Open Journal of Genetics
Vol.1 No.3(2011), Article ID:8895,10 pages DOI:10.4236/ojgen.2011.13014
Implications of double-stranded DNA structure for development, cancer and evolution
Neo-Morgan Laboratory Inc., Biotechnology Research Center, Kawasaki, Japan.
Email: furusawa@neo-morgan.com
Received 21 September 2011; revised 30 October 2011; accepted 12 November 2011.
Keywords: Blast; Graph Theory; Redundant Sequences; CD-HIT
ABSTRACT
Genomes consist of DNA and the genetic information is encoded in a linear form of DNA. According to the central dogma of molecular biology, the genetic information is transcribed into mRNA, and mRNA translated into a polypeptide. Gene expression should be precisely regulated in order to create progeny. Unlike RNA, DNA has double-stranded structure. Is there any specific biological reason why DNA has evolved to possess double-stranded structure? In this presentation, biological implications of the double-stranded structure of the DNA molecule will be reviewed. In eukaryotes, it has been reported that cells might have the machinery that distinguishes one DNA-strand from the other, and that the strand-recognition mechanism might control development, cancer and evolution. Three prominent models concerning biological implications of replication of double-stranded DNA will be discussed: 1) Klar’s “somatic strand-specific imprinting and selective chromatid segregation model” for differential gene regulation, 2) Cairns’ “immortal strand inheritance model” for cancer prohibition, and 3) the “disparity mutagenesis model” for the acceleration of evolution proposed by the present author.
1. INTRODUCTION
The double-stranded model of DNA postulated by Watson and Crick in 1953 clearly explained how precisely equivalent daughter cells are produced when genetic material of the parental cell is replicated [1]. Furthermore, the organisms must correctly regulate the expression of genes. In bacteria, gene expression is regulated by the “so-called” Jacob-and-Monod system, using watersoluble molecules; an inducer and a repressor.
Compared to prokaryotes, eukaryotes have a considerably larger number of genes and the genetic constitution must provide for vast arrays of differentiated cells at the exact places with exact timing in development. Therefore, new strategies, e.g., epigenetic gene-controlling mechanisms including somatic recombination by means of a specific recognition of DNA strand should have evolved [2].
For multi-cellular organisms the most troublesome problem is of cancer. Especially, cells that are subject to highest risk of cancer development will be stem cells. This is because stem cells continue to replicate throughout the organisms life span. Thus, errors accompanying DNA replication of stem cells are thought to be the main cause of cancer. For instance, mouse intestinal ectoderm stem cells multiply about 1000 times in a life span. According to Cairns, when nonequivalent mitosis occurs in the stem cells, “oldest” DNA strands are always kept in the stem cells. This biased segregation of DNA strands suggests the existence of a strand-recognition mechanism, which guarantees the effective avoidance of carcinogenesis in self-renewing intestinal cells [3].
Double-stranded DNA is used not only for differentiation or prevention of cancer, but also it might be used for controlling the speed of evolution. Without exception genomic DNA is duplicated by leadingand laggingstrand mode of replication. This troublesome way of replication may result in biased mutagenesis in the lagging strand replication complexes that differs in biological activities from that of the leading strand replication complex. This universal way of DNA replication may result in biased mutagenesis of one strand over the other. The nonequivalent distribution of spontaneous mutations to both strands during replication can cause increase in mutation rates. Accordingly, organisms can produce unchanged progeny maintaining the species, and at the same time, higher mutation rates can provide raw material for accelerated evolution [4]. This idea was tested by computer simulations and also proved experimentally using living organisms of Escherichia coli and yeasts [5,6].
Although the above-mentioned concepts have been independently proposed in different laboratories, there exists a basic thread common to all of them; i.e., these three lines of research have tried to seek for additional biological implications of the double-stranded structure of the DNA molecule.
2. KLAR’S “SOMATIC STRAND-SPECIFICIMPRINTING AND SELECTIVE CHROMATID SEGREGATION MODEL (SSIS)”
2.1. Mating-Type Switching in Fission Yeasts
The basic idea of the SSIS model [7] seems to be confirmed by research on the phenomenon of mating-type switching in fission yeast (Schizosaccharomyces pombe) [2].
A yeast diploid cell produces two P (plus sex)-cells and two M (minus sex)-cells, called ascospores, after meiosis. A remarkable feature of fission yeasts, is the phenomenon of mating-type switching, a process that spontaneously changes the cell type during mitosis. Three genes are essential for mating-type twitching; mat1, mat2 and mat3 (Figure 1). The mat2 and mat3 loci are located near the mat1 locus. When the mat1-P gene is located at the mat1 locus, the haploid cell exhibits a P-cell type. When the mat1-M gene occupies the mat1 locus, however, it becomes M-cell. Under nutrient starvation condition, a P-cell mates with an M-cell and makes a diploid cell. For the mating-type switching to occur, an intrachromosomal translocation of one donor gene from the mat2 or the mat3 locus to the mat1 locus is required. How does such a dramatic “cassette” change take place?
In Figure 2, DNA of the mat1 locus region in the parent P-cell is shown. The cell-type studies in cell pedigrees have shown that after two mitotic divisions, one parental haploid cell (P) makes only one switched cell (M) out of four granddaughters. This biased segregation of chromatids is nicely explained by Klar’s unique idea that proposed a DNA strand-specific imprinting at mat1 locus.
Two daughter cells are produced after replication of a parent Pu (un-switchable) cell. One of the daughter cells (left) becomes Ps (switchable), because of the imprinting (or perhaps “nick”: a star-mark) in the newly-synthesizing lagging strand, while the other cell inheriting the leading strand of the parental cell is of Pu type. That is, asymmetric cell division results as a consequence of the usual leadingversus lagging-strand replication at mat1. To generate four granddaughter cells, both daughter cells are required to undergo additional cell division. When Ps replicates, one of the granddaughter cells remains as Ps, but the other cell switches to Mu. Instead, the Pu daughter cell makes Ps and Pu cells, as seen in the first division. The coupling of strand-specific imprinting and its recognition in the following cell cycle explains the pattern of mating-type switching in cell pedigrees [8].
The mat2 and mat3 loci are epigenetically “silent” so that transcription, epigenetic conversion and recombination in the intervening interval are prohibited. Thus, selffertilization in sibling cells readily takes place when cells are starved for nutrients. Surprisingly, mating-type switching occurs at a high rate (~80%), meaning that there is an unknown mechanism of directionality, by which the opposite mating-type is preferably chosen from one of the donor genes.
