Open Journal of Endocrine and Metabolic Diseases
Vol.3 No.1(2013), Article ID:28033,8 pages DOI:10.4236/ojemd.2013.31009
RU486 Reversal of Cortisol Repression of 1,25-Dihydroxyvitamin D3 Induction of the Human Osteocalcin Promoter
School of Medical Science, Griffith University, Gold Coast Campus, Gold Coast, Australia
Email: N.Morrison@griffith.edu.au
Received November 8, 2012; revised December 1, 2012; accepted January 12, 2013
Keywords: RU486; Osteocalcin; Vitamin D; Glucocorticoid
ABSTRACT
In conditions of corticosteroid excess, such as Cushing’s syndrome, a reduction in serum osteocalcin is observed and bone loss occurs. The human osteocalcin gene is induced by 1,25-dihydroxyvitamin D3 derivatives and repressed by glucocorticoids. In this paper we show that cortisol, a natural glucocorticoid, represses both basal and vitamin D induced activity of the human osteocalcin promoter. Furthermore, we address the specific question as to whether the anti-progestin anti-glucocorticoid RU486 is able to antagonize the inhibitory effect of cortisol on osteocalcin gene expression. We show that RU486 has agonist activity alone, in that it is able to repress the basal promoter activity of the osteocalcin gene and antagonist activity, reversing incompletely the cortisol mediated repression of 1,25-dihydroxyvitamin D3 induction.
1. Background
The compound 11β-[p-(Dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one, known as RU486 or mifepristone, is a synthetic anti-progesterone compound that also has strong anti-glucocorticoid properties [1] and is used occasionally for treatment for Cushing’s syndrome, a disease characterized by high circulating corticosteroid levels [2,3]. One of the clinical manifestations of corticosteroid excess is the development of osteopenia and osteoporosis [4,5]. A humoral marker of bone formation, the protein osteocalcin, is substantially reduced in corticosteroid osteoporosis [6]. Glucocorticoids are differentiation stimulators in cultured osteoblasts [5] and result in more mineralized matrix over reasonable time spans, using in vitro cell models [6]. In contrast, glucocorticoids can repress the ability of vitamin D3 metabolites to induce the osteocalcin gene in osteoblast-like cells in culture, presumably mimicking the situation of repressed osteocalcin in vivo [7]. Longerterm effects of glucocorticoids in vivo create negative effects in bone, including inhibition of osteoblast proliferation [8] and apoptosis of both osteoblasts and osteocytes [9]. Finally, Brennan-Speranza et al. [10], using osteoblast-specific gene knockout, show that the negative effects of glucocorticoids on energy balance in the entire body are mediated through osteoblast glucocorticoid receptor (GR), making the mode of action of anti-glucocorticoids in osteoblasts of particular relevance to glucocorticoid action in general.
Steroid hormones act as ligands by associating with nuclear receptor proteins that are members of the steroid-thyroid-retinoic acid and vitamin D receptor super family. The glucocorticoid receptor-ligand complex localizes in the promoter regions of target genes at specific sequences known as glucocorticoid responsive elements (GRE), and increased transcription of the gene results, leading to increased levels of the particular protein in question [11]. Although most of the studied receptorDNA interactions to date involve gene induction phenomena, a number of genes are thought to be repressed by the glucocorticoid receptor binding to promoter sites that overlap other strong activators: the osteocalcin promoter is thought to have a GRE that overlaps the TATA box [7,12], resulting in a model of repression by blocking of the TATA box site by GR. In such a hypothetical model, it is not yet established if the TATA box GRE is actively negative, or simply weakly positive (blocking the TATA box should result in net repression if binding of a powerful factor is replaced by a weaker factor). Recently, Surjit et al. [13] reported a previously unexpected mechanism of GR repression via negative glucocorticoid response elements (nGRE) that are quite distinct from the consensus positive GRE, suggesting that a reappraisal of all negative GR effects is warranted. Given the importance and evident complexity of glucocorticoid action in bone, it is reasonable to address highly focused specific hypotheses in building understanding of glucocorticoids: this study is directed at assessing in an acute treatment model of only 36 hours, whether the anti-hormone RU486 can reverse the glucocorticoid repression of 1,25- dihydroxyvitamin D3 (1,25(OH)2D3) mediated induction of osteocalcin using an in vitro model.
