Journal of Cosmetics, Dermatological Sciences and Applications
Vol.1 No.4(2011), Article ID:8840,7 pages DOI:10.4236/jcdsa.2011.14025
Xeroderma Pigmentosum with Desquamative Gingivitis a Rare Case Report and Detailed Review of Literature
![]()
1(Oral Medicine & Radiology) Faculty of Dentistry JMI, New Delhi, India; 2Z. A. Dental College & Hospitals, A. M. U. Aligarh, India.
E-mail: {shamim0571, mohdabbaskhan}@gmail.com
Received July 23rd, 2011; revised September 4th, 2011; accepted September 21st, 2011.
Keywords: Oral Genodermatosis, Xeroderma Pigmentosum, Desquamative Gingivitis, Developmental Tongue Lesions
ABSTRACT
In xeroderma pigmentosum, a rare genodermatosis, transmitted as an autosomal recessive disorder, excessive solar damage to the skin develops at an early age. The disease is characterized by cutaