Open Journal of Genetics
Vol.4 No.1(2014), Article ID:44043,6 pages DOI:10.4236/ojgen.2014.41009
Predictive Analysis of Microarray Data*
Paulo C. Marques F., Carlos A. de B. Pereira
Departamento de Estatística, Instituto de Matemática e Estatística, Universidade de São Paulo, São Paulo, Brasil
Email: pmarques@ime.usp.br, cpereira@ime.usp.br
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 11 February 2014; revised 3 March 2014; accepted 23 March 2014
ABSTRACT
Microarray gene expression data are analyzed by means of a Bayesian nonparametric model, with emphasis on prediction of future observables, yielding a method for selection of differentially expressed genes and the corresponding classifier.
Keywords:Bayesian Nonparametrics; Dirichlet Process; Microarray Data; Differential Gene Expression; Classification

1. Introduction
DNA microarrays are devices used to determine the expression (activity level) of a set of genes contained in a tissue sample. Briefly, they consist of small arrays of thousands of probes on which surfaces are deposited many copies of single stranded DNAs sequences corresponding to specific genes, or pieces of genes. Reverse transcription of messenger RNAs extracted from the tissue produces a solution of DNAs whose sequences are complementary to those found on the microarray probes. This solution is colored and put into contact with the microarray surface. Sequences present in the solution hybridize with their complementary pairs on the microarray probes. Subsequent illumination of the microarray surface provides an image in which the intensity of each probe spot is related to the corresponding amount of messenger RNAs present in the tissue. Digital processing of this image outputs for each probe a positive number which measures the relative expression of the corresponding genes (see [1] and references therein for a detailed description of microarray technology).
Data from a typical microarray experiment consist of positive numbers representing the expression levels of the genes associated with the microarray probes for a group of individuals. Because the convoluted nature of the numeric values describing the expression levels makes it difficult to commit to a specific family of probability distributions in their modeling, our proposal is to approach this problem by means of a Bayesian nonparametric analysis. The emphasis placed by De Finetti [2] on prediction guides us, in the sense that both products of our analysis, a subset of differentially expressed genes and the corresponding classifier, are derived from probabilities of events related to values of future observables, with (unobservable) parameters playing only a subsidiary role.
2. Microarray Data Model
Our microarray data consist of the expression levels of 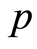 gene probes for
gene probes for 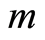 case patients that have been diagnosed with a certain disease or show some physiological alteration, and
case patients that have been diagnosed with a certain disease or show some physiological alteration, and 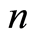 healthy control individuals. The expression level of the
healthy control individuals. The expression level of the  -th microarray probe for the
-th microarray probe for the 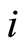 -th case patient is denoted by
-th case patient is denoted by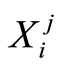 . Similarly, expression levels for controls are denoted by
. Similarly, expression levels for controls are denoted by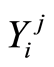 . The expression levels of the
. The expression levels of the 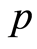 gene probes for the
gene probes for the 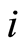 -th case patient are abbreviated by
-th case patient are abbreviated by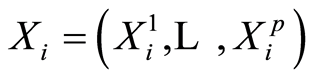 . For controls, we define similarly
. For controls, we define similarly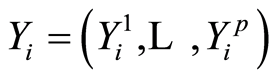 .
.