2. Materials and Methods
2.1. Hormone Reagents
RU486 is also known as mifepristone, and has a systematic International Union of Pure and Applied Chemistry (IUPAC) name of 11β-[p-(Dimethylamino)phenyl]-17β- hydroxy-17-(1-propynyl)estra-4,9-dien-3-one. Cortisol is also known as hydrocortisone and has the IUPAC name of (11β)-11,17,21-trihydroxypregn-4-ene-3,20-dione. Corticosterone has the IUPAC name of (11β)-11,21-dihydroxypregn-4-ene-3,20-dione. 1,25-dihydroxy-vitamin D3, (abbreviated in this paper as 1,25(OH)2D3) is also known as 1a,25 dihydroxyvitamin D3, 1a,25-Dihydroxycholecalciferol and calcitriol, and has the IUPAC name of (1R,3S)-5-[2-[(1R,3aR,7aS)-1-[(2R)-6-hydroxy-6-methyl-hetan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1H-inden-4-ylidene]ethylidene]-4-methylidene-cyclohexane-1,3-diol. Dexamethasone, also known as prednisolone F, has the IUPAC name of (8S,9R,10S,11S,13S,14S,16R,17R)-9- Fluoro-11,17-dihydroxy-17-(2-hydroxya-cetyl)-10,13,16- trimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one.
RU486 was obtained from Roussel-UCLAF (France). Cortisol, corticosterone, dexamethasone and 1,25(OH)2D3 were obtained from Sigma-Aldrich. Hormones were stored under nitrogen in ethanol at −20˚C in the dark and diluted appropriately prior to use.
2.2. Transfection and Cell Culture Procedures
Rat osteoblastic sarcoma cells ROS 17/2.8 were cultured using Ham’s F12 medium or Dulbecco’s modification of Eagle’s medium (DMEM) (Flow Laboratories) supplemented with 10% fetal calf serum (FCS) in a 5% carbon dioxide atmosphere at 37˚C. The plasmid construct pOSCAT2 [7] contains the promoter of the human osteocalcin gene driving the chloramphenicol acetyl transferase (CAT) gene. pSV2NEO contains the neomycin phosphotransferase gene driven by the SV40 virus major late promoter. pOSCAT2 and pSV2NEO were mixed equimolar and precipitated with calcium phosphate according to Gorman et al. [14]. Fifty mg of precipitated DNA was added per flask to five 150 cm2 flasks of ROS17/2.8 cells at 2 × 106 cells per flask, grown in DMEM augmented with 10% FCS. After six hours the cells were shocked with 10% glycerol in DMEM (plus 10% FCS) for one minute and the original medium replaced. Cells were left undisturbed for three days before the addition of antibiotic G418 (Life technologies) at 500 mg/ml, a concentration established to be toxic to ROS17/2.8 cells. Cells were mixed and re-plated after six days into Ham’s F12 medium (plus 10% FCS) supplemented with ROS17/2.8 conditioned medium (50%), containing G418. Cells were re-fed conditioned selective medium every three days to remove dead cells and replated into smaller flasks at weekly intervals. After three weeks, a pool of G418 resistant cells had developed which had a high plating efficiency in the presence of G418. These cells have a high level of CAT activity that was inducible by 1,25(OH)2D3 and were used in hormone treatment experiments. In the pool of selected cells, G418 selection was not necessary to maintain either G418 resistance or CAT activity, which has remained constant for over a year in the absence of G418 selection.
2.3. Hormone Treatment of Cultured Cells
Transfected cells were expanded in Hams-F12 medium (plus 10% FCS) in 150 cm2 flasks. Two days prior to harvesting for hormone treatments, medium was replaced with DMEM supplemented with 2% charcoal stripped FCS (CS-FCS, Life technologies). Cells were harvested, washed and re-plated into six well plates at a density of 2.5 × 105 cells per well in 2.5 ml of DMEM (plus 2% CS-FCS) medium. Hormone treatments were compared to appropriate controls of the vehicle (ethanol) at the same concentration. 2.5 ml of hormone concentrate or vehicle was added to the well after the cells had plated down. Cells were incubated at 37˚C in 5% CO2 for 36 hours prior to harvest.
2.4. Chloramphenicol Acetyl Transferase Assays
CAT activity assays were based on the transfer of carbon-14 labeled acetyl groups from 14C acetyl-CoA (Perkin Elmer) to unlabelled chloramphenicol [15]. Cells were harvested by trypsinization, recovered in DMEM (plus 10% FCS) medium, washed in phosphate buffered saline and lysed by three cycles of freeze-thaw for CAT assay in a volume of 100 ml of which 10 ml was used to assay CAT activity for one hour at 37˚C in a final reaction volume of 100 ml. Three sequential organic extractions in 100 ml ice-cold ethyl acetate were used to separate labeled chloramphenicol from 14C acetyl-CoA. Prior to counting, an aqueous wash of 100 ml phosphate buffered saline was done and the organic phase carefully separated and counted in scintillation fluid using the carbon- 14 channel of a Packard liquid scintillation counter. All hormone treatments were for 36 hours (in triplicate) and data is expressed as the mean ± standard error of the mean of disintegrations per minute (dpm) of radioactive acetyl groups transferred from 14C acetyl-CoA to chloramphenicol per hour per 106 cells.
2.5 In Silico Sequence Analysis
The web based transcription element search system (TESS) computer program [16] was used to search for motifs related to glucocorticoid receptor binding sites. In order to identify nGRE similar to those described by Surjit et al. [13] the simple motifs CTCC and GAGG were used.
3. Results
3.1. Corticosteroid and RU486 Repression of Osteocalcin Promoter Basal Activity
Dexamethasone is a powerful synthetic glucocorticoid known to repress vitamin D induction of human osteocalcin promoter activity [7]. Cortisol is a natural glucocorticoid and the primary circulating glucocorticoid in humans, while corticosterone is the primary physiological glucocorticoid in rodents. These three agents were compared with RU486 in order to establish if basal, un-induced osteocalcin promoter activity could be affected within a short (36 hours) treatment window. Transfected cells were treated with increasing concentrations of these agents (Figure 1) and CAT activity measured as a surrogate for promoter activity. The three glucocorticoids resulted in sensitive repression of osteocalcin basal promoter activity in a manner consistent with dexamethasone having a higher affinity for the GR than cortisol or corticosterone. Despite the differences in the concentration dependence of the repression of basal activity, all three steroids attained the same extent of suppression at maximal concentrations, reducing osteocalcin basal promoter activity to about 40% of the vehicle treated control. The anti-hormone RU486, which might have been expected to have no effect, had a suppressive effect as well, that was detected at reasonably low concentrations (10–8 and 10–9 M). In contrast to dexamethasone and cortisol, the repression effect of RU486 on the osteocalcin promoter did not attain the same magnitude as the other authentic agonists (Figure 1).
3.2. Cortisol Repression of 1,25(OH)2D3 Induction of the Osteocalcin Promoter
Considering that cortisol represses osteocalcin promoter basal activity in transfected cells, the effect of cortisol on
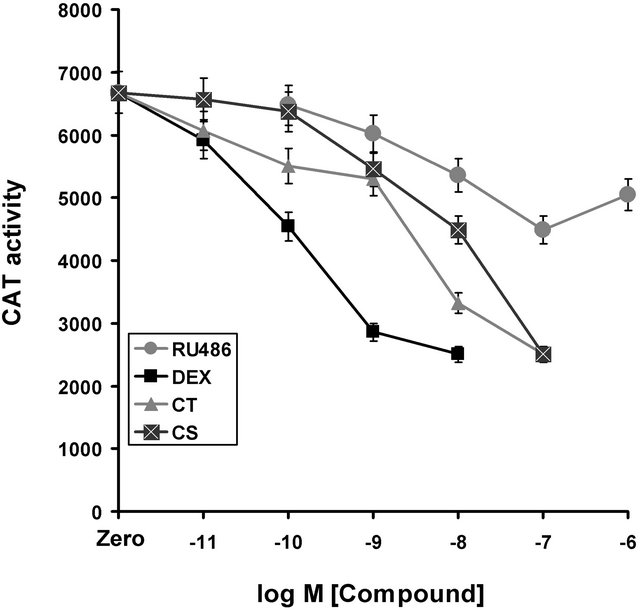
Figure 1. Repression of osteocalcin promoter basal activity by glucocorticoids and RU486. Zero indicates CAT activity in the presence of appropriate vehicle. Dexamethasone (Dex) and cortisol (CT) have significant repression at very low concentrations (10−11 M) whereas higher concentrations of corticosterone (CS) are required for appreciable repression (10−9 M). In addition, DEX, CT and CS all achieve the same final extent of repression. On the other hand, RU486 does not achieve the same final extent of repression, despite having a high affinity for the glucocorticoid receptor and showing progressive repression at reasonably low concentrations (10−10 M), suggesting that the mechanism of repression is fundamentally different and self limiting.
1,25(OH)2D3 induction was investigated. Dose response curves of 1,25(OH)2D3 mediated induction (with concentrations from 10−12 to 10−8 M) were established at increasing cortisol concentrations with hormones added simultaneously. The control experiment of 1,25(OH)2D3 induction (Figure 2) shows around five-fold induction within 36 hours of treatment with 1,25(OH)2D3 and a saturated induction at 10−9 M, with a half maximal dose of between 10–11 and 10–10 M (Figure 2). Increasing concentrations of cortisol have a profound repressive effect on 1,25(OH)2D3 induction, in keeping with represssion of basal activity seen already (Figure 1). Cortisol at 10–8 M results in an effective halving of the 1,25(OH)2D3 mediated promoter induction, meaning that physiologically sensible levels of cortisol within a short time frame could suppress 1,25(OH)2D3 mediated promoter induction. Furthermore, cortisol at 10–7 and 10–6 M results in virtual shut down of vitamin D effects on the osteocalcin promoter. At the lower concentrations of 1,25(OH)2D3, the repression overcomes induction to such an extent that the level of CAT activity is reduced below the control. Therefore, in the presence of 10–7 M cortisol a concentration of 1,25(OH)2D3 of 10–10 M is required merely to compensate the gene promoter activity to the normal level.
3.3. RU486 Effect on Cortisol Repression
RU486 had a repressive effect on osteocalcin promoter basal activity: therefore the action of RU486 on cortisolmediated repression of vitamin D induction could not be
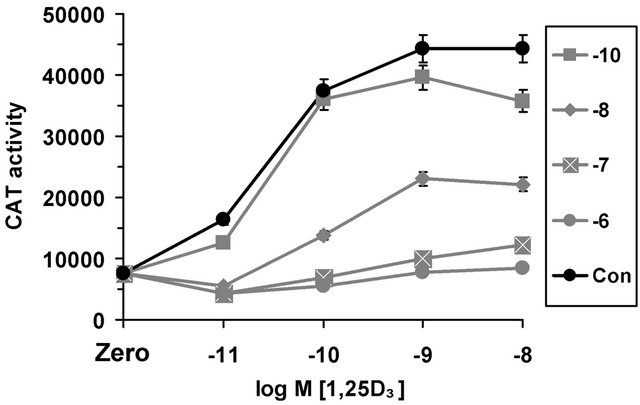
Figure 2. Cortisol repression of 1,25(OH)2D3 (1,25D3) induction of the human osteocalcin promoter. The horizontal axis indicates increasing concentrations of 1,25D3. Zero indicates no treatment with cortisol or 1,25D3, representing the basal promoter activity in the presence of the appropriate vehicle concentration. Con indicates no treatment with cortisol, but in the presence of 1,25D3 and appropriate vehicle. Vitamin D mediated induction in the presence of increasing concentrations of cortisol are indicated on the graph by different lines. Symbols –10, –8, –7, –6 in the boxed graph legend indicate the concentration of cortisol used for each associated line as log M [cortisol].
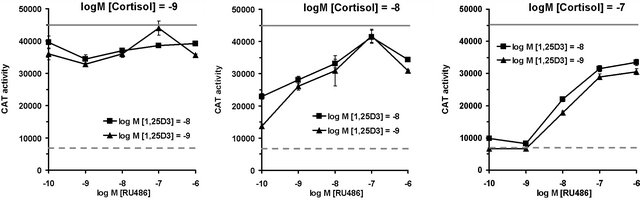
predicted. Although RU486 exhibited weak agonist activity on osteocalcin basal activity (Figure 1), there was virtually no repression of 1,25(OH)2D3 induced CAT activity by RU486 at 10–8 M and 10–7 M concentrations and only a minor effect (activity at 91%) when used at 10–6 M, in marked contrast to dexamethasone and cortisol (Figure 3). Multiple dose response curves of the interacttion between RU486, cortisol and 1,25(OH)2D3 were established (Figure 4) at the following concentrations: 1,25(OH)2D3 was at 10–10 and 10–9 M; RU486 varied from 10–10 M to 10–6 M; cortisol varied from 10–10 M to 10–6 M. As expected, low cortisol concentrations (10–9 M), resulted in weak repression of 1,25(OH)2D3 induction (Figure 4(a)): surprisingly addition of a small amount of RU486 (compare 10–10 to 10–9 M RU486 at both concentrations of 1,25(OH)2D3 resulted in slightly more inhibition. As RU486 concentration was increased, however, repression by cortisol was relieved. At higher concentrations of cortisol (10–8 M, Figure 4(b)) the reversal of cortisol repression mediated by RU486 is more obvious, since repression was a more potent. Increasing RU486 results in dose-dependent steady increase in CAT activity, due to reversal of cortisol repression of 1,25(OH)2D3 induction (Figure 4(b)). However, it is notable that the relief of repression curve alters direction at 10–6 M RU486, becoming inhibitory again (Figure 4(b)). This suggests that lower concentrations of RU486 act as a pure antagonist while very high levels of RU486 may act as a GR agonist. At the highest concentrations of cortisol tested in this experiment (10–7 M, Figure 4(c)), RU486 shows a sigmoidal curve of relief of cortisol repression with apparent saturation of the effect at 10–7 to 10–6 M RU846. However, a notable feature of this graph (Figure 4(c)) is that RU486 does not compensate the osteocalcin promoter activity back to 100% activity: the
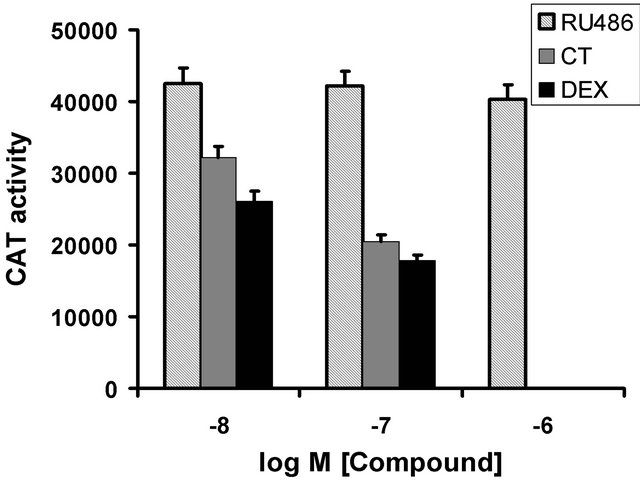
Figure 3. Effect of RU486 compared to dexamethasone (DEX) and cortisol (CT) on osteocalcin promoter activity induced by 1,25(OH)2D3 (10–8 M). All compounds were added si- multaneously. DEX and CT were not done at 10–6 M. RU486 has a weak effect on the induced promoter.
 (a) (b) (c)
(a) (b) (c)
Figure 4. Incomplete RU486 mediated reversal of cortisol repression of 1,25(OH)2D3 (1,25D3) induction of the human osteocalcin promoter measured by CAT activity in transfected cells. Cortisol at 10–9 M (graph (a)), 10–8 M (graph (b)) or 10–7 M (graph (c)) was used to repress 1,25D3 (10–10 M and 10–9 M shown by squares and triangles, respectively) mediated induction of osteocalcin promoter activity. In order to provide a reference for basal and induced activities, in each graph the basal promoter activity in the un-induced state is shown by the dashed line while the maximal level of induction of the osteocalcin promoter represented by treatment with 1,25D3 at 10–9 M in the absence of any other treatment is shown by the gray complete line. Graph (c) shows that in the presence of cortisol at 10–7 M, RU486 at any concentration is unable to compensate the promoter back to the original level of 1,25D3 mediated induction (gray line) indicating that the relief of repression is limiting as RU486 concentrations increase. In other words, the concentrations of RU486 required to antagonize such high levels of cortisol are themselves inhibitory.
level expected for complete reversal of cortisol repression. Rather, the osteocalcin promoter activity in the presence of RU486 at 10–6 M, cortisol at 10–7 M and 1,25(OH)2D3 at 10–9 M does not exceed approximately 75% of the activity of 1,25(OH)2D3 alone. Although speculative, the inability of RU486 to totally relieve the repression effected by cortisol at 10–7 M may indicate that the system is limited to a new set point.
3.4. In Silico Derived Promoter Sequence Candidates for nGRE Activity
The osteocalcin promoter sequence was searched for a classical GRE (motif shown in Figure 5(a)) and recently defined active nGRE sequences (based on the motif CTCC or GGAG, Figure 5(b)). Simple search motifs were used search the construct sequence for both canonical GRE and recently described active nGRE aided a transcription element search system (TESS) computer program [16]. A total of 31 perfect CTCC and GAGG motifs were identified in the osteocalcin promoter sequence present in the construct. None of these were arranged where half-sites were in proximity as inverted repeats as described by Surjit et al. [13]. Of these, a single half site exists within a sequence adjacent to a defined RUNX2 binding site. Sequences that matched consensus glucocorticoid or progesterone receptor binding sites (GRE/PRE) were found using TESS (Figure 5), including the sequence overlapping the TATA box that matched prior data [7,12]. The vitamin D response element (VDRE) did not overlap or contain candidate nGRE motifs (Figure 5(b)).
4. Discussion
This study shows that cortisol has profound negative effects on the human osteocalcin promoter, when assayed in the rat osteoblast model cell line ROS17/2.8. Most research has focused on synthetic glucocorticoids such as dexamethasone, since these as prescribed, have longer half-lives and some are more resistant to metabolism. Cortisol is the natural glucocorticoid that is the primary driver of Cushing’s syndrome and other forms of natural glucocorticoid excess. These data confirm that the human osteocalcin promoter is a good model for anti-hormone effects in the study of physiologically relevant glucocorticoids, such as cortisol and corticosterone, in bone cells. Under the conditions used here, profound gene expression changes were seen after a reasonably short exposure to hormone (36 hours in this case). Significant repression was observed at nanomolar cortisol levels, certainly within the range of circulating concentrations of glucocorticoid excess syndromes such as Cushing’s syndrome. Under these conditions, with the natural glucocorticoid cortisol, RU486 was able to exert relief of cortisol repression of the 1,25(OH)2D3 induction of the osteocalcin promoter.
Despite relieving cortisol repression of 1,25(OH)2D3 induction, RU486 could not compensate the promoter to full activity. RU486 itself at high concentrations appeared to have a repressive effect, where a partial agonist activity of RU486 may overcome antagonist function.

Figure 5. Potential glucocorticoid/progesterone receptor (GR/PR, respectively) binding sites in the osteocalcin promoter. (a) An example of the sequence of glucocorticoid response element (GRE) consensus motif used to search the osteocalcin promoter with the program TESS: the relative height of the particular base signifies the relative abundance in GR/PR binding sites. (b) Sequence of the human osteocalcin promoter construct used in this study with the location of GRE consensus candidate sites identified by TESS (dashed lines) and nGRE half-site motifs (either CTCC or GAGG, over shaded in gray). The size of the putative GRE binding site sequence and the strength of the TESS score is indicated by the single and double (stronger score) dashed lines. The half sites of the direct repeat vitamin D response element (VDRE) with a three base gap are underlined: note that no nGRE candidates occur within the VDRE. A canonical RUNX2 binding site is in underlined in italics: note that a CTCC motif is immediately upstream of the RUNX2 binding site. The TATA box is over-boxed and an extended GRE sequence overlaps the TATA box. Sequence downstream of the TATA box is given in lower case. GR, PR and GR_a mean glucocorticoid receptor, progesterone receptor and glucocorticoid receptor alpha, respectively. Those sites marked GR/PR are detected by TESS sequence matrix I00104 and/or Q00076, representing six nucleotide hormone response element half sites: sites marked GR are detected by TESS matrix I00279 representing a 9-mer sequence; the site marked GR, GR_a is detected by matrix M00205, shown graphically in (a) which is a 16-mer sequence.
Such partial agonist activity of RU486 has been demonstrated on the progesterone receptor [17] and GR [18]. In our data, partial agonist activity was reflected in repression of basal promoter activity (in the absence of 1,25(OH)2D3) in a manner similar to that observed in cortisol treatment although requiring higher concentrations of RU486 and having a lower final effect. Despite the low final repression of basal promoter activity attained by RU486 treatment alone, the agonist activity was apparent at quite low concentrations, such as 10–10 M, presumably associated with the fact that RU486 has a high affinity for the glucocorticoid receptor, reported to be three times greater than that of dexamethasone [1]. Therefore, the action of RU486 on the osteocalcin promoter may be self-limiting in that the agonist activity ultimately reduces the capacity of RU486 to fully reverse cortisol repression of osteocalcin promoter induction by 1,25(OH)2D3. Furthermore, the GR exists in two prominent isoforms in humans, a canonical GRa and a dominant negative GRb that are products of differential splicing; however, GRb binds RU486 but apparently no other of 57 tested potential ligands [19]. The GRb:RU486 complex is an active inducer of some target genes [19] suggesting that agonist activity may be derived from GRb. The status of GRb in the rat cell line ROS17/2.8 is not yet confirmed, although since a mouse GRb has been identified that is functionally equivalent to the human GRb, it seems likely that GRb will be a universal GR variant [20]. A human osteoblast model system is an alternative for further study; unfortunately a convenient and well-characterized human model similar to the rat ROS17/2.8 model is currently not available [21]. Experiments in primary human osteoblasts are a logical next step in understanding the action of RU486 on bones.
It seems reasonable that in rats as well as humans, agonist activity of RU486 is mediated through a GRblike isoform, and that antagonist action is through the equivalent GRa. Since cortisol at 10–7 M was maximally effective at suppressing the osteocalcin promoter (see Figure 2) we conclude that the glucocorticoid receptor (rat GRa) is saturated by cortisol at concentrations (10–7 M) maximally effective for repression of osteocalcin promoter activity. Human GRb does not bind cortisol, but is able to bind RU486 [19]. If rat GRb is similar the human GRb, then at high RU486 concentrations, it may be possible to observe antagonist activity mediated by RU486 through GRa and partial agonist activity through GRb. This may explain why RU486 does not fully recover cortisol mediated repression of 1,25(OH)2D3 induction of the osteocalcin promoter. Therefore the action of RU486 on reversing cortisol repression may be selflimiting through the presence of partial agonist activity. This question needs to be resolved in further work on the contribution of GRa and GRb to RU486 relief of repression of osteocalcin induction by 1,25(OH)2D3.
A new paradigm for DNA based glucocorticoid repression was described recently, directed by specific negative glucocorticoid receptor response elements (nGRE) that act in an active manner, with a response element sequence unrelated to the classic positive GRE [13]. Although a number of these newly defined nGRE half-site motifs (CTCC and GAGG) exist within the osteocalcin promoter, none are of the configurations described by Surjit et al. [13], where an inverted repeat of the half-sites with a 0, 1 or 2 base pair spacing was proposed as the active nGRE. Using computer searches of the osteocalcin promoter sequence, a number of candidate half sites were identified. However, none of these predicted canonical GRE half-sites or the newly defined nGRE sites, presents as a better candidate mechanism than the original hypothesis of the glucocorticoid receptor binding site overlapping the TATA box [7,12,22]. In any case, a reasonable hypothesis of steric hindrance related to the activity of glucocorticoid receptor on the TATA box of the osteocalcin promoter has remained unchallenged through time. In the data presented here, the anti-hormone was able to cause repression of basal activity; a condition consistent with direct binding of RU486 to the GR in an agonist mechanism (possibly GRb). Regardless of how cortisol represses 1,25(OH)2D3 induced promoter activity, RU486 was able to relieve repression of low to reasonable concentrations of cortisol, but when cortisol concentrations were sufficiently high the RU486 concentrations required for antagonism were self limiting. These data suggest benefit from RU486 as a selective anti-glucocorticoid in the treatment of bone related problems of cortisol excess syndromes might have the greatest effects on milder cases.
5. Acknowledgements
This study was supported by the National Health and Medical Research Council of Australia.
REFERENCES
- I. Jung-Testas and E. E. Baulieu, “Inhibition of Glucocorticosteroid Action in Cultured L-929 Mouse Fibroblasts by RU 486, a New Anti-Glucocorticosteroid of High Affinity for the Glucocorticosteroid Receptor,” Experimental Cell Research, Vol. 147, No. 1, 1983, pp. 177-182. doi:10.1016/0014-4827(83)90282-3
- F. Castinetti, T. Brue and B. Conte-Devolx, “The Use of the Glucocorticoid Receptor Antagonist Mifepristone in Cushing's Syndrome,” Current Opinion in Endocrinology, Diabetes and Obesity, Vol. 19, No. 4, 2012, pp. 295-299.
- M. Fleseriu and S. Petersenn, “Medical Management of Cushing’s Disease: What Is the Future?” Pituitary, Vol. 15, No. 3, 2012, pp. 330-341. doi:10.1007/s11102-012-0397-5
- T. Mancini, M. Doga, G. Mazziotti and A. Giustina, “Cushing’s Syndrome and Bone,” Pituitary, Vol. 7, No. 4, 2004, pp. 249-252. doi:10.1007/s11102-005-1051-2
- G. Mazziotti, A. Angeli, J. P. Bilezikian, E. Canalis and A. Giustina, “Glucocorticoid-Induced Osteoporosis: An Update,” Trends in Endocrinology and Metabolism, Vol. 17, No. 4, 2006, pp. 144-149. doi:10.1016/j.tem.2006.03.009
- S. Minisola, R. D. Fiacco, S. Piemonte, M. Iorio, M. L. Mascia, F. Fidanza, C. Cipriani, I. Raso, M. L. Porfiri, C. M. Francucci, E. D’Erasmo and E. Romagnoli, “Biochemical Markers in Glucocorticoid-Induced Osteoporosis,” Journal of Endocrinological Investigation, Vol. 31, No. 7, 2008, pp. 28-32.
