International Journal of Clinical Medicine
Vol. 3 No. 7 (2012) , Article ID: 26039 , 7 pages DOI:10.4236/ijcm.2012.37110
Hereditary Leukemia Due to Rare RUNX1c Splice Variant (L472X) Presents with Eczematous Phenotype
![]()
1Department of Pediatrics, City Hope National Medical Center, Duarte, USA; 2Division of Clinical Cancer Genetics, City Hope National Medical Center, Duarte, USA; 3Division of Population Sciences, City Hope National Medical Center, Duarte, USA; 4Division of Hematology and Transplantation, City Hope National Medical Center, Duarte, USA; 5Department of Molecular Genetics, City Hope National Medical Center, Duarte, USA.
Email: asorrell@coh.org
Received September 15th, 2012; revised October 11th, 2012; accepted November 25th, 2012
Keywords: Hereditary Leukemia; Eczema; FPDMM; L472X; RUNX1c Isoform
ABSTRACT
Deleterious mutations in the RUNX1 gene cause hereditary leukemia due to a rare syndrome called Familial platelet Disorder with Associated Myeloid Malignancy (FPDMM). We describe the characteristics of a family with FPDMM due to a novel RUNX1 mutation (L472X), located in the most 3-prime end of the gene reported to date. Our 36-year-old proband presented with incidentally detected thrombocytopenia and a family history suggestive of FPDMM. Contrary to previously described families, affected members of our kindred express an eczematous phenotype, reportedly most severe in members who develop leukemia. Pedigree analysis shows that the L472X mutation tracks with thrombocytopenia, acute leukemia, and eczema. The L472X mutation produces a stably expressed RUNX1 protein product with a corresponding decrease in wild type RUNX1 expression. Our data supports the inclusion of eczema in the FPDMM phenotype and suggests the possibility that the RUNX1 L472X mutant causes the type of dominant negative affect that is associated with an elevated risk of leukemia in FPDMM families.
1. Introduction
Deleterious mutations in RUNX1 (Runt-related transcription factor 1; MIM ID *151385) are associated with a rare Autosomal Dominant hereditary cancer syndrome (linked to 21q22.12) that is characterized by qualitative and quantitative platelet defects with a high-propensity for myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and lymphoid malignancies [1]. At least 20 different causative RUNX1 gene mutations have been described in the more than 30 reported families with Familial platelet Disorder with Associated Myeloid Malignancy Syndrome (FPDMM; MIM 601399) [2,3]. Most of these reports, summarized in Figure 1(a), describe heterozygous germline mutations that cluster in the amino-terminal (N-terminal) region of the gene within the DNA-binding runt homology domain [3-31].
We describe a North American family with FPDMM syndrome caused by an unusually located RUNX1 frameshift mutation at the 3-prime end of exon 8 (L472X). This RUNX1 L472X variant tracks with an atypical clinical history of early-onset eczema; reported in symptomatic members of our kindred across four consecutive generations. Consistent with the phenotype seen in other families with FPDMM syndrome, the RUNX1 L472X variant presents with a bleeding diathesis, variable degrees of thrombocytopenia, adult-onset myelodysplastic syndrome/acute myeloid leukemia, and juvenile-onset acute lymphoblastic leukemia. The eczematous phenotype, reportedly most severe in the affected family members who develop leukemia, is a unique clinical characteristic of our kindred.
2. Materials and Methods
2.1. Patient Selection
The proband and his mother were referred to physicians at the City of Hope for clinical consultation. Peripheral blood samples from each patient were sent to CLIA-approved laboratories for diagnostic RUNX1 gene mutation analysis. Subsequently the proband, his mother, and family members were invited to participate in a COH Institutional Review Board (IRB)-approved Hereditary Cancer Research registry. The study participants provided personal and family health data for the construction of a five
 (a)
(a)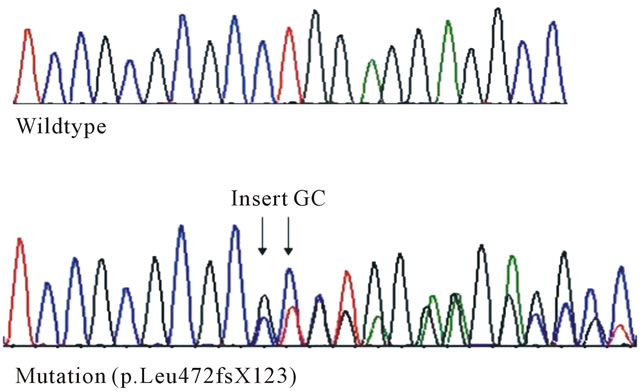 (b)
(b)
Figure 1. (a) RUNX1 mutations associated with familial platelet disorder with associated myeloid malignancy phenotype; (b) RUNX1 L472X mutation. DNA sequence analysis: Sequencing chromatograms showing a GC insertion in Exon 8 of the RUNX1 gene.
generation pedigree. All studies were approved by the City of Hope IRB, consistent with the Declaration of Helsinki, and informed consent was obtained from all subjects.
2.2. Mutation Analysis
Genomic DNA from peripheral blood lymphocytes (and paraffin embedded non-malignant spleen tissue from one deceased member of the kindred; Figure 2 case IV-20) was extracted by standard procedures. Polymerase chain reaction amplification and bidirectional sequencing of exons one through eight (and flanking regions) of the RUNX1 gene was performed using DNA from the proband and his mother. Site-specific mutation analysis was performed for all other at-risk family members tested.
2.3. Western Blot Analysis
Western Blot Analysis was performed using total protein isolated from peripheral blood and bone marrow mononuclear cells from the proband, his mother, and two healthy controls. Equal amounts of protein (25 μg) were loaded onto a 4% - 20% SDS-PAGE gel, transferred onto a nitrocellulose membrane, and probed with rabbit polyclonal RUNX1 antibody (Cell Signaling). The membrane was stripped, washed, and reprobed with mouse monoclonal anti-Actin antibodies (Sigma-Aldrich). Antibody detection was done using enhanced chemiluminescence (Super Femto kit, Pierce Biotechnology). The levels of Actin served as loading controls.
3. Results
3.1. Phenotype
Our 36-year-old proband is a healthy, non-dysmorphic, male with mild eczema and asymptomatic thrombocytopenia (range 94 to 120,000), initially detected during a routine physical exam. A bone marrow evaluation showed
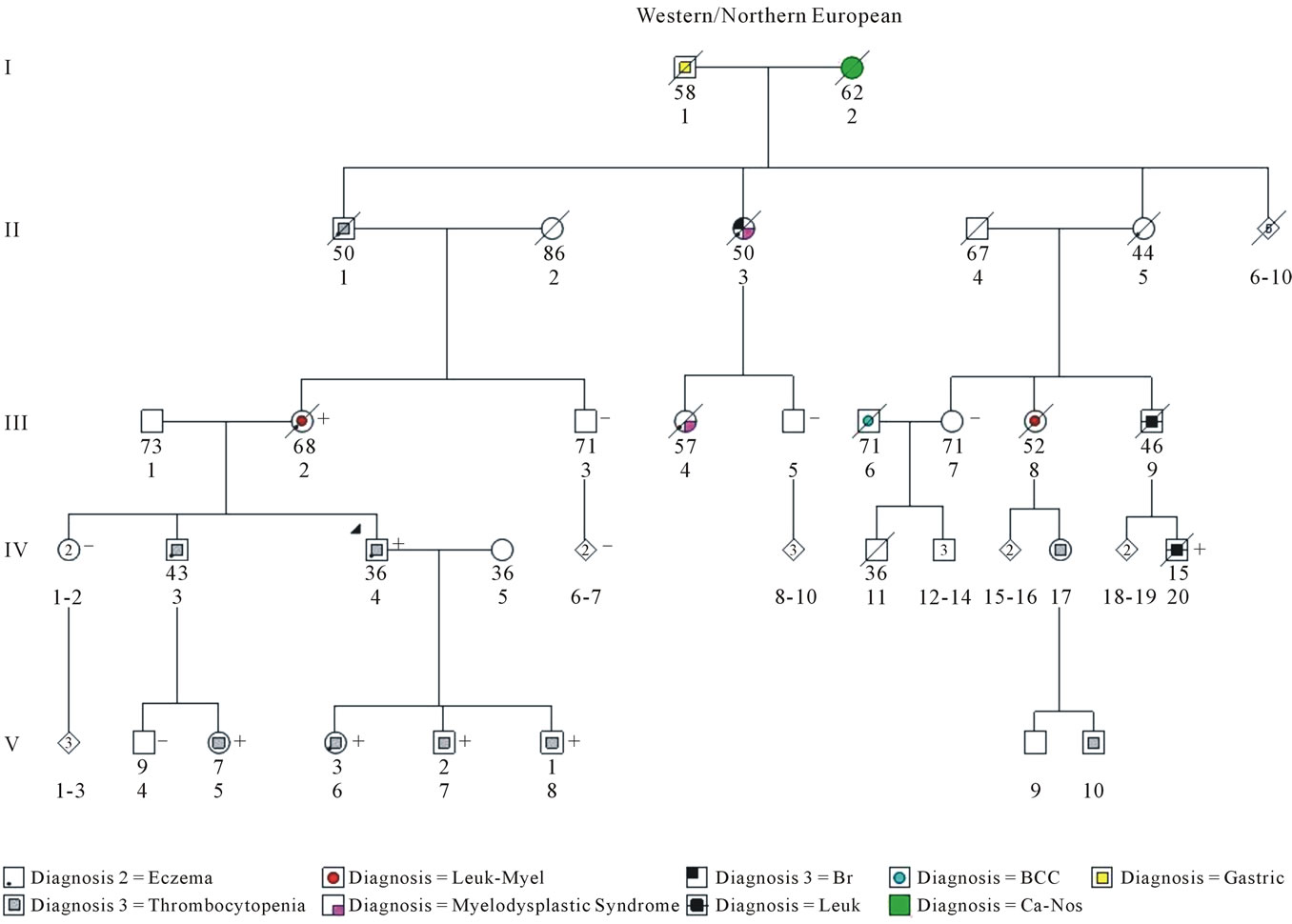
Figure 2. COH fpdmm kindred. A five generation (I-V) pedigree of a City of Hope (COH) family with Familial Platelet Disorder with Associated Myeloid Malignancy (FPDMM) is shown. The index patient (proband) is indicated by an arrow. The family members with confirmed RUNX1 L472X gene mutations are identified by the plus sign (+); negative cases are identified with a (−) sign. The square symbols represent males; the circles represent females; the ◊ represents gender which is not specified or unknown. Symbols that contain numbers represent more than one family member. The