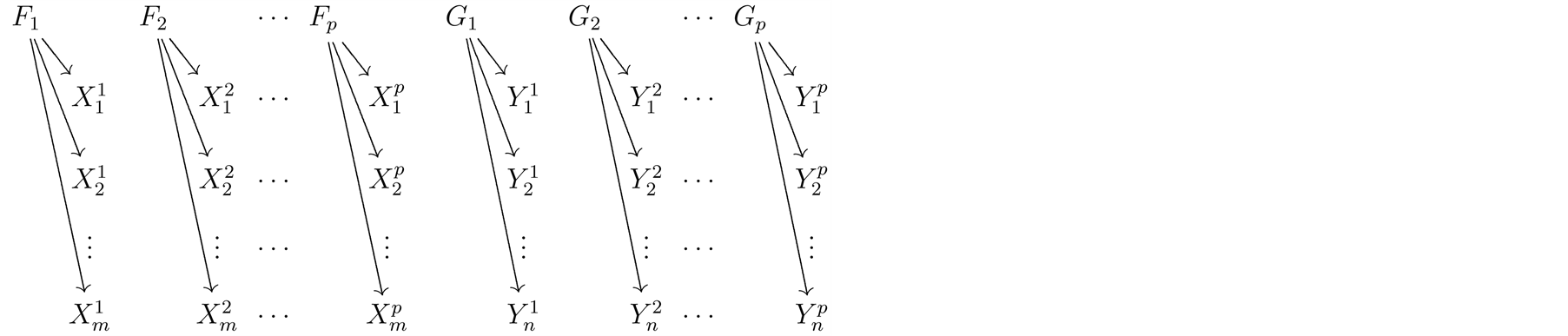
The graph below depicts the microarray data model. Absence of an arrow connecting two random objects means that they are conditionally independent given their parents. In this graph, the orphan vertexes are independent Dirichlet processes distributed as 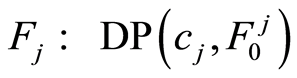 and
and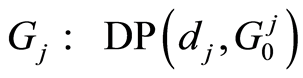 , for gene probes
, for gene probes . Necessary Dirichlet process properties and notations are collected in the first Appendix.
. Necessary Dirichlet process properties and notations are collected in the first Appendix.
3. Predictive Selection of Differentially Expressed Genes
Suppose that we have microarray gene expression data for 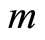 case patients and
case patients and 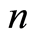 healthy control individuals. Following the notations introduced in the previous section and using the convention that upper case letters represent random variables and small case letters their realizations, we denote this data by
healthy control individuals. Following the notations introduced in the previous section and using the convention that upper case letters represent random variables and small case letters their realizations, we denote this data by 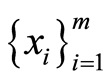 and
and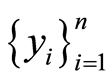 . Our first goal is to use the information contained in this data to establish which genes are expected to be less or more active for a future case patient, making these differentially expressed genes a subset of disease markers.
. Our first goal is to use the information contained in this data to establish which genes are expected to be less or more active for a future case patient, making these differentially expressed genes a subset of disease markers.
For each gene probe, the relative expression of the corresponding gene can be determined by our posterior opinion that for a future case patient the expression level of this probe will be smaller than the expression level of the same probe for a future healthy control individual, given all the information contained in the data. This posterior opinion is quantified by the posterior predictive probabilities
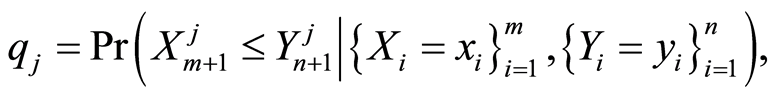
for gene probes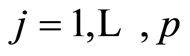 . Using the nonparametric data model of the previous section, these posterior predictive probabilities can be computed using the results given in the first Appendix.
. Using the nonparametric data model of the previous section, these posterior predictive probabilities can be computed using the results given in the first Appendix.
The values of these probabilities determine an ascending ranking of relative expression of the gene probes. We denote by 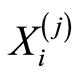 the expression level of the gene probe occupying the
the expression level of the gene probe occupying the  -th position in this ranking for the
-th position in this ranking for the 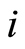 -th case patient. To refer to the gene probes at the end of the ranking, we use the notation
-th case patient. To refer to the gene probes at the end of the ranking, we use the notation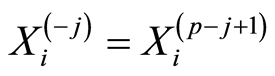 . Similar notations,
. Similar notations, 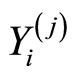 and
and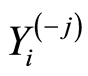 , are used for healthy control individuals.
, are used for healthy control individuals.
The criterion for the choice of the subset of differentially expressed genes is to select from this ranking the first 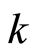 (down regulated) gene probes and the last
(down regulated) gene probes and the last 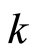 (up regulated) ones, for some integer
(up regulated) ones, for some integer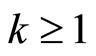 . In the last section we show how
. In the last section we show how 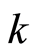 can be selected by cross validation.
can be selected by cross validation.
4. Predictive Classification
With the subset of 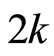 differentially expressed gene probes obtained in the previous section, we construct a classification rule that allows us to pick microarray data for a new individual and classify him as unhealthy or healthy. Let us denote case patients and healthy controls together as
differentially expressed gene probes obtained in the previous section, we construct a classification rule that allows us to pick microarray data for a new individual and classify him as unhealthy or healthy. Let us denote case patients and healthy controls together as

Table 1. Gene probes and cross validation.
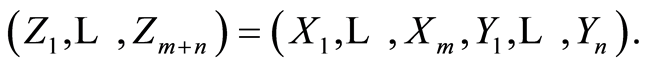
Defining the statistic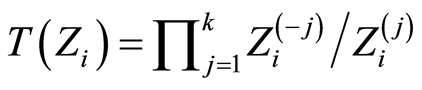 , which is an increasing function of the expression level of the up regulated gene probes and a decreasing function of the down regulated ones, we expect case patients to exhibit values for this statistic that are larger than the corresponding values for healthy controls.
, which is an increasing function of the expression level of the up regulated gene probes and a decreasing function of the down regulated ones, we expect case patients to exhibit values for this statistic that are larger than the corresponding values for healthy controls.
The classification rule is based on this one dimensional statistic and takes into account the different sample sizes of the two groups, cases and controls. Given a new individual for which we have microarray data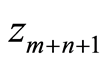 , the rule is to classify him as healthy when
, the rule is to classify him as healthy when 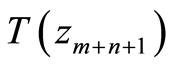 is less than the critical value
is less than the critical value  for which
for which
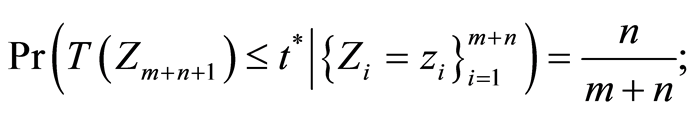
otherwise, we classify him as unhealthy. The critical value 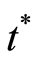 is computed using the results given in the first Appendix.
is computed using the results given in the first Appendix.
5. Example
The publicly accessible Gene Expression Omnibus (GEO) database [3] provides microarray data from a study [4] of peripheral circulating B cells for 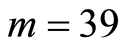 smoking and
smoking and 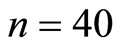 non-smoking healthy american white women.
non-smoking healthy american white women.
We proceed the analysis of this dataset using the results of the previous sections and considering the case of weak prior information about the distribution of the expression levels of the gene probes, which means that within the nonparametric model we compute all the desired probabilities taking the limit to zero of the concentration parameters of the Dirichlet processes.
Table 1 shows the identifiers for the 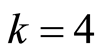 pairs of down and up regulated gene probes computed for this dataset. This table also presents a leave one out cross validated study of the sensitivity and specificity of the predictive classifier for this dataset using the
pairs of down and up regulated gene probes computed for this dataset. This table also presents a leave one out cross validated study of the sensitivity and specificity of the predictive classifier for this dataset using the 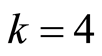 pairs of down and up regulated gene probes. For this dataset,
pairs of down and up regulated gene probes. For this dataset, 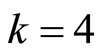 is the smallest number of pairs of up and down regulated gene probes that gives us the best balance between cross validated fractions of false negatives and false positives. Computer code in the Perl language [5] is presented in the second appendix.
is the smallest number of pairs of up and down regulated gene probes that gives us the best balance between cross validated fractions of false negatives and false positives. Computer code in the Perl language [5] is presented in the second appendix.
References
- Friend, S.H. and Stoughton, R.B. (2002) The Magic of Microarrays. Scientific American, 286, 34-41. http://dx.doi.org/10.1038/scientificamerican0202-44
- De Finetti, B. (1974) Theory of Probability (Two Volumes). John Wiley & Sons, Hoboken.
- GEO Dataset GDS3713. http://www.ncbi.nlm.nih.gov/geo
- Pan, F., Yang, T.L., Chen, X.D., Chen, Y., Gao, G., Liu, Y.Z., Pei, Y.F., Sha, B.Y., Jiang, Y., Xu, C., Recker, R.R. and Deng, H.W. (2010) Impact of Female Cigarette Smoking on Circulating B Cells in Vivo: The Suppressed ICOSLG, TCF3, and VCAM1 Gene Functional Network May Inhibit Normal Cell Function. Immunogenetics, 62, 237-251. http://dx.doi.org/10.1007/s00251-010-0431-6
- Wall, L., Christiansen, T. and Orwant, J. (2000) Programming Perl. 3rd Edition, O’Reilly Media, Sebastopol.
- Ferguson, T. (1972) A Bayesian Analysis of Some Nonparametric Problems. The Annals of Statistics, 1, 209-230. http://dx.doi.org/10.1214/aos/1176342360
- Schervish, M.J. (1997) Theory of Statistics. Springer-Verlag, Berlin.
Appendix 1: The Dirichlet Process
We are concerned with the representation of our uncertainties about some observable properties assuming values in a sampling space , with sigma-field
, with sigma-field , by means of a probability measure defined over an underlying measurable space
, by means of a probability measure defined over an underlying measurable space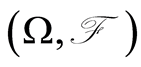 . The probability of an event
. The probability of an event 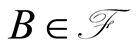 is denoted by
is denoted by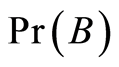 .
.
The map 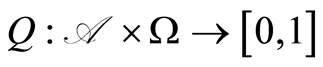 is a random probability measure over
is a random probability measure over 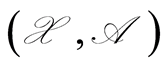 if
if 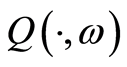 is a probability measure over this measurable space for every
is a probability measure over this measurable space for every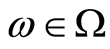 , and
, and 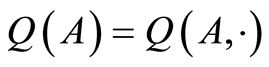 is a random variable for each
is a random variable for each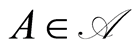 .
.
Ferguson [6] defined a random probability measure 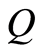 as follows. Let
as follows. Let 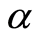 be a finite nonzero measure over
be a finite nonzero measure over 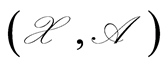 and specify that for each
and specify that for each 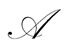 -measurable partition
-measurable partition 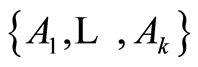 of
of  the random vector
the random vector
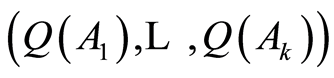
has the usual Dirichlet distribution with parameters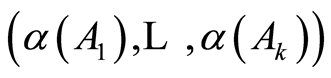 . One such
. One such 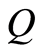 is denominated a Dirichlet process with base measure
is denominated a Dirichlet process with base measure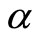 .
.
Ferguson proved that this definition entails the following facts. First, 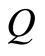 is a properly defined random process in the sense of Kolmogorov’s consistency theorem [7] . Second, the expectation of
is a properly defined random process in the sense of Kolmogorov’s consistency theorem [7] . Second, the expectation of 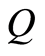 has the simple expression
has the simple expression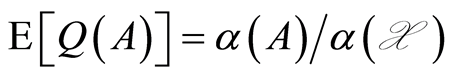 , for each
, for each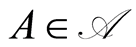 . Third, if measurable observables
. Third, if measurable observables 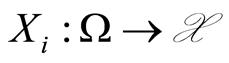 are conditionally independent and identically distributed, given
are conditionally independent and identically distributed, given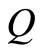 , with
, with 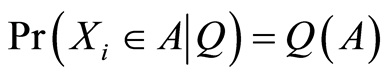 almost surely, for
almost surely, for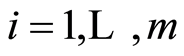 , then a posteriori
, then a posteriori 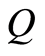 is again a Dirichlet process with base measure
is again a Dirichlet process with base measure 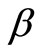 defined almost surely by
defined almost surely by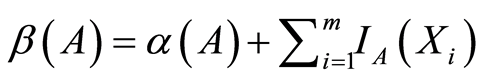 , for each
, for each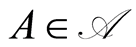 .
.
If we add a new observable 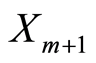 to the just described conditional model, its posterior predictive probability is
to the just described conditional model, its posterior predictive probability is
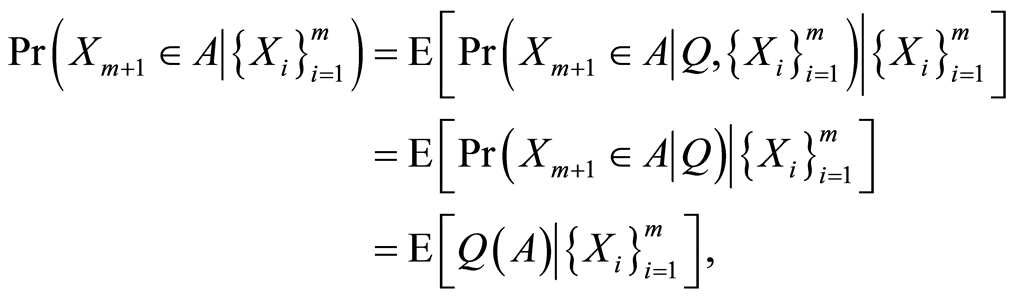
almost surely, for every , in which the second equality follows from the conditional independence of the observables.
, in which the second equality follows from the conditional independence of the observables.
For microarray data the sampling space can be taken as the real line with Borel sigma-field. If 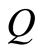 is a Dirichlet process with base measure
is a Dirichlet process with base measure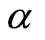 , it is convenient to work with the random distribution function defined by
, it is convenient to work with the random distribution function defined by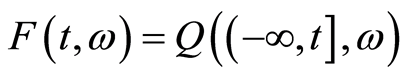 . We abbreviate
. We abbreviate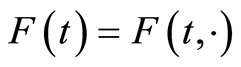 . Defining
. Defining 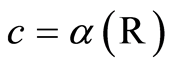 and
and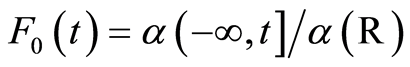 , we denote the distribution of the random distribution function by
, we denote the distribution of the random distribution function by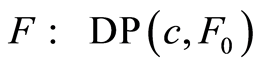 . Since
. Since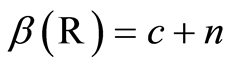 , the posterior expectation of
, the posterior expectation of 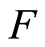 is almost surely
is almost surely
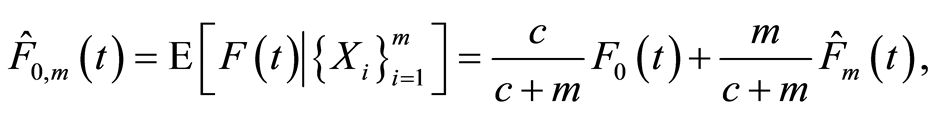
in which 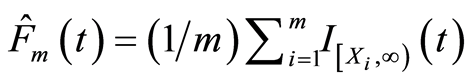 is the empirical distribution function. This gives us an interpretation of the base measure of the Dirichlet process. The total measure
is the empirical distribution function. This gives us an interpretation of the base measure of the Dirichlet process. The total measure 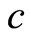 works as a concentration parameter: for fixed sample size
works as a concentration parameter: for fixed sample size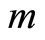 , if we make
, if we make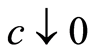 , the posterior expectation reduces to the empirical distribution function. Also, this expression of
, the posterior expectation reduces to the empirical distribution function. Also, this expression of 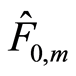 shows that prior information contained in
shows that prior information contained in 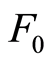 is washed out when, for fixed
is washed out when, for fixed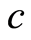 , we let
, we let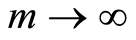 .
.
Finally, suppose that we have a second sample: let 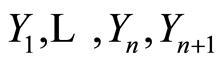 be conditionally independent and identically distributed, given
be conditionally independent and identically distributed, given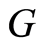 , each one of them having conditional distribution
, each one of them having conditional distribution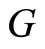 , and
, and 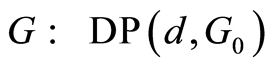 is independent of
is independent of . The posterior expectation is almost surely
. The posterior expectation is almost surely
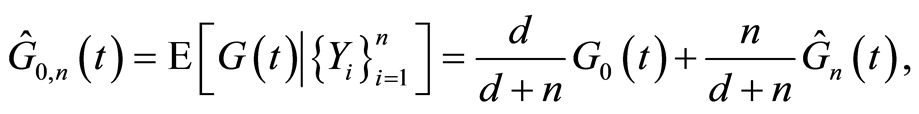
in which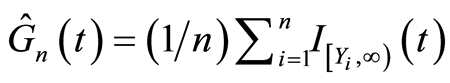 . If
. If 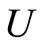 and
and 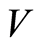 are independent random variables with distribution functions
are independent random variables with distribution functions 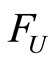 and
and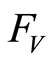 , respectively, a simple computation shows that
, respectively, a simple computation shows that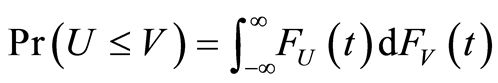 . Therefore, since
. Therefore, since 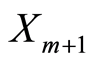 and
and 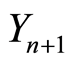 are conditionally independent, given
are conditionally independent, given 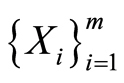 and
and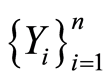 , and almost surely
, and almost surely
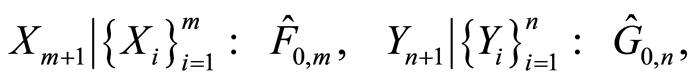
it follows that almost surely
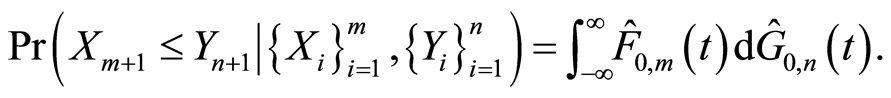
If we let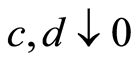 , this conditional probability reduces to
, this conditional probability reduces to
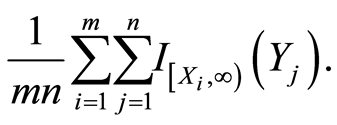
Appendix 2: Computer Code
#!/usr/bin/perl
# predictive.pl -
use strict;
use warnings;
my $k = 4;
print "\nSelecting k = $k pairs of Down/Up regulated gene probes.\n\n";
my @cases = (42..80);
my @controls = (2..41);
my @all = (@cases, @controls);
my $ua;
open(DATA, "./GDS3713.soft") or die $!;
while () {
next unless $. >= 118 && $. <= 22400;
chomp;
push @$ua, [split(/\t/, $_)];
}
close(DATA);
my @pr;
foreach my $probe (@$ua) {
push @pr, pr_next_case_leq_next_control($probe, \@cases, \@controls);
}
my @ranking = sort {$pr[$b] <=> $pr[$a]} (0.. @$ua - 1);
print "Down regulated|Up regulated\n";
print "---------------+--------------\n";
for (my $i = 0; $i < $k; $i++) {
printf (" $ua->[$ranking[$i]]->[1]$ua->[$ranking[-($i + 1)]]->[1]);
}
print "\n";
printf ("Critical t = %. 4f\n\n"
critical_t($ua, \@ranking, $k, scalar @controls, \@all));
print "Cross validated sensitivity and specificity.\n\n";
my ($case_unhealthy, $case_healthy$control_unhealthy, $control_healthy) = (0, 0, 0, 0);
foreach my $group (\@cases, \@controls) {
foreach my $off (@$group) {
my $s = T_statistic($ua, \@ranking, $k, $off);
my @T;
foreach my $individual (@all) {
next if $individual == $off;
push @T, T_statistic($ua, \@ranking, $k, $individual);
}
@T = sort {$a <=> $b} @T;
if ($group == \@cases) {
if ($s <= $T[@controls - 1]) { $case_healthy++ }
else { $case_unhealthy++ }
} else {
if ($s <= $T[@controls - 2]) { $control_healthy++}
else { $control_unhealthy++ }
}
}
}
print "-" x 29, "\n Unhealthy|Healthy\n";
print "-------------------+---------\n";
printf ("Case %. 4f | %. 4f\n"$case_unhealthy/@cases, $case_healthy/@cases);
print "-" x 29, "\n";
printf (" Control %. 4f | %. 4f\n"$control_ unhealthy/@controls, $control_healthy/@controls);
print "-" x 29, "\n";
exit 1;
sub pr_next_case_leq_next_ control {
my ($probe, $cases, $controls) = @_;
my $leq = 0;
foreach my $case (@$cases) {
foreach my $control (@$controls) {
$leq++ if $probe->[$case] <= $probe->[$control];
}
}
return $leq/(@$cases * @$controls);
}
sub T_statistic {
my ($ua, $ranking, $k, $individual) = @_;
my $t = 1;
for (my $i = 0; $i < $k; $i++) {
$t *= $ua->[$ranking->[-($i + 1)]]->[$individual];
$t/= $ua->[$ranking->[$i]]->[$individual];
}
return $t;
}
sub critical_t {
my ($ua, $ranking, $k, $n, $all) = @_;
my @T;
foreach my $individual (@$all) {
push @T, T_statistic($ua, $ranking, $k, $individual);
}
@T = sort {$a <=> $b} @T;
return $T[$n - 1];
}
NOTES

*This paper is dedicated to Paulo Cilas Marques in memoriam. We thank Professor Luiz Eugênio Barbosa de Oliveira for his critical reading of the manuscript. Work partially supported by CAPES.