Open Journal of Obstetrics and Gynecology
Vol.3 No.1(2013), Article ID:26502,4 pages DOI:10.4236/ojog.2013.31009
Vulvar sarcomas: A 25 plus-year experience from Queensland
![]()
1Queensland Centre for Gynaecological Cancer, University of Queensland, Brisbane, Australia
2Women’s and Newborn Services, Royal Brisbane and Women’s Hospital, Herston, Australia
Email: *ian_jones@health.qld.gov.au
Received 23 July 2012; revised 25 August 2012; accepted 2 September 2012
Keywords: Vulvar Sarcoma; Diagnosis; Management; Outcome; Follow up
ABSTRACT
Objective: To review the characteristics of cases of vulvar sarcomas (VS) referred to the Queensland Centre for Gynaecological Cancer (QCGC) between mid 1984 and mid 2012. Methods: VS case data from QCGC were reviewed and analysed using the computer software Statistical Package for the Social Sciences (SPSS) 11.0. Results: Of the nine cases three died of their disease, one died of ovarian cancer and five are still alive and disease free. The mean age at diagnosis was 54.8 years. Time from onset of symptoms to diagnosis averaged one month. All diagnoses were confirmed histologically. The most common presenting complaint was a lump. Treatment in all but one case involved surgical excision, one case had chemotherapy alone and two cases had surgery followed by radiotherapy. Conclusions: To talk of five year survival for patients with such a rare cancer, so many histological types with variable cell differentiation plus the unknown of when sarcomatous cells metastasize from even early FIGO staged lesions is in our opinion taking generalisation to far. A multidisciplinary approach to management achieves the best results for each individual and pooling multicentre data will improve our understanding of this rare disease and benefit future patients.
1. INTRODUCTION
Primary soft tissue sarcomas form less than two to three percent of all female genital tract cancers [1] and one to three percent of all vulvar cancers [2]. In general, vulvar sarcomas are characterised by local recurrences and distant haematogenous metastases [3].
The aim of the study was to review the clinical features, diagnosis, management and outcomes from the patient records of cases of SV held in the Queensland Centre for Gynaecological Cancer (QCGC) data base and determine if the type of lesion influenced management and altered patient outcome.
2. MATERIALS AND METHODS
Case records, including pathology reports from the state wide QCGC data base for patients referred for registration and management of vulvar sarcoma (VS) between July 1984 and July 2012 were reviewed and abstracted relative to past surgical, medical and family history for cancer plus presenting symptoms, duration of symptoms prior to the commencement of treatment, clinical findings, FIGO staging, treatment, histological findings and long term follow-up. In addition information was sought for conditions that are known to be associated with vulvar carcinomas to see if they were also associated with VS. Such conditions included the changes of human papilloma virus, vulvar in-situ carcinoma, cervical carcinoma in-situ, vulvar dystrophy, cigarette smoking, past history of cancer and a family history of cancer for each case. This information was analysed using the computer software Statistical Package for the Social Sciences (SPSS) 11.0. Where data was found to be incomplete attempts were made to collect this information by contacting referring doctors, reviewing patient case notes and pathology department records.
Ethics approval for the review of case records was obtained from the Clinical Research Ethics Committee of the Royal Brisbane and Women’s Hospital.
3. RESULTS
The incidence of VS was 1.2% of all vulvar carcinomas recorded in the QCGC register between mid 1984 and mid 2012. Nine cases of vulvar sarcoma (VS) were registered with QCGC during the study period. The frequency of cases registered each year ranged from zero to two. Of the nine cases three died of their disease, one died
Table 1. Vulvar sarcomas—Type, treatment and outcome.
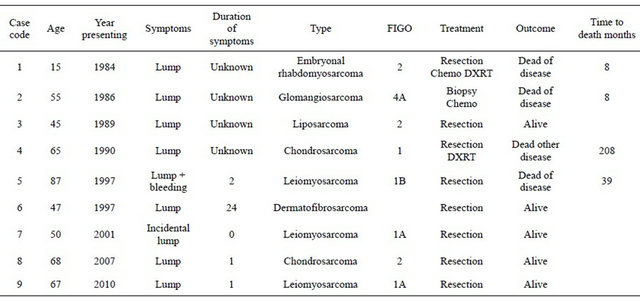
of a non related condition with no sign of VS, and five are still alive and disease free. The mean age at diagnosis was 54.8 years (range 15 - 87). If the 15 year old patient’s data was excluded the mean age at diagnosis was 65.5 years. The recorded time from onset of symptoms to diagnosis averaged one month. The most common presenting complaint was a lump. Three cases had leiomyosarcomas, two chondrosarcomas and there was one case each of embryonal rhabdomyosarcoma, glomangiosarcoma, liposarcoma and dermatofibrosarcoma. All diagnoses made histologically.
Presenting complaints were a painless lump in six cases, painful vulvar lump in one case, a bleeding lump in one case and an incidental finding of a lump in another case. The size of the lesions where documentation had occurred ranged from 2.4 to 8 cm (mean 4.5 cm). Management was dictated by the extent of each patient’s disease. In one case of extensive disease following biopsy she was treated with chemotherapy but died of disease eight months later. All the other cases underwent radical surgical excision of their lesions. Subsequent treatment regimes were again determined by the type and extent of disease. The 15-year-old patient with the embryonal rhabdomyosarcoma received chemo-radiation therapy following surgical excision and another patient received a surgical excision followed by radiation therapy.
Histological type and patient outcome is shown in Table 1. Three cases of VS died of their disease at eight, eight and 39 months after diagnosis. Their lesions were in order, embryonal rhabdomyosarcoma, glomangiosarcoma and leiomyosarcoma.
One case of leiomyosarcoma also had an area of lichen sclerosus found histologically but no other condition known to be associated with patients with vulvar carcinoma was found in any of the eight remaining cases of VS.
In the more recent cases immunohistochemical stains were used to assist in differentiating between types of sarcoma. Table 2 lists the outcome for the various types of stain used to confirm the diagnoses of leiomyosarcoma in three cases. Reverse transcriptase-polymerase chain reaction was not used in any of the nine cases described.
Follow up consisted of a post operative review between three to six weeks later, third monthly reviews for at least a year, extending to six months for two years and
Table 2. Results from immunohistochemistry testing of three leiomyosarcomas.
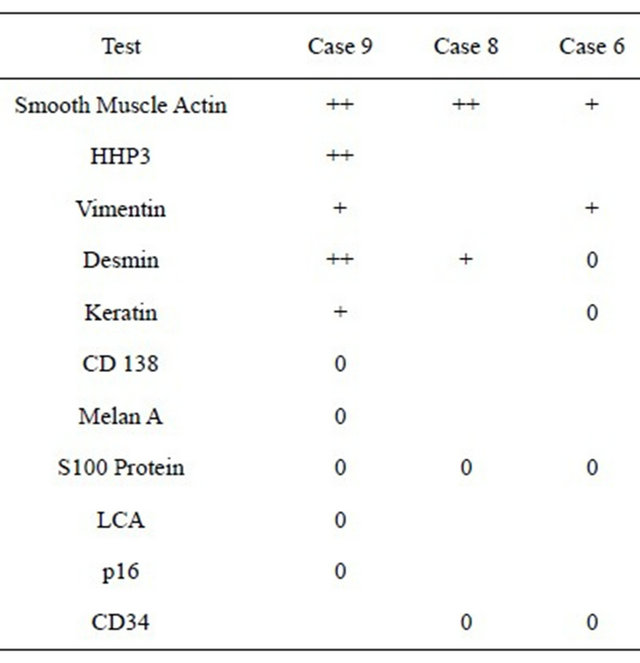
Key: Empty spaces in Table 2 mean that the test was not performed.
then yearly reviews for five years with a total of ten years follow up before being discharged from specialist care. At each visit an up dated history was followed by a general and gynaecological examination. Abnormal findings were investigated using appropriate radiology (Chest X-Ray, Computerised Tomography, and Magnetic Resonance Imaging) and pathology including Fine Needle Biopsy of suspicious masses. Initially follow up was provided in Brisbane or at a satellite gynaecological oncology centre to be referred back to a generalist gynaecologist if the patient lived a long way from such centres.
4. DISCUSSION
Vulvar sarcomas (VS) are rare tumors of mesenchymal origin that form a heterogeneous and complex group of lesions. Lesions can show a broad range of differentiation where histological classification is based upon morphologic demonstration of a specific line of differentiation. This results in the assignment of lesions to one of many categories. Categories include leiomyosarcomas, embryonal rhabdomyosarcomas (also known as sarcoma botryoides), liposarcomas, chondrosarcomas, epithelioid sarcomas, dermatofibrosarcomas, angiosarcomas, neurosarcomas, glomangiosarcomas and malignant fibrous histocytoma to mention most types found. Of these leiomyosarcomas, rhabdomyosarcomas, angiosarcomas, neurofibrosarcomas, malignant fibrohistiocytomas and aggressive angiomyxomas are the types most frequently found [4]. In a study of 47 vulvar sarcomas 25 were leiomyosarcomas, nine dermatofibrosarcomas, eight epithelioid sarcomas and five malignant fibrous histocytomas [5].
Immunohistochemistry was used in three cases to assist the pathologist to rule out nonmesenchymal tumors and to define the mesenchymal cell lineage. A useful reference detailing how immunohistochemistry assists the pathologist to make the correct diagnosis is provided by Heim-Hall and Yohe [6].
Medically induced immunosuppression with for example patients with renal transplants are known to predispose patients to some types of cancer [7]. None of the study patients had a past history of previous treatment with immunosuppressives. Apart from the one case of leiomyosarcoma who was found histologically to have an area of lichen sclerosus found on the excision specimen no other condition known to be associated with patients with vulvar carcinoma was found in any of the eight remaining cases of VS. One of the two cases of chondrosarcoma died of an unrelated non malignant condition. Hence we were unable to find any condition that increased a patients’ chance of developing a VS, which was not surprising considering the small study sample.