current age/age at death (years) for each family member is noted directly below the symbol. Family members affected by cancer [myelodysplastic syndrome, acute myeloid leukemia (Leuk-Myel), acute lymphoblastic leukemia (Leuk), basal cell carcinoma (BCC), breast (Br), gastric, and cancer not otherwise specified (Ca-NOS)] or other disease-related characteristics [thrombocytopenia and eczema] are identified by symbols. Pathological and/or medical record documentation is available for all family members with leukemia, myelodysplastic syndrome, and thrombocytopenia. Dashed symbols represent deceased individuals. Five generations are represented with Roman numerals (I, II, III, IV, and V). Identification numbers representing family members within each generation are located immediately under each person’s current age/age at death. The proband is case IV-4. The proband’s mother is case III-2.
normal trilineage hematopoiesis without evidence of leukemia or myelodysplasia. He was referred for a cancer genetics risk evaluation after his mother was diagnosed with myelodysplastic syndrome. Our proband’s mother was a 68-year old who had a ~3-year-history of progressive anemia. Sequential bone marrow evaluations confirmed myelodysplastic syndrome with the subsequent development of acute myeloid leukemia. Despite chemotherapy, she eventually died from progressive, refractory disease. Our proband provided an extensive family history of hematological disorders and leukemia involving four generations of family members along his maternal lineage (Figure 2). Interestingly, our proband’s 7- year-old niece was clinically diagnosed by a pediatric hematologist with an MYH9-related disorder known as Sebastian syndrome (OMIM 605249), due to her history of thrombocytopenia, easy bruisability, and a confirmed aspirin-like platelet defect. No causative MYH9 mutation could be identified by DNA sequence analysis of genomic DNA from this child; testing was performed by a commercially available diagnostic laboratory. Our proband’s maternal grandfather died after a ~6-month-long illness characterized by easy bruisability, mucosal membrane bleeding, and progressive splenomegaly.