The precise mechanism of mating-type switching remains unclear. It is, however, proven that swi(for switch)1 and swi3 gene products temporally stop the replication fork at the mat1 locus and imprinting (nicking) at a specific site of nucleotide takes place [9]. Using donor mat2-P or mat3-M genes as DNA template, one strand is copied into the mat1 locus. Subsequently, the opposite strand is synthesized by using the newly synthesized strand as the template. For the details of switching mechanism, see Klar’s review [2]. It is very well established that asymmetric cell division in yeast results from the inherent asymmetry of replication of
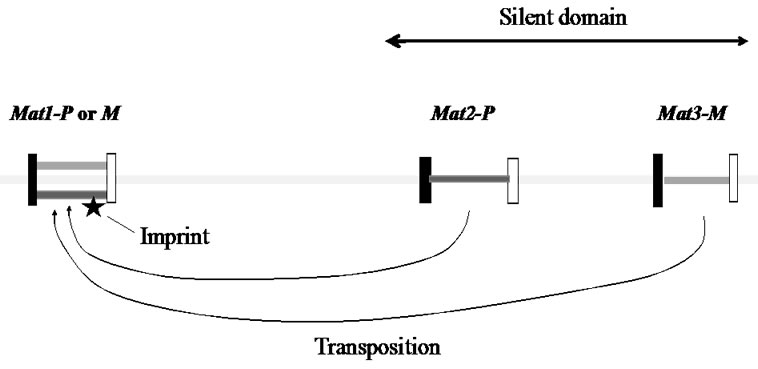
Figure 1. The mating-type region in fission yeast chromosome.
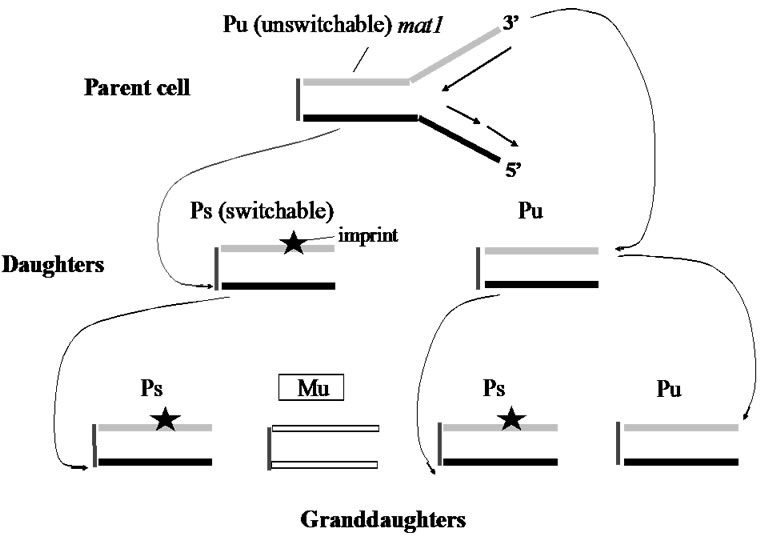
Figure 2. Mating-type switching in fission yeast.
DNA strands. However, yeast is a haploid organism, can such a mechanism operate in diploid organisms where non-random distribution of two sets of chromosomes to specific progeny cells will be additionally required? Discussion of such a theory will be presented in the following section.
2.2. Support for SSIS Hypothesis: Determination of the Left-Right Body Axis in Mice
Determination of mouse laterality is regulated by the lrd1 gene (left-and-right dynein, LRD; microtubule-based motor protein) [10]. In mutant mice with lrd1-/lrd1-, 50 percent of mice develop with normal arrangement and 50 percent with mirror-image placement of visceral organs. Concerning the determination of body laterality, the most prominent “nodal flow hypothesis” in mice has been postulated. Anti-clockwise rotation of mono-cilia on the cell surface of the node (the mid embryonic structure) directs the flow of extra-cellular fluid carrying putative “morphogen” molecules to one side of the node. This directed flow is proposed to determine mouse laterality [11].
The fundamental question of what mechanism initially determines the body laterality, however, remains to be answered. Before findings concerning the mating-type switching mechanism, Klar had already proposed his chromosomal-based SSIS model (Figure 3) as a mechanism for initially breaking the embryonic symmetry [7].
“The SSIS model proposed a cell-lineage based developmental mechanism whereby a single laterality-generating progenitor cell in the embryo produces one leftand another right-side-generating daughter cell through an asymmetric cell division.” A left/right axis determining gene (lra1) was hypothesized. As shown in Figure 3, usually, the lra1 of the progenitor cell is “off” by epigenetic mechanism according to the SSIS model. When the progenitor cell undergoes asymmetrical cell division, resulting in one daughter cell having “old” W-strand becomes “on” and contributes to make left visceral organs, while the lra1 of the other daughter cell with “old” C-strand is kept in the “off” condition to make right visceral organs. LDR is hypothesized to selectively distribute differentiated sister chromatids to specific daughter cells to constitute an asymmetrical cell division. In the lrd1-/lrd1-mutant mice, however, random visceral situs would result from random distribution of sister chromatids owing to the LRD defect (see the right of Figure 3). As a result, 25 percent of offspring develop normal situs and another 25 percent with inverted situs, and the remaining 50 percent are lethal because of LL or RR isomers are predicted to be produced. The LL or RR isometric embryos should die. Consequently, among the live homozygous embryos, 50 percent should be of normal situs and 50 percent should be of inverted type [12]. These predictions of 50 percent inverted-visceral mice in alive mice and 50% embryonic death had been reported in the original study that described the lrd1-mutation [13].
It has been suggested that the phenomenon of selective segregation of chromosome 7 in mouse cell mitoses exists, and that segregation requires the lrd1 function [13, 14,15]. Judging from these data, it can be said that chromosome-specific and cell-type regulated selective strandspecific segregation may comprise one of the mechanisms of cellular development in mammals as well.
2.3. Breast Cancer Preposition and Brain Hemispheric Likely Share a Common Genetic Cause
In 1992, a noteworthy report on the relationship between women breast-cancer incidence and brain laterality in a group of white persons was published; 18 percent of reversed brain asymmetry was observed in healthy subjects, while 49 percent of the reversed asymmetry occurred in the breast cancer subjects [16]. The study concluded that the increased rates of reversed brain asymmetry in breast cancer patients was because that embryos might be exposed to higher concentrations of hormones in the uterus to effect to both traits.