- N. A. Morrison, J. Shine, J. C. Fragonas, V. Verkest, M. L. McMenemy and J. A. Eisman, “1,25-Dihydroxy-Vitamin D-Responsive Element and Glucocorticoid Repression in the Osteocalcin Gene,” Science, Vol. 246, No. 4934, 1989, pp. 1158-1161. doi:10.1126/science.2588000
- D. Hong, H. X Chen, H. Q. Yu, C. Wang, H. T. Deng, Q. Q. Lian and R. S. Ge, “Quantitative Proteomic Analysis of Dexamethasone-Induced Effects on Osteoblast Differentiation, Proliferation, and Apoptosis in MC3T3-E1 Cells Using SILAC,” Osteoporosis International, Vol. 22, No. 7, 2011, pp. 2175-2186. doi:10.1007/s00198-010-1434-8
- R. S. Weinstein, R. L. Jilka, A. M. Parfitt and S. C. Manolagas, “Inhibition of Osteoblastogenesis and Promotion of Apoptosis of Osteoblasts and Osteocytes by Glucocorticoids. Potential Mechanisms of Their Deleterious Effects on Bone,” Journal of Clinical Investigation, Vol. 102, No. 2, 1998, pp. 274-282. doi:10.1172/JCI2799
- T. C. Brennan-Speranza, H. Henneicke, S. J. Gasparini, K. I. Blankenstein, U. Heinevetter, V. C. Cogger, D. Svistounov, Y. Zhang, C. J. Cooney, F. Buttgereit, C. R. Dunstan, C. Gundberg, H. Zhou and M. J. Seibel, “Osteoblasts Mediate the Adverse Effects of Glucocorticoids on Fuel Metabolism,” Journal of Clinical Investigation, Vol. 122, No. 11, 2012, pp. 4172-4189. doi:10.1172/JCI63377
- N. C. Nicolaides, Z. Galata, T. Kino, G. P. Chrousos and E. Charmandari, “The Human Glucocorticoid Receptor: Molecular Basis of Biologic Function,” Steroids, Vol. 75, No. 1, 2010, pp. 1-12. doi:10.1016/j.steroids.2009.09.002
- P. E. Strömstedt, L. Poellinger, J. A. Gustafsson and J. Carlstedt-Duke, “The Glucocorticoid Receptor Binds to a Sequence Overlapping the TATA Box of the Human Osteocalcin Promoter: A Potential Mechanism for Negative Regulation,” Molecular and Cellular Biology, Vol. 11, No. 6, 1991, pp. 3379-3383.
- M. Surjit, K. P. Ganti, A. Mukherji, T. Ye, G. Hua, D. Metzger, M. Li and P. Chambon, “Widespread Negative Response Elements Mediate Direct Repression by Agonist-Liganded Glucocorticoid Receptor,” Cell, Vol. 145, No. 2, 2011, pp. 224-241. doi:10.1016/j.cell.2011.03.027
- C. M. Gorman, B. H. Howard and R. Reeves, “Expression of Recombinant Plasmids in Mammalian Cells Is Enhanced by Sodium Butyrate,” Nucleic Acids Research, Vol. 11, No. 21, 1983, pp. 7631-7648. doi:10.1093/nar/11.21.7631
- M. J. Sleigh, “A Nonchromatographic Assay for Expression of the Chloramphenicol Acetyltransferase Gene in Eucaryotic Cells,” Analytical Biochemistry, Vol. 156, No. 1, 1986, pp. 251-256.
- J. Schug and G. C. Overton, “TESS: Transcription Element Search Software on the WWW. Technical Report CBIL-TR-1997-1001-v0.0,” Computational Biology and Informatics Laboratory, School of Medicine University of Pennsylvania, 1997. http://www.cbil.upenn.edu/tess
- S. E. Wardell, R. Narayanan, N. L. Weigel and D. P. Edwards, “Partial Agonist Activity of the Progesterone Receptor Antagonist RU486 Mediated by an Amino-Teminal Domain Coactivator and Phosphorylation of Serine400,” Molecular Endocrinology, Vol. 24, No. 2, 2010, pp. 335- 345. doi:10.1210/me.2008-0081
- M. Schulz, M. Eggert, A. Baniahmad, A. Dostert, T. Heinzel and R. Renkawitz, “RU486-Induced Glucocorticoid Receptor Agonism Is Controlled by the Receptor N Terminus and by Corepressor Binding,” Journal of Biological Chemistry, Vol. 277, No. 29, 2002, pp. 26238- 26243. doi:10.1074/jbc.M203268200
- L. J. Lewis-Tuffin, C. M. Jewell, R. J. Bienstock, J. B. Collins and J. A. Cidlowski, “Human Glucocorticoid Receptor Beta Binds RU-486 and Is Transcriptionally Active,” Molecular and Cellular Biology, Vol. 27, No. 6, 2007, pp. 2266-2282. doi:10.1128/MCB.01439-06
- T. D. Hinds Jr., S. Ramakrishnan, H. A. Cash, L. A. Stechschulte, G. Heinrich, S. M. Najjar and E. R. Sanchez, “Discovery of Glucocorticoid Receptor-Beta in Mice with a Role in Metabolism,” Molecular Endocrinology, Vol. 24, No. 9, 2010, pp. 1715-1727. doi:10.1210/me.2009-0411
- E. M. Czekanska, M. J. Stoddart, R. G. Richards and J. S. Hayes, “In Search of an Osteoblast Cell Model for in Vitro Research,” European Cells and Materials Journal, Vol. 24, 2012, pp. 1-17.
- N. Morrison and J. Eisman, “Role of the Negative Glucocorticoid Regulatory Element in Glucocorticoid Repression of the Human Osteocalcin Promoter,” Journal of Bone and Mineral Research, Vol. 8, No. 8, 1993, pp. 969- 975. doi:10.1002/jbmr.5650080810