3.2. Genotype
A heterozygous c.1413_1414insGC (p. L472X) germline mutation was identified in exon 8 of the RUNX1 gene in our proband, his mother, and five additional family members; a total of 7 mutation carriers out of 14 family members studied (Figure 1(b)). Each of the RUNX1 L472X mutation carriers in this family were reported to have mild to severe eczema, which directly correlated to their degree of thrombocytopenia and/or clinical bleeding difficulties. There were no reports of eczema in family members without clinical or genotype evidence of FPDMM syndrome. The probability that our proband’s daughter (Figure 2; case V-6) and his cousin (case IV-20) share the RUNX1 L472X germline mutation by chance alone is 1.6% (1/64), according to Mendelian probability estimates.
3.3. Protein Expression
Because the L472X mutation causes a frameshift near the stop codon of the gene, it is predicted to bypass the normal termination codon and produce a run-on RNA product. If stable, this run-on RNA product could be translated into a substantially extended protein product (p.Leu472fsX123), which would be detectable by Western Blot analysis.
Peripheral blood cells from the proband (case IV-4) and his mother (case III-2) demonstrated low levels of expression of a >55 kD RUNX1 variant protein as well as the expected 55kD wild type RUNX1 protein, which was expressed at reduced levels (Figure 3). The 55 kD RUNX1 variant protein was also detected in samples from the proband’s bone marrow (data not shown) and was not detected in healthy control samples (normal 1 and 2). These results indicate that the heterozygous RUNX1 L472X germline mutation produces a stably expressed RUNX1 protein product, but that expression of wild type RUNX1 is reduced.
4. Discussion
Genotype characteristics of RUNX1 germline mutations are known to have clinically significant functional consequences that may predict leukemia risk in germline carriers. Families carrying mutations that retain greater degrees of biological activity of the wild type RUNX1 protein (e.g. deletions and frameshift mutations that act via haploinsufficiency) have an ~20% risk of MDS/AML as compared to a ~60% risk in families that carry mutations with little residual functional activity (e.g. dominant negative or biallelic nonsense mutations) [15]. Our data suggests that the RUNX1 L472X mutant may inhibit the transcription or translation of wild type RUNX1 protein,
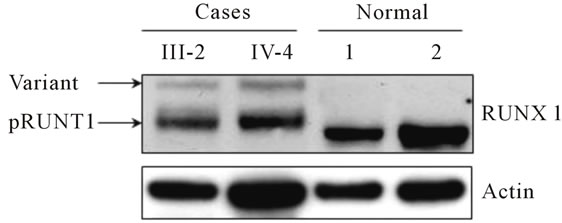
Figure 3. RUNX1 L472X expression. Protein Expression: Western Blot analysis of total protein from the proband (case IV-4) and his mother (case III-2) showed low expression of the normal 55 kD RUNX1 protein and identified the presence of an abnormal RUNX1 band (variant).
possibly leading to the type of dominant negative effect that has been associated with a higher risk of leukemia in some FPDMM families. Although speculated to have the functional capacity to cause leukemia in humans, the tumorigenesis potential of the RUNX1c isoform has not been well established.
Expression of functional RUNX1 protein is tightly regulated at the level of transcription, mRNA processing (splicing), and translation. At least three splice variants of the RUNX1 gene have been identified to date: RUNX1a (250 amino acids, aa), RUNX1b (453 aa), and RUNX1c (480 aa) [NCBI NP_001745; UniProt Q01196] [32]. The RUNX1c variant represents the longest and least abundant isoform of the RUNX1 protein; it is transcribed from the distal P1 promoter, encoding for its own distinct N terminal region [33,34]. The RUNX1b variant is transcribed from the proximal P2 promoter and is 27 amino acids shorter than RUNX1c. The RUNX1a variant is also transcribed from the P2 promoter; but, it differs in the 3-prime coding and UTR regions (compared to variants 1b and 1c) and it lacks the RUNX1 transactivation domain. The distal exon 8 location of the L472X mutation suggests that alternative splicing of the RUNX1 protein occurs in (at least) the mutation carrying members of our kindred who, presumably, express the RUNX1c variant.
In addition to demonstrating that the novel L472X RUNX1 mutation associates with FPDMM clinical characteristics, our report expands the FPDMM phenotype by adding eczematous skin rashes to the clinical findings associated with this disease. To our knowledge, this is the first report to confirm an association between eczema and FPDMM.
Leukemia-associated skin rashes are typically due to infection, leukemic infiltration, or paraneoplastic phenomenon. A prior history of eczema has been associated with a ~30% decreased risk of childhood acute lymphoblastic leukemia, an increased risk of viral (esp. human T-cell lymphotrophic virus) and non-virus associated forms of lymphoma, but no clear alterations in risk for acute myeloid leukemia [35-38]. The only clear link between eczema and a hereditary cancer syndrome is Wiskott-Aldrich syndrome (WAS). WAS is an X-linked disease that often presents with a classic triad of severetreatment resistant eczema (that persists into adulthood), thrombocytopenia (with small platelets), immunodeficiency, and a high-risk of lymphoma (occasionally presenting with bone marrow dysplasia in young people) [39]. Eczema is not a reported characteristic of other chromosome 21-linked disorders with propensity for leukemia and/or impaired erythroid and megakaryocyte hematopoiesis (e.g. Down syndrome and X-linked (macro) thrombocytopenia) [40-42]. Functional investigations of our L472X variant protein may provide novel insights into the biological consequences of distal Cterminal RUNX1 mutations and the genetic pathways that control skin manifestations of immune dysfunction, autoimmunity, and megakaryocytopoiesis [39].
In conclusion, our data supports the inclusion of eczema in the FPDMM phenotype and suggests the possibility that the RUNX1 L472X mutant causes the type of dominant negative affect that is associated with an elevated risk of leukemia in FPDMM families.
5. Acknowledgements
The authors sincerely thank all of the family members who participated in this research. A.D.S. was supported by a Cancer Genetics Career Development Award (5R25CA085771-10) and the Altschul foundation.
REFERENCES
- C. Owen, “Insights into Familial Platelet Disorder with Propensity to Myeloid Malignancy (FPD/AML),” Leukemia Research, Vol. 34, No. 2, 2010, pp. 141-142. doi:10.1016/j.leukres.2009.07.037
- M. Horwitz, E. L. Goode and G. P. Jarvik, “Anticipation in Familial Leukemia,” American Journal of Human Genetics, Vol. 59, No. 5, 1996, pp. 990-998.
- W. J. Song, M. G. Sullivan, R. D. Legare, S. Hutchings, X. Tan, D. Kufrin, et al., “Haploinsufficiency of CBFA2 Causes Familial Thrombocytopenia with Propensity to Develop Acute Myelogenous Leukaemia,” Nature Genetics, Vol. 23, No. 2, 1999, pp. 166-175. doi:10.1038/13793
- P. Ganly, L. C. Walker and C. M. Morris, “Familial Mutations of the Transcription Factor RUNX1 (AML1, CBFA2) Predispose to Acute Myeloid Leukemia,” Leukemia & Lymphoma, Vol. 45, No. 1, 2004, pp. 1-10. doi:10.1080/1042819031000139611
- C. Preudhomme, A. Renneville, V. Bourdon, N. Philippe, C. Roche-Lestienne, N. Boissel, et al., “High Frequency of RUNX1 Biallelic Alteration in Acute Myeloid Leukemia Secondary to Familial Platelet Disorder,” Blood, Vol. 113, No. 22, 2009, pp. 5583-5587. doi:10.1182/blood-2008-07-168260
- M. C. Jongmans, R. P. Kuiper, C. L. Carmichael, E. J. Wilkins, N. Dors, A. Carmagnac, et al., “Novel RUNX1 Mutations in Familial Platelet Disorder with Enhanced Risk for Acute Myeloid Leukemia: Clues for Improved Identification of the FPD/AML Syndrome,” Leukemia, Vol. 24, No. 1, 2010, pp. 242-246. doi:10.1038/leu.2009.210
- S. van der Crabben, E. van Binsbergen, M. Ausems, M. Poot, M. Bierings and A. Buijs, “Constitutional RUNX1 Deletion Presenting as Non-Syndromic Thrombocytopenia with Myelodysplasia: 21q22 ITSN1 as a Candidate Gene in Mental Retardation,” Leukemia Research, Vol. 34, No. 1, 2010, pp. e8-e12. doi:10.1016/j.leukres.2009.06.030
- B. Lages, S. J. Shattil, D. F. Bainton and H. J. Weiss, “Decreased Content and Surface Expression of AlphaGranule Membrane Protein GMP-140 in One of Two Types of Platelet Alpha Delta Storage Pool Deficiency,” Journal of Clinical Investigation, Vol. 87, No. 3, 1991, pp. 919-929. doi:10.1172/JCI115099
- S. B. Dowton, D. Beardsley, D. Jamison, S. Blattner and F. P. Li, “Studies of a Familial Platelet Disorder,” Blood, Vol. 65, No. 3, 1985, pp. 557-563.
- C. Y. Ho, B. Otterud, R. D. Legare, T. Varvil, R. Saxena, D. B. DeHart, et al., “Linkage of a Familial Platelet Disorder with a Propensity to Develop Myeloid Malignancies to Human Chromosome 21q22.1-22.2,” Blood, Vol. 87, No. 12, 1996, pp. 5218-5224.
- J. M. Gerrard, E. D. Israels, A. J. Bishop, M. L. Schroeder, L. L. Beattie, A. McNicol, et al., “Inherited PlateletStorage Pool Deficiency Associated with a High Incidence of Acute Myeloid Leukaemia,” British Journal of Haematology, Vol. 79, No. 2, 1991, pp. 246-255. doi:10.1111/j.1365-2141.1991.tb04529.x
- G. Arepally, T. R. Rebbeck, W. Song, G. Gilliland, J. M. Maris and M. Poncz, “Evidence for Genetic Homogeneity in a Familial Platelet Disorder with Predisposition to Acute Myelogenous Leukemia (FPD/AML),” Blood, Vol. 92, No. 7, 1998, pp. 2600-2602.
- R. D. Legare, D. Lu, M. Gallagher, C. Ho, X. Tan, G. Barker, et al., “CBFA2, Frequently Rearranged in Leukemia, Is Not Responsible for a Familial Leukemia Syndrome,” Leukemia, Vol. 11, No. 12, 1997, pp. 2111-2119. doi:10.1038/sj.leu.2400852
- A. Buijs, P. Poddighe, R. van Wijk, W. van Solinge, E. Borst, L. Verdonck, et al., “A Novel CBFA2 SingleNucleotide Mutation in Familial Platelet Disorder with Propensity to Develop Myeloid Malignancies,” Blood, Vol. 98, No. 9, 2001, pp. 2856-2858. doi:10.1182/blood.V98.9.2856
- J. Michaud, F. Wu, M. Osato, G. M. Cottles, M. Yanagida, N. Asou, et al., “In Vitro Analyses of Known and Novel RUNX1/AML1 Mutations in Dominant Familial Platelet Disorder with Predisposition to Acute Myelogenous Leukemia: Implications for Mechanisms of Pathogenesis,” Blood, Vol. 99, No. 4, 2002, pp. 1364-1372. doi:10.1182/blood.V99.4.1364
- L. C. Walker, J. Stevens, H. Campbell, R. Corbett, R. Spearing, D. Heaton, et al., “A Novel Inherited Mutation of the Transcription Factor RUNX1 Causes Thrombocytopenia and May Predispose to Acute Myeloid Leukaemia,” British Journal of Haematology, Vol. 117, No. 4, 2002, pp. 878-881. doi:10.1046/j.1365-2141.2002.03512.x
- I. Appelmann, T. Linden, A. Rudat, C. Mueller-Tidow, W. E. Berdel and R. M. Mesters, “Hereditary Thrombocytopenia and Acute Myeloid Leukemia: A Common Link Due to a Germline Mutation in the AML1 Gene,” Annals of Hematology, Vol. 88, No. 10, 2009, pp. 1037-1038. doi:10.1007/s00277-009-0722-x
- M. Osato, N. Asou, E. Abdalla, K. Hoshino, H. Yamasaki, T. Okubo, et al., “Biallelic and Heterozygous Point Mutations in the Runt Domain of the AML1/PEBP2- Alphab Gene Associated with Myeloblastic Leukemias,” Blood, Vol. 93, No. 6, 1999, pp. 1817-1824.
- M. Beri-Dexheimer, V. Latger-Cannard, C. Philippe, C. Bonnet, P. Chambon, V. Roth, et al., “Clinical Phenotype of Germline RUNX1 Haploinsufficiency: From Point Mutations to Large Genomic Deletions,” European Journal of Human Genetics, Vol. 16, No. 8, 2008, pp. 1014-1018. doi:10.1038/ejhg.2008.89
- T. Taketani, T. Taki, J. Takita, R. Ono, Y. Horikoshi, Y. Kaneko, et al., “Mutation of the AML1/RUNX1 Gene in a Transient Myeloproliferative Disorder Patient with Down Syndrome,” Leukemia, Vol. 16, No. 9, 2002, pp. 1866-1867. doi:10.1038/sj.leu.2402612
- C. J. Owen, C. L. Toze, A. Koochin, D. L. Forrest, C. A. Smith, J. M. Stevens, et al., “Five New Pedigrees with Inherited RUNX1 Mutations Causing Familial Platelet Disorder with Propensity to Myeloid Malignancy,” Blood, Vol. 112, No. 12, 2008, pp. 4639-4645. doi:10.1182/blood-2008-05-156745
- M. Shinawi, A. Erez, D. L. Shardy, B. Lee, R. Naeem, G. Weissenberger, et al., “Syndromic Thrombocytopenia and Predisposition to Acute Myelogenous Leukemia Caused by Constitutional Microdeletions on Chromosome 21q,” Blood, Vol. 112, No. 4, 2008, pp. 1042-1047. doi:10.1182/blood-2008-01-135970
- T. A. Peterson, A. Adadey, I. Santana-Cruz, Y. Sun, A. Winder and M. G. Kann, “DMDM: Domain Mapping of Disease Mutations,” Bioinformatics, Vol. 26, No. 19, 2010, pp. 2458-2459.
- C. Preudhomme, D. Warot-Loze, C. Roumier, N. GrardelDuflos, R. Garand, J. L. Lai, et al., “High Incidence of Biallelic Point Mutations in the Runt Domain of the AML1/PEBP2 Alpha B Gene in Mo Acute Myeloid Leukemia and in Myeloid Malignancies with Acquired Trisomy 21,” Blood, Vol. 96, No. 8, 2000, pp. 2862-2869.
- Y. Imai, M. Kurokawa, K. Izutsu, A. Hangaishi, K. Takeuchi, K. Maki, et al., “Mutations of the AML1 Gene in Myelodysplastic Syndrome and Their Functional Implications in Leukemogenesis,” Blood, Vol. 91, No. 9, 2000, pp. 3154-3160.
- J. R. Downing, M. Higuchi, N. Lenny and A. E. Yeoh, “Alterations of the AML1 Transcription Factor in Human Leukemia,” Seminars in Cell & Developmental Biology, Vol. 11, No. 5, 2000, pp. 347-360. doi:10.1006/scdb.2000.0183
- S. E. Langabeer, R. E. Gale, S. J. Rollinson, G. J. Morgan and D. C. Linch, “Mutations of the AML1 Gene in Acute Myeloid Leukemia of FAB Types M0 and M7,” Genes, Chromosomes and Cancer, Vol. 34, No. 1, 2002, pp. 24- 32. doi:10.1002/gcc.10031
- J. E. Churpek, J. S. Garcia, J. Madzo, S. A. Jackson, K. Onel and L. A. Godley, “Identification and Molecular Characterization of a Novel 3’ Mutation in RUNX1 in a Family with Familial Platelet Disorder,” Leukemia & Lymphoma, Vol. 51, No. 10, 2010, pp. 1931-1935. doi:10.3109/10428194.2010.503821
- N. Shiba, D. Hasegawa, M. J. Park, C. Murata, A. Matsubara, C. Ogawa, et al., “CBL Mutation in Chronic Myelomonocytic Leukemia Secondary to Familial Platelet Disorder with Propensity to Develop Acute Myeloid Leukemia (FPD/AML),” Blood, Vol. 119, No. 11, 2011, pp. 2612-2614.