The SSIS model, however, can explain this data by a completely different mechanism. It is proposed that brain laterality and right-handed (RH) are regulated by a single gene, RGHT1 (R1). The R1 may cause dominant hemisphere development in the left hemisphere of RH persons. The SSIS hypothesis proposes that a single asymmetric cell division in the fetus results in brain laterality specification, much like for visceral laterality development in mice discussed above. Namely, DNA
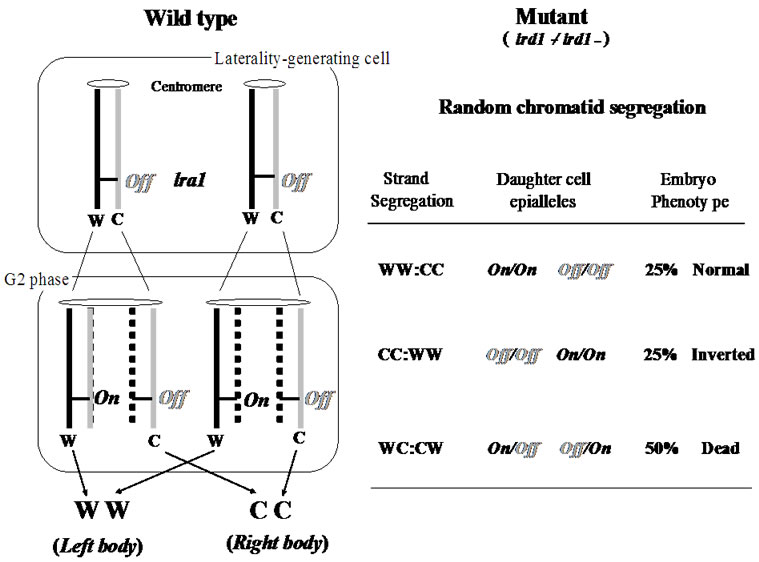
Figure 3. The strand-specific imprinting and selective chromatid segregation model concerning lrd1-/lrd1-mutant mice.
strand-specific epigenetic imprinting differentiates sister chromatids, followed by selective chromatid segregation in mitosis produces differentiated sister cells [14,15]. Accordingly, left/right-hemisphere-generating progenitor cells are produced during embryo development [17].
In persons carrying homozygous r/r, both traits of dominant-hand and left/right-hemisphere of the brain may be distributed independently of each other [18]. Accordingly the group of patients of breast-cancer having random brain laterality is predicted to be of r/r homozygous genotype. Therefore, it was proposed that the basic cause of breast cancers in the white females examined, the so-called sporadic cases of disease, would be of the r/r genotype. He speculated that “the normally operating developmental noise resulting from stochastic errors in the SSIS processes might cause cancer in a minority of r/r individuals”. “In other words, predisposition to cancer likely originates as an anomaly of the mechanism of asymmetric cell division”. The apparent association of two seemingly unrelated traits, the laterality of brain right-and-left hemisphere and predisposition to breast cancer, might indicate the existence of the same genetic cause responsible for the expression of these traits [17].
After all, as he says, the SSIS model proposes a new paradigm, of mitotic genetics (“mitogenetic”), which explains the mechanism of differentiation of somatic cells in development.
3. CAIRN’S “IMMORTAL STRAND HYPOTHESIS MODEL”
In multi-cellular organisms, matured cells are continuously supplied from organ-specific immortal stem cells, so that, stem cells continue to replicate throughout of life. Consequently, stem cells are endowed a high risk for carcinogenesis. More than thirty-five years ago, Cairns proposed a fantastic idea, which is still working today, i.e., how stem cells could avoid becoming cancer cells, irrespective of their ceaseless replicating performance [3].
The location of stem cells of mammalian small intestine is morphologically identified. In mice, the stem cells replicate about 1000 times in a life span. Sixteen stem cells are located in a limited place, at intervals of four to five cells from the very bottom of the crypt of villus. The stem cell is asymmetrically replicated and produces two daughter cells, a stem cell and a differentiating cell. The latter replicates eleven times and makes a cell group consisting of non-replicating fully-differentiated cells, which move upward, just like an escalator-system, to the tips of neighboring villi, followed by shedding into the gut lumen accompanying apoptosis. Thus, even if the differentiated epithelial cells become cancer cells, there is almost no possibility that the individual is killed by the cancer. Instead, the stem cells are always located at the same place and never stop to replicate. Nevertheless, the occurrence of intestinal stem-cell cancer is very low, suggesting that there might be some hidden mechanism with which carcinogenesis is prevented in the stem cells.
In order to explain a reasonable mechanism for avoiding carcinogenesis in stem cells, Cairns proposed an excellent idea, well-known as “immortal-strand model”. When stem cells of the intestine, blood cells or skin are carrying out asymmetrical cell divisions, “oldest” DNA strands might be exclusively segregated into stem cells. This biased segregation of strands might occur, most probably through centromeres, in all chromosomes in a stem cell. Accordingly, in the stem-cell chromosomes, newly-synthesized strands are always synthesized from the “oldest” strands as a template (Figure 4). Consequently, the occurrence of errors accompanying DNAreplications, which are seemed to be the main cause of cancer, is considerably diminished [3].
In principle, Cairns’ model can be experimentally proven as follows. 3H-labelled thymidine (or nucleotideanalogue, BrDU) were injected into young mice to label newly-synthesizing DNA strands. After chromosomal DNA in divided cells was sufficiently labeled due to the uptake of labeled-thymidine, free labeled-thymidine molecules were removed from the body by the injection of a saline containing a sufficient amount of cold thymidine.