There were four cases worthy of further discussion. In the case with the rhabdomyosarcoma surgery was initially a wide local excision but histology demonstrated incomplete excision. A week later a hemi-vulvectomy was performed with no further persistence of disease being detected histologically. A month later chemotherapy was commenced followed five weeks later by radiotherapy. Despite all this treatment she died six months later. In the case of the patient with the glomangioma she had disease extending to the pelvic side wall and so after biopsy she was treated with chemotherapy. The 1997 case (case five), who had a leiomyosarcoma had been successfully treated for a stage 1B adenocarcinoma of the endometrium and a stage 1B ovarian carcinoma in 1982, 15 years before being diagnosed with her VS. Her VS was treated with a radical vulvectomy and bilateral groin dissections. No lymph nodes were involved and the excision margins were clear. Follow up was uneventful for three years and then she died of cerebral metastases. The 1990 case (case four) of chondrosarcoma had been treated for a 1B endometrial carcinoma in 2001 and had made an uneventful recovery. Routine follow up found a mobile multilobulated tumour in the mons pubis. Initially this was thought to be a recurrence of her endometrial cancer but histology proved otherwise. She was treated by a wide local excision and continues to be followed up and to date there is no sign of recurrence from either cancer. In summary in some cases the type and extent of the lesion did influence management but the small size of this study did not allow us to determine if these treatment variations altered patient outcome.
The finding of two cases of VS and endometrial cancer occurring before the development of VS in one case and afterwards in the other case is interesting but because of the small numbers is probably not an association. However future studies may be able to answer this question.
VS have been characterised by local recurrences and distant haematogenous metastases [3]. Commencing treatment with wide radical excision has been reported as improving outcome [8]. The addition of radiation [9] and chemo-radiation in selected cases has been beneficial [10]. We agree with the suggested management of VS by others quoted in this article that early diagnosis and wide local excision is the optimum management and that if the original excision is found to be incomplete a more radical local excision is performed subsequently. In selected cases radiation and chemo-radiation have a place.
To talk of five year survival for patients with such a rare cancer, so many histological types with variable cell differentiation plus the unknown of when sarcomatous cells metastasize from even early FIGO staging is in our opinion taking generalisation to far. However, we do agree with others that to obtain the best results for each individual patient a multidisciplinary approach from the time of diagnosis to plan and carry out the treatment, follow up and documenting progress.
Data collection over long time spans is problematic as we found with some data from cases two to five being destroyed after 15 years according to hospital policy for the management of hospital records. We recommend that data for rare cancers be collected at either a national or international centre, pooled and published to improve our understanding of these diseases and so benefit all future patients with these conditions.
5. ACKNOWLEDGEMENTS
We gratefully acknowledge all those doctors who referred cases to the QCGC and the gynaecological oncologists who have updated QCGC records, especially doctors Ward, Nicklin and Perrin.
![]()
![]()
REFERENCES
- Sleijfer, S., Seynaeve, C. and Verweij, J. (2007) Gynaecological sarcomas. Current Opinion Oncology, 19, 492- 496. doi:10.1097/CCO.0b013e3282748eaa
- Behranwala, K.A., Latifaj, B., Blake, P., Barton, D.P.J., Shepherd, J.H. and Thomas, J.M. (2004) Soft tissue tumors. International Journal Gynecologic Cancer, 14, 94- 99.
- Davos, I. and Abell, M.R. (1976) Soft tissue sarcomas of vulva. Gynecologic Oncolology, 4, 70-86. doi:10.1016/0090-8258(76)90009-3
- Nirenberg, A., Ostor, A.G., Slavin, J., Riley, C.B. and Rome, R.M. (1995) Primary vulvar sarcomas. International Journal Gynecological Pathology, 14, 55. doi:10.1097/00004347-199501000-00010
- Aartsen, E.J. and Albus-Lutter, C.E. (1994) Vulvar sarcoma: Clinical implications. European Journal Obstetrics & Gynecology Reproductive Biology, 56, 181-189. doi:10.1016/0028-2243(94)90168-6
- Heim-Hall, J. and Yohe, S.L. (2008) Application of immunohistochemistry to soft tissue neoplasms. Archives Pathology Laboratory Medicine, 132, 476-489.
- Lutz, J. and Heemann, U. (2003) Tumours after kidney transplant. Current Opinion in Urology, 13, 105-109. doi:10.1097/00042307-200303000-00004
- Di Saia, P.J., Rutledge, F. and Smith, J.P. (1971) Sarcoma of the vulva: Report of 12 patients. Obstetrics and Gynecology, 38, 180-184.
- Barakut, R.R., Markman, M. and Randall, M. (2009) Principles and practice of gynecologic oncology. Lippincott Williams and Wilkins Publishers, Baltimore, Chapter 20, 585.
- Holloway, C.L., Russell, A.H., Muto, M. and Albert, M. (2007) Synovial cell sarcoma of the vulva: Multimodality treatment incorporating preoperative external-beam radiation, hemivulvectomy, flap reconstruction, interstitial brachytherapy and chemotherapy. Gynecologic Oncology, 104, 253-256. doi:10.1016/j.ygyno.2006.09.018
NOTES
*Corresponding author.