mathematical content and LLOD values 0.054, 0.083 and 0.073 μg∙mL−1 in methanol, buffer of pH 6.8 and buffer of pH 9 respectively and is more reliable than other spectrophotometric methods and strongly encourages us to apply these calibration models for a routine analysis and quality control of commercial products.
The presented method was found to be simple, accurate and precise which gives an acceptable recovery of the analyte, which can be directly and easily applied to the analysis of the pharmaceutical tablet formulations of NpSd.
REFERENCES
- S. Mohideen, M. Shivakanth, P. SureshKumar, S. N. Krishnan, Y. Surendranath and T. Satyanarayana, “Development and Validation of Analytical Method for Naproxen and Pantoprazole in Capsule Dosage Form,” Der Pharmacia Sinica, Vol. 2, No. 6, 2011, pp. 114-121.
- G. Kanoute, E. Nivaud, B. Paulet and P. Boucly, “Dosage de Derives de l’acide Phenylpropionique: A Activite Pharmacologique par Titrage Coulometrique,” Talanta, Vol. 31, No. 2, 1984, pp. 144-146. http://dx.doi.org/10.1016/0039-9140(84)80035-1
- M. S. Mahrous, M. M. Abdel-Khalek and M. E. AbdelHamid, “Quantitation of Indomethacin, Naproxen, and Ibuprofen in Pharmaceutical Dosage Forms by First and Second Derivative Ultraviolet Spectrometry,” JournalAssociation of Official Analytical Chemists, Vol. 68, No. 3, 1985, pp. 535-539.
- S. Kulsum, M. Padmalatha, K. Sandeep, B. Saptasila and G. Vidyasagar, “Spectrophotometric Methods for the Determination of Naproxen Sodium in Pure and Pharmaceutical Dosage Forms,” International Journal of Biomedical and Pharmaceutical Sciences, Vol. 2, 2011, pp. 1303- 1307.
- R. P. Gondalia and A. P. Dharamsi, “Spectrophotometric Simultaneous Estimation of Naproxen Sodium and Sumatriptan Succinate in Tablet Dosage Forms,” International Journal of Biomedical and Pharmaceutical Sciences, Vol. 1, 2011, pp. 24-26.
- T. Haque, M. M. U. Talukder, S. Laila, K. Fatema and A. K. L. Kabir, “Simultaneous Estimation of Naproxen and Ranitidine HCl by Using UV Spectrophotometer,” Stamford Journal of Pharmaceutical Sciences, Vol. 1, 2008, pp. 18-24.
- A. Segura Carretero, C. Cruces-Blanco, M. I. Ramirez Garcia, B. Canabate Diaz and A. Fernandez Gutierrez, “Simple and Rapid Determination of the Drug Naproxen in Pharmaceutical Preparations by Heavy Atom-Induced Room Temperature Phosphorescence,” Talanta, Vol. 50, No. 2, 1999, pp. 401-407. http://dx.doi.org/10.1016/S0039-9140(99)00126-5
- P. Venkatarao, M. N. Kumar and M. R. Kumar, “Novel Validated Stability-Indicating UPLC Method for the Estimation of Naproxen and Its Impurities in Bulk Drugs and Pharmaceutical Dosage Form,” Scientia Pharmaceutica, Vol. 80, 2012, pp. 965-976. http://dx.doi.org/10.3797/scipharm.1207-12
- I. W. Wainer and T. D. Doyle, “Application of HighPerformance Liquid Chromatographic Chiral Stationary Phases to Pharmaceutical Analysis Structural and Conformational Effects in the Direct Enantiomeric Resolution of α-Methylarylacetic Acid Antiinflammatory Agents,” Journal of Chromatography A, Vol. 284, 1984, pp. 117- 124. http://dx.doi.org/10.1016/S0021-9673(01)87807-9
- R. Kumar, P. Singh and H. Singh, “Development and Validation of Rp-hplc Method for Simultaneous Estimation of Naproxen and Pantoprazole in Pharmaceutical Dosage Form,” International Journal of Pharma Research and Development, Vol. 2, 2011, p. 6.
- Y. H. Hsu, Y. B. Liou, J. A. Lee, C. Y. Chen and A. B. Wu, “Assay of Naproxen by High Performance Liquid Chromatography and Identification of Its Photoproducts by LC-ESI MS,” Biomedical Chromatography, Vol. 20, No. 8, 2006, pp. 787-793. http://dx.doi.org/10.1002/bmc.598
- X. Yu-Jia, “Determination of Related Substances in Compound Naproxen Suppositories by RP-HPLC,” Central South Pharmacy, Vol. 5, 2005, p. 006.
- L. Monser and F. Darghouth, “Simultaneous Determination of Naproxen and Related Compounds by HPLC Using Porous Graphitic Carbon Column,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 32, No. 4-5, 2003, pp. 1087-1092. http://dx.doi.org/10.1016/S0731-7085(03)00213-9
- W. R. G. Baeyens, G. Van der Weken and M. Schelkens, “Diclofenac and Naproxen Analysis by Microbore Liquid Chromatography (LC) with Native Fluorescence Detection,” Journal of Fluorescence, Vol. 5, No. 2, 1995, pp. 131-134. http://dx.doi.org/10.1007/BF00727529
- I. C. H. H. T. Guideline, “Validation of Analytical Procedures: Text and Methodology Q2 (R1),” International Conference on Harmonization, Geneva, November 2005.
NOTES
*Corresponding author.