- K. Kirito, K. Sakoe, D. Shinoda, Y. Takiyama, K. Kaushansky and N. Komatsu, “A Novel RUNX1 Mutation in Familial Platelet Disorder with Propensity to Develop Myeloid Malignancies,” Haematologica, Vol. 93, No. 1, 2008, pp. 155-156. doi:10.3324/haematol.12050
- D. Bluteau, L. Gilles, M. Hilpert, I. Antony-Debre, C. James, N. Debili, et al., “Down-Regulation of the RUNX1-Target Gene NR4A3 Contributes to Hematopoiesis Deregulation in Familial Platelet Disorder/Acute Myelogenous Leukemia,” Blood, Vol. 118, No. 24, 2011, pp. 6310-6320. doi:10.1182/blood-2010-12-325555
- D. Levanon, G. Glusman, T. Bangsow, E. Ben-Asher, D. A. Male, N. Avidan, et al., “Architecture and Anatomy of the Genomic Locus Encoding the Human LeukemiaAssociated Transcription Factor RUNX1/AML1,” Gene, Vol. 262, No. 1-2, 2001, pp. 23-33. doi:10.1016/S0378-1119(00)00532-1
- Y. Fukushima-Nakase, Y. Naoe, I. Taniuchi, H. Hosoi, T. Sugimoto and T. Okuda, “Shared and Distinct Roles Mediated through C-Terminal Subdomains of Acute Myeloid Leukemia/Runt-Related Transcription Factor Molecules in Murine Development,” Blood, Vol. 105, No. 11, 2005, pp. 4298-4307. doi:10.1182/blood-2004-08-3372
- P. Sroczynska, C. Lancrin, V. Kouskoff and G. Lacaud, “The Differential Activities of Runx1 Promoters Define Milestones during Embryonic Hematopoiesis,” Blood, Vol. 114, No. 26, 2009, pp. 5279-5289. doi:10.1182/blood-2009-05-222307
- S. Shankar, E. Rytina and N. P. Burrows, “Acute Myeloid Leukaemia Presenting with Eczema,” Clinical and Experimental Dermatology, Vol. 31, No. 4, 2006, pp. 593-594. doi:10.1111/j.1365-2230.2006.02136.x
- A. M. Hughes, T. Lightfoot, J. Simpson, P. Ansell, P. A. McKinney, S. E. Kinsey, et al., “Allergy and Risk of Childhood Leukaemia: Results from the UKCCS,” International Journal of Cancer, Vol. 121, No. 4, 2007, pp. 819-824. doi:10.1002/ijc.22702
- A. M. Linabery, A. M. Jurek, S. Duval and J. A. Ross, “The Association between Atopy and Childhood/Adolescent Leukemia: A Meta-Analysis,” American Journal of Epidemiology, Vol. 171, No. 7, 2010, pp. 749-764. doi:10.1093/aje/kwq004
- J. Rudant, L. Orsi, F. Menegaux, A. Petit, A. Baruchel, Y. Bertrand, et al., “Childhood Acute Leukemia, Early Common Infections, and Allergy: The ESCALE Study,” American Journal of Epidemiology, Vol. 172, No. 9, 2010, pp. 1015-1027. doi:10.1093/aje/kwq233
- R. Escher, P. Wilson, C. Carmichael, R. Suppiah, M. Liu, M. Kavallaris, et al., “A Pedigree with Autosomal Dominant Thrombocytopenia, Red Cell Macrocytosis, and an Occurrence of t(12:21) Positive Pre-B Acute Lymphoblastic Leukemia,” Blood Cells, Molecules, and Diseases, Vol. 39, No. 1, 2007, pp. 107-114. doi:10.1016/j.bcmd.2007.02.009
- K. A. Alford, K. Reinhardt, C. Garnett, A. Norton, K. Böhmer, C. von Neuhoff, et al., “Analysis of GATA1 Mutations in Down Syndrome Transient Myeloproliferative Disorder and Myeloid Leukemia,” Blood, Vol. 118, No. 8, 2011, pp. 2222-2238.
- M. Daneshpazhooh, T. Mohammad-Javad Nazemi, L. Bigdeloo and M. Yoosefi, “Mucocutaneous Findings in 100 Children with Down Syndrome,” Pediatric Dermatology, Vol. 24, No. 3, 2007, pp. 317-320. doi:10.1111/j.1525-1470.2007.00412.x
- Q. Zhu, M. Zhang, R. Blaese, J. Derry, A. Junker, U. Francke, et al., “The Wiskott-Aldrich Syndrome and X-Linked Congenital Thrombocytopenia Are Caused by Mutations of the Same Gene,” Blood, Vol. 86, No. 10, 1995, pp. 3797-3804.