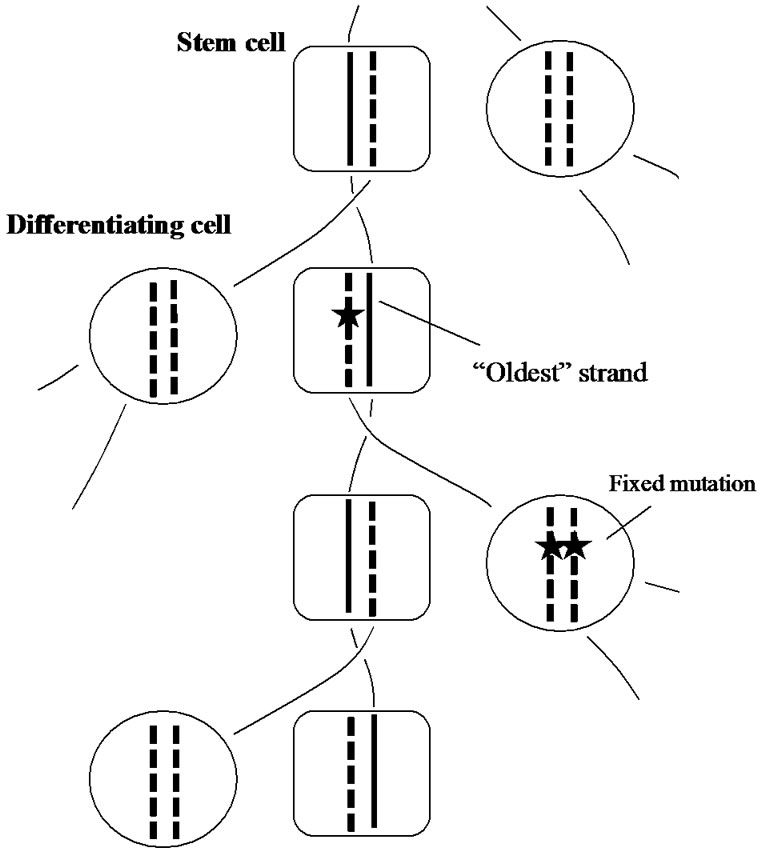
Figure 4. The immortal-strand inheritance model. “Oldest” strand is segregated in the stem cell lineage. Mutations are indicated by black stars.
When the treated mice grew up, they were sacrificed and thin sections of intestine were prepared, followed by autoradiography. Strong radio-activities, black dots, were observed only in the stem cells under a microscope, but other intestinal cells were unstained. These results clearly indicate that “oldest” DNA-strands had been retained in the stem cells during the experiment. In other divided cells, however, the labeled thymidine molecules once trapped in the DNA might be diluted out step by step with repeating cellular replications, and finally disappeared from their cell body.
Cairns’ model has been proven by different researchers and many supporting data have been reported [19-26]. On the other hand, a report which denied the model was also published [27]. Using other somatic cells than stem cells, random segregations [28,29] and also nonrandom segregations were reported [30,31]
In the present state, although the mechanism of biased segregation of “oldest” chromatids in stem cells remains to be examined, it is believed that there might be a molecular mechanism for recognizing and anchoring “oldest” DNA-stands in stem cells.
After all, it should be emphasized here that Cairns’ “immortal strand hypothesis” is the first concept that takes notice of the biological meaning of the doublestranded structure of DNA.
4. BIASED-MUTAGENESIS IN LAGGING STRANDS ACCELERATES EVOLUTION: “DISPARITY MODEL OF EVOLUTION”
4.1. Leading/Lagging-Strands Replication System Providing an Evolutionary Advantage
No evolution theory has been proposed so far in which the molecular mechanism of cell replication is considered. Without exceptions, organisms use leading and lagging strands for the replication of genomic DNA. Practically, however, as seen in PCR, DNA can be synthesized by using only leading strands, but this is not the case in nature. So, it is likely that organisms have to pay a higher cost, because considerably more enzymes are required for the lagging strand syntheses than for the leading strand. Why nature has chosen such a laborious system? There might be some evolutional advantages.
Figure 5 shows a pedigree of a linear chromosomal DNA with a single origin of replication (ori) at its upper end, i.e., indicating a single “replicore” (a limited region of DNA replicated by a single DNA-polymerase-complex). Thus, a single replicon consists of two replicors in a chromosome with multiple oris. It is roughly estimated that several genes, on average, exist in a single replicore in higher organisms.
According to our disparity-mutagenesis model, mutations occur preferentially in the lagging strand [4]. The pedigree in Figure 5 implies several interesting features; 1) the ancestor has been kept forever; 2) a genotype that once appeared in the past, has been precisely guaranteed in any generation; 3) the threshold of mutation rates is increased; 4) even if circumstances changed dramatically, the fittest individual is selected as a new ancestor and starts again to produce a new pedigree as well. These outstanding features appear to be beneficial for evolution.
In the conventional model of DNA pedigree, mutagenesis occurs stochastically and evenly in both strands (not shown). Thus, when mutation rates are higher than the threshold value, all individuals would be eventually killed by the excess accumulation of deleterious mutations.
We compared the distribution of mutations in a paritymutagenesis model (average 2 mutations/replication being introduced into both strands) with that of a disparitymutagenesis model (average 1.99 mutations into the lagging-strand and 0.01 mutations in the leading strand). For the simulation, a binominal distribution was used. Figure 6 summarizes the results of twelve trials at the 10th generation. Figure 6(a) shows the distribution of mutants in the parity model. There is no individual with zero-mutations. Contrastingly, the disparity shows a very flat distribution. The ancestral individuals with zeromutations are observable and highly mutated mutants comparing with the parity model are produced [4]. This flat distribution of mutants including ancestral individuals in the disparity model is expected from the pedigree of the disparity model shown in Figure 5.
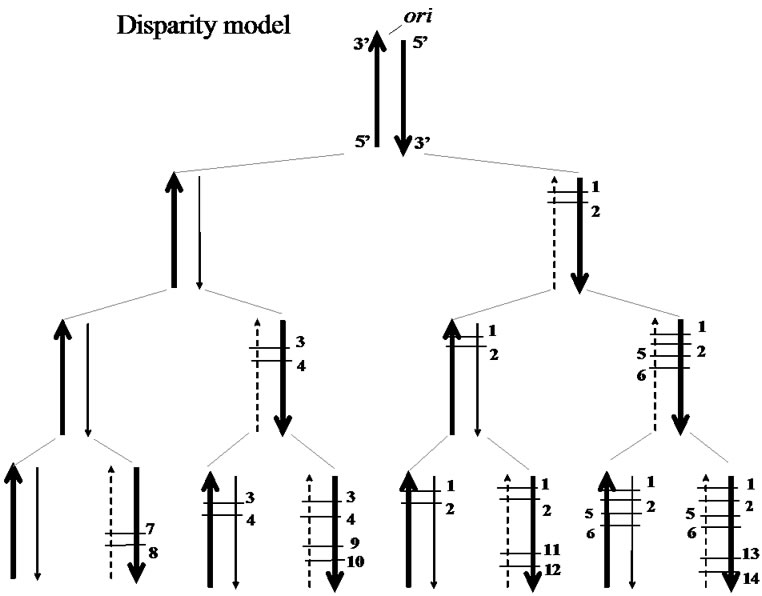
Figure 5. A DNA pedigree in the disparity mutagenesis model. Template strand (broad arrow), newly-synthesized leading strand (fine arrow), newly-synthesized lagging strand (dotted fine arrow), point mutations (short cross bars with numbers; different numbers indicating different sites of mutation).
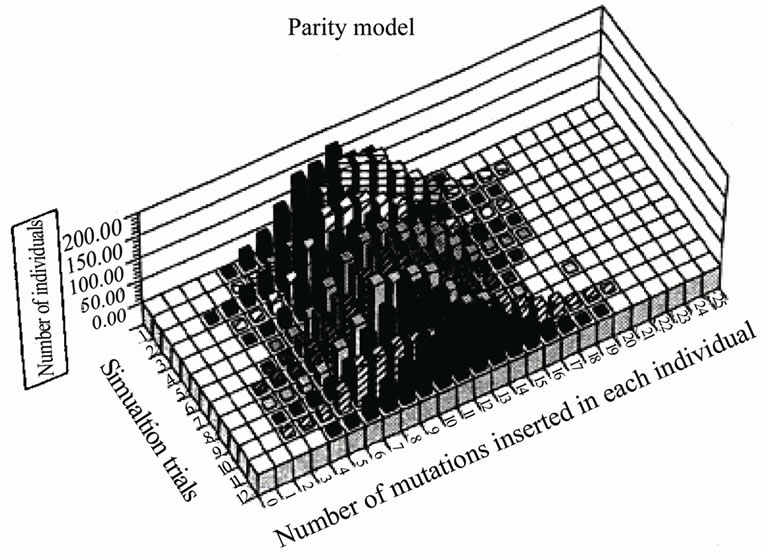 (a)
(a)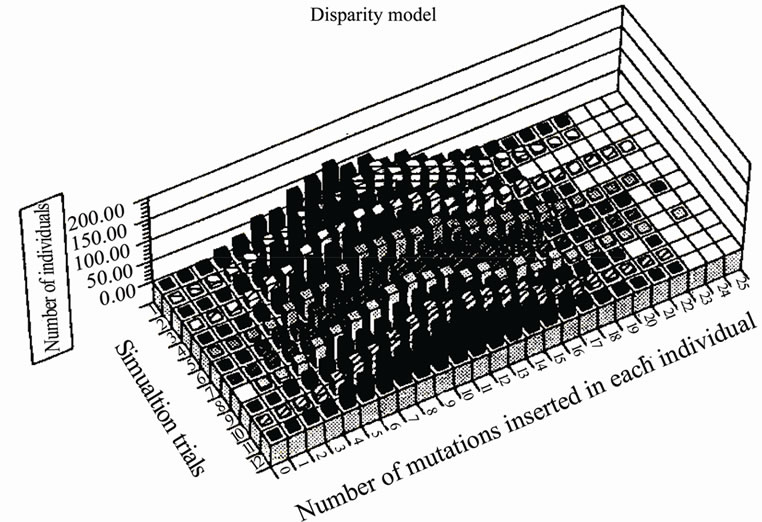 (b)
(b)
Figure 6. Comparison of the distribution of mutations in the parity model and that of the disparity model at the 10th generation.
4.2. Computer Simulations of Disparity Mutagenesis Showing Promotion of Evolution
Effects of disparity mutagenesis on adaptive evolution were simulated. The “Neo-Darwinian genetic algorithm” was constructed, in which genetic information was located on a double-stranded linear “DNA”, and a “knapsack problem” was resolved [32]. The results are summarized as follows: 1) advantageous conditions for the disparity model; small population, strong selectionpressure, high mutation rates, diploid sexual, and competitive world; 2) advantageous conditions for the conventional parity model; large population, weak selection-pressure, low mutation rates, haploid asexual, and non-competitive world. In addition, in both models, appropriate rates of crossing-over (recombination) increased fitness scores.
In conclusion, living organisms in natural fields might replicate their genomes according to the disparity-mutagenesis-system. Especially when environments changed dramatically, they might decrease the fidelity of the lagging-strand synthesis and adapt to new environments. On the other hand, a parity-mutagenesis-system might be useful for bacteria or yeasts cultured on an agar-plate with sufficient nutrients.
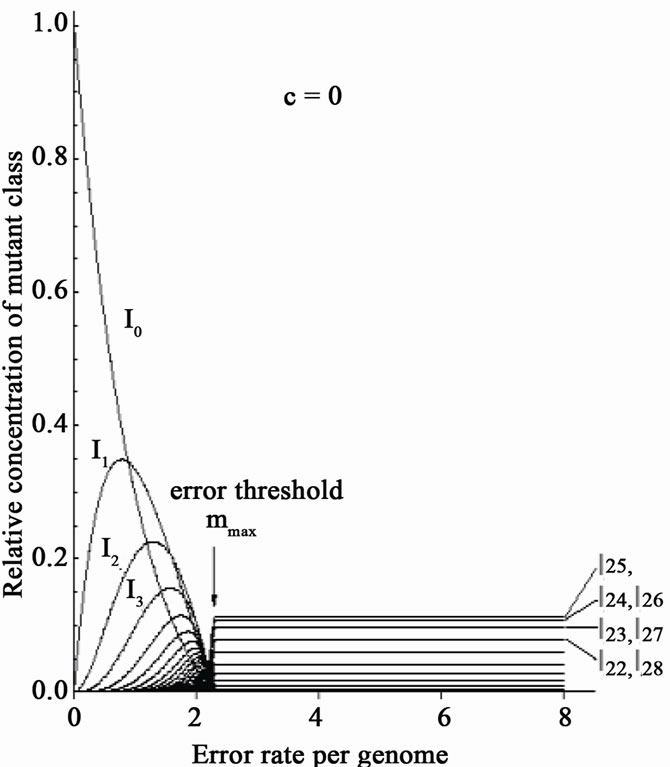
Eigen’s group showed the existence of a critical error-threshold using a model RNA [33]. Different kinds of “RNA polymerase” having various fidelities were provided, and each of them was added into a reactor respectively. When the replication reaction reached a stable state, the number of mutations in each RNA molecule was calculated. Needless to say, when no-errorpolymerase was used, no mutant was found in the reactor. With increasing error-rates of error-prone polymerase, the number of the RNA molecules carrying more mutations increased (Figure 7(a)). Finally, the error-rate increased to a critical point just before the genetic information being melted away (“edge of chaos”). If the mutation-rate exceeds the critical value, the population immediately falls into a deep chaotic sea, i.e., death (Figure 7(a)).
When a mixture of error-free and error-prone polymerases was used, however, the extinction of the population was avoided, even if the average mutation rates thoroughly exceeded the threshold value (Figure 7(b)) [34]. This result is expected from the above-mentioned simulation data (Figures 5-6) [32]. This is because, biased-mutagenesis in lagging strands means that the lagging strand is synthesized by error-prone polymerase, while the leading-strand by error-less polymerase [6, 32]. In other words, co-existence of error-less (or error-free) and error-prone polymerase in a living cell would result in the increase of the error-threshold; consequently, evolution might be accelerated. If so, it can be predicted that the efficiency of in vitro directed evolution of DNA molecules might be considerably increased by adding error-less and error-prone polymerases simultaneously in the reaction medium of PCR.
4.3. Acceleration of Evolution Using Living Organisms
Based on the results of computer simulations, we tried to accelerate evolution using a special mutator of E. coli and of budding yeasts (Saccharomyces cerevisiae). A short life-span is advantageous to evolution experiments. An E. coli mutant was used that has a mutated gene of dnaQ encoded 3’-5’exonuclease (editing enzyme). The dnaQ49 strain shows high mutation rates at 37℃, but nearly normal mutation rates at 24℃. So, we can control mutation rates at will. We showed that mutations were preferentially introduced into lagging strands. We estimated that the average mutation rate of dnaQ49
 (a)
(a)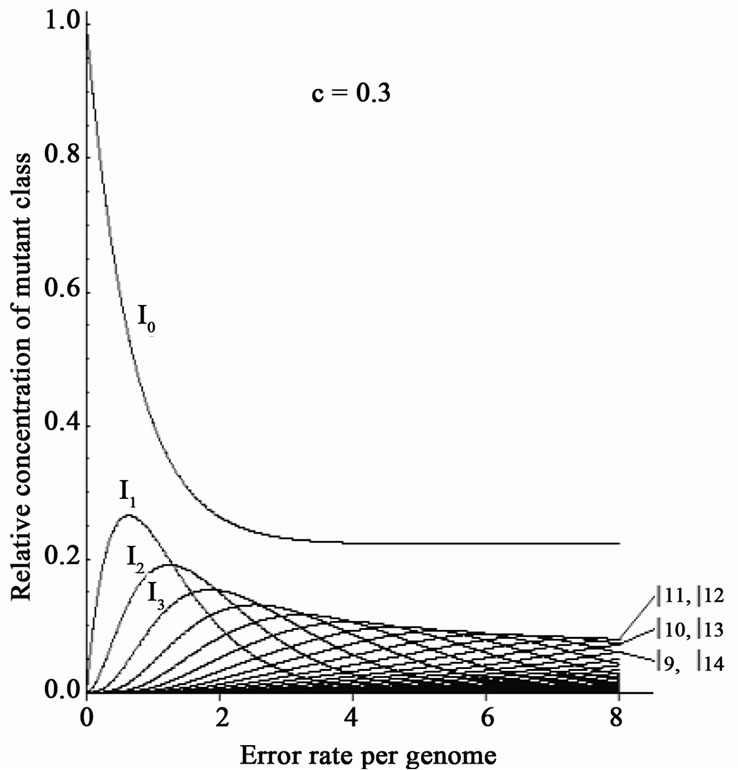 (b)
(b)
Figure 7. Disappearance of the error threshold by the co-existence of error-free and errorprone polymerase. The original figure [33] is used. One kind of polymerase was added into the reactor. In: Relative concentration of n-error mutants. One kind of polymerase was added into the reactor; (a) A mixture of error-free and error-prone polymerase was added into the reactor; (b) Ratio of error-free polymerase; (c) For details, see text.
sufficiently exceeded the threshold value, and that the relative mutation rate of the lagging strand was at least 100 times higher than the wild-type [35]. Irrespective of its high mutation rate, dnaQ49 replicates normally.
Most surprisingly, danQ49 was able to survive at the super-saturation of different kinds of antibiotics tested (e.g., penicillin: 30 mg/ml, streptomycin: 20.6 mg/ml, ofloxacin, a kind of quinolone: 3 mg/ml). For instance, the resultant penicillin-super-resistant dnaQ49 was highly sensitive to other antibiotics, suggesting that dnaQ49 was exclusively adapted to the antibiotics used for selection-pressure. Quinolone has been clinically used for such a long time that mutant E. coli strains having acquired-resistance to this drug in patients have been isolated. Analysis of ofloxacin-resistant danQ49 showed that the sites and the history of point mutations introduce into gyrA and topoIV gene, which are target genes of quinolone, were coincident with those of the samples from the patients. Furthermore, no other mutation was found in the limited regions of these genes as far as sequenced [5]. These results appear to indicate that E. coli (dnaQ49) evolution is accelerated in vitro, somehow in natural way.
Eukaryotic budding yeast, S. cerevisiae, has much more complex structures compared to bacteria. Haploid yeast has 16 chromosomes and each chromosome consists of a tandem array of many replicores. Replicores synthesized by lagging or leading-strand might be alternately arranged along a chromosome after repeated replications. Crossing-over (sister chromatid exchange) will complicate the situation. Forgetting these troublesome features, first of all, we decided to start an adaptation experiment using a mutator yeast, in which the editing-activity of polδ was deleted by one or two aminoacids substitutions. As lagging strands in budding yeasts are thought to be exclusively synthesized by polδ, this mutator might accumulate mutations in replicores synthesized by mutated polδ. This mutant yeast proliferated as well as wild-type.
The results were satisfying for us. Our mutator yeasts well adapted to high temperature. We quickly isolated adapted mutants that proliferated well at 40˚C. Then, mutated polδ gene was replaced by the wild-type one by mating with the wild-type yeasts. Genetic analyses indicated that two or three genes were concerned with temperature-resistant phenotype, and we identified one of them, named hot1 [6].
In 1999, it was predicted that yeasts and mice might be the best candidates for the experiment of accelerating evolution [36]. In deed, mice having the deletion of editing-activity of polδ can produce offspring normally, without accompanying severe deleterious phenotypes, indicating that mice actually are useful animals for our purpose [37,38]. Theoretically, mutation-rates can be increased roughly ~10,000 times using mutators having editing-activity-deleted polδ.
4.4. “Disparity Model of Evolution” Supported by the Study of Molecular Evolution
Katoh and his colleagues have found that the molecular clock of mammals runs faster than other vertebrates. In order to find out the reason, they examined the aminoacid substitution rates of polα, polε, and polδ, which mainly contribute to chromosomal DNA replications, obtained from fishes including coelacanth, amphibians, reptiles, birds and mammals. The result was striking for us in that only mammalian polδ showed high substitution rates. (It was recently found out that bird polδ also has a higher substitution rates; Katoh K, personal communication). Thus, it was speculated that the fast run of the mammalian molecular clock might be due to the lower fidelity of polδ. More surprisingly, amino-acid substitutions occurred intensively in the editing (exonuclease)-domain of polδ. They also showed that the physicochemical nature of amino-acids was changed by those substitutions; strongly suggesting that in the process of mammalian evolution, the fidelity of polδ might be occasionally changed. This change in key amino-acids for the editing activity would cause an occasional up-anddown of the speed of evolution [39]. This unexpected coincidence of our experimental results with those findings of Katoh and his colleagues appears to indicate that polδ, especially its editing domain, may play a key role for controlling the speed of evolution.
Polδ may contribute to lagging-strand synthesis in multi-cellular organisms, though the precise replication mechanism of the genomic DNA is still unclear. In the process of evolution, we can speculate that similar distribution of mutations shown in Figure 5 might be displayed in each replicore. Because of complex chromosomal constitutions in eukaryotes, the effect of biasedmutagenesis on evolution has not yet been examined. As stated above, we have obtained, however, two experimental results: 1) the mutator in budding yeast can normally replicate and quickly adapt to high temperature; 2) the mouse mutators can normally produce descendents without accompanying severe deleterious phenotypes, while cancer predisposition is increased. From these facts, we can imagine that organisms might accelerate evolution by means of decreasing the fidelity of editing activity of polδ. Or, at least, we can say that evolution might be experimentally accelerated by deleting the editing activity of polδ.
It is however unlikely, that evolution is successfully accelerated by artificially-decreasing the fidelity of polymerase domain of polδ. This is because most mutations introduced by the polymerase domain of intact polδ may not be introduced at random but at the so-called “hot spots”. Accordingly, artificial destructions of the polymerase-domain by gene-manipulation may mean the disturbance of the natural cause of mutations. After all, the best target to be manipulated for attaining experimental acceleration of evolution would be the editing (exonuclease)-domain of polδ gene.
5. CONCLUDING REMARKS
In the present review, three different concepts; 1) determination of cell differentiation by means of selective segregation of chromatids, 2) preventing mechanism of cancer in stem cells, 3) the acceleration of evolution by biased-mutagenesis in lagging-strand synthesis, were introduced. Although these three concepts were constructed from different philosophies respectively, they share a common point of view. Namely, three researchers introduced here are advancing the same paradigm; in that higher organisms recognize and utilize the “doublestranded structure of DNA” for the works of life.
As Klar pointed out, his model is generally expanded into the explanation of the causal genes of human diseases that seem to relate “laterality”; e.g., the etiology of schizophrenia and bipolar psychiatric disorders, etc. [18].
On the other hand, Cairns’ model not only would give us a new aspect of understanding cell differentiation, but also offer a new way of diagnosis and the treatment of cancer. Cairns’ concept is a developing one. To prove his concept, more accumulations of evidence that support his idea would be required.
The most noticeable feature of our disparity mutagenesis model would be that parental genotypes are guaranteed by error-less leading strands for realizing more reliable inheritance, while lagging strands make a venture for future evolution by accumulating mutations. Our disparity-mutagenesis model is also underdeveloped. First of all, using living organisms, we have to check whether strand-biased-mutagenesis does take place in germ-line cells. Computer simulations are also necessary using more complex genetic algorithms copying the genetics systems of multi-cellular organisms.
The new paradigm discussed in the present review seems to involve implications far beyond conventional molecular biology.
6. ACKNOWLEDGEMENTS
The present author thanks Drs. Klar, A., Conway Morris, S., Ko, M., Yagi, T., Uchimura A. and Mr. Noda, M. for their illuminating discussions about general aspects of evolution, and also Drs. Komiya, T., Shimoda, C., Yoshida, M and Mr. Suwa, N. for useful discussions about the disparity model of evolution. The author thanks Drs. Rueker, F. and K. A. for their critical reading of the manuscript and for useful suggestions.
REFERENCES
- Watson, J. and Crick, F. (1953) Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature, 171, 737-738. doi:10.1038/171737a0
- Klar. A.J. (2007) Lessons learned from studies of fission yeast mating-type switching and silencing. Annual Review of Genetics, 41, 213-236. doi:10.1146/annurev.genet.39.073103.094316
- Cairns, J. (1975) Mutation selection and the natural history of cancer. Nature, 255, 197-200. doi:10.1038/255197a0
- Furusawa, M. and Doi, H. (1992) Promotion of evolution: disparity in the frequency of strand-specific misreading between the lagging and leading strands enhances disproportionate accumulation of mutations. Journal of Theoretical Biology, 157, 127-133. doi:10.1016/S0022-5193(05)80761-1
- Tanabe, K., Kondo, T., Onodera, Y. and Furusawa, M. (1999) A conspicuous adaptability to antibiotics in the Escherichia coli mutator strain, dnaQ49. FEMS Microbiology Letters, 176, 191-196. doi:10.1111/j.1574-6968.1999.tb13661.x
- Shimoda, C., Itadani, A., Sugino, A. and Furusawa, M. (2006) Isolation of thermotolrant mutants by using proofreading-deficient DNA polymerase δ as an effective mutator in Saccharomyces serevisiae. Genes & Genetic Systems, 81, 391-397. doi:10.1266/ggs.81.391
- Klar, A.J. (1994) A model for specification of the leftright axis in vertebrates. Trends in Genetics, 10, 392-396. doi:10.1016/0168-9525(94)90055-8
- Dalgaard, J. and Klar, A.J. (2001) Does S. pombe exploit the intrinsic asymmetry of DNA synthesis to imprint daughter cells for mating-type switching? Trends in Genetics, 17, 153-157. doi:10.1016/S0168-9525(00)02203-4
- Dalgaard, J. and Klar, A.J. (2000) Swi 1 and swi 3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell, 102, 745-751. doi:10.1016/S0092-8674(00)00063-5
- Supp, D., Witte. D., Potter. S. and Bruekner (1997) Mutation of an exonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature, 389, 963-966. doi:10.1038/40140
- Hirokawa, N., Tanaka, Y., Okada, Y. and Takeda, S. (2006) Nodal flow and the generation of left-right asymmetry. Cell, 125, 33-45. doi:10.1016/j.cell.2006.03.002
- Klar, A.J. (2008) Support for the selective chromatid segregation hypothesis advanced for the mechanism of leftright body axis development in mice. Breast Disease, 29, 47-56.
- Layton, W. Jr. (1978) Heart malformations in mice homozygous for a gene causing situs inversus. Birth Defects Original Article Series, 14, 277-293.
- Armakolas A. and Klar, A.J. (2006) Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science, 311, 1146-1149. doi:10.1126/science.1120519
- Armakolas A. and Klar, A.J. (2007) Left-light dynein motor implicated in selective chromatid segregation in mouse cells. Science, 315, 100-101. doi:10.1126/science.1129429
- Sandon, T., Wen, P. and LeMay, M. (1992) Reversed cerebral asymmetry in women with breast cancer. Lancet, 339, 523-524. doi:10.1016/0140-6736(92)90341-Y
- Klar. A.J. (2010) A hypothesis: Breast cancer predisposition and brain hemispheric laterality specification likely share a common genetic cause. Breast Disease, 31, 1-4.
- Klar, A.J. (2004) An epigenetic hypothesis for human brain laterality, handedness and psychosis development. Cold Sprig Harbor Symposium on Quantitative Biology, 69, 499-406.
- Potten. C., Hume, W., Reid, P. and Cairns, J. (1978) The segregation of DNA in epithelial stem cells. Cell, 15, 899-906. doi:10.1016/0092-8674(78)90274-X
- Potten, C., Owen, G. and Booth, D. (2002) Intestinal stem cells protect their genome by selective segregation of template DNA strands. Journal of Cell Science, 115, 2381-2388.
- Merok, J. Lansita, J., Tunstead, J. and Sherly, J. (2002) Cosegregation of chromosomes containing immortal DNA strands in cells that cycle with asymmetric stem cell kinetics. Cancer Research, 62, 66791-6795.
- Potten, C. (2004) Keratinocyte stem cells, label-retaining cells and possible genome protection mechanisms. Journal of Investigative Dermatology Symposium Proceedings, 9, 183-195. doi:10.1111/j.1087-0024.2004.09305.x
- Karpowicz, P., Morshead, C., Kam, A., Jervis, E., Ramunas, J., Cheng, V. and van der Kooy, D. (2005) Support for the immortal strand hypothesis: Neural stem cells partition DNA asymmetrically in vitro. Journal of Cell Biology, 170, 721-734. doi:10.1083/jcb.200502073
- Smith, G. (2005) Label-retaining epithelial cells in mouse mammary gland divide asymmetrically and retain their template DNA strands. Development, 132, 681-687. doi:10.1242/dev.01609
- Rambhatla, L., Ram-Mohan, S., Cheng, J. and Sherly, J. (2005) Immortal DNA strand cosegregation requires p53/IMPDH-dependent asymmetric self-renewal associated with adult stem cells. Cancer Research, 65, 3155- 3161.
- Conboy, M., Karasov, A. and Rando, T. (2007) High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biology, 5, e102. doi:10.1371/journal.pbio.0050102
- Kuroki. T and Murakami, Y. (2007) Random segregation of DNA strands in epidermal basal cells. Japanese Journal of Cancer Research, 80, 637-642. doi:10.1111/j.1349-7006.1989.tb01690.x
- Ito, K and McGhee, J. (1987) Parental DNA strands segregate randomly during embryonic development of Caenorhabditis elegance. Cell, 49, 329-336. doi:10.1016/0092-8674(87)90285-6
- Ito, K., McGhee, J. and Shultz, G. (1988) Parental DNA strands segregate to both trophectoderm and inner cell mass of the developing mouse embryo. Genes & Development, 2, 929-936. doi:10.1101/gad.2.8.929
- Lark, K., Consigli, R. and Minocha, H. (1966) Segregation of sister chromatids in mammalian cells. Science, 154, 1202-1205. doi:10.1126/science.154.3753.1202
- Rosenberger, R. and Kessel, M. (1968) Nonrandom sister chromatid segregation and nuclear migration in hyphae of Asperigillus nidulans. Journal of Bacteriology, 96, 1208-1213.
- Wada, k., Doi, H., Tanaka, S., Wada, Y. and Furusawa, M. (1993) A neo-Darwinian algorithm: Asymmetrical mutations due to semiconservative DNA-type replication promote evolution. Proceedings of the National Academy of Sciences of the United States of America, 90, 11934- 11938.
- Eigen, M., McCaskill, J. and Schuster, P. (1989). The mo lecular quasispecies. Advance in Chemistry and Physics, 75,149-263. doi:10.1002/9780470141243.ch4
- Aoki, K. and Furusawa, M. (2003) Increase in error threshold for quasispecies by heterogeneous replication accuracy. Physical Review E, 68, 031904-1-031904-6.
- Iwaki, T., Kawamura, A., Ishino, Y., Kohno, K., Kano, Y., Goshima, N., Yara, M., Furusawa, M., Doi, H. and Imamoto. F. (1996). Preferential replication-dependent mutagenesis in the lagging DNA strand in Escherichia coli. Molecular & General Genetics, 251, 657-664.
- Furusawa, M. (1999) DNA’s exquisite evolutionary strategy. Kodansha Ltd., Tokyo.
- Albertson, T., Ogawa, M., Bugni, J., Hays, L., Chen, Y., Wang, Y., Treuting, P., Heddle, J., Goldsby, R. and Preston, B. (2009) DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proceedings of the National Academy of Sciences of the United States of America, 106, 17101-17104. doi:10.1073/pnas.0907147106
- Uchimura, A., Hidaka, Y., Hirabayashi, T., Hirabayashi, M. and Yagi, T. (2009) DNA polymerase delta is required for early mammalian embryogenesis. PLoS One, 4, e4184. doi:10.1371/journal.pone.0004184
- Katoh, K., Kuma, K., Iwabe, N. and Miyata, T. (2005) Putative decline of DNA polymerase δ in mammalian lineage. Proceedings of the 28th Conference on the Molecular Biology Society of Japan, 7-10 December 2005, Fukuoka, 2P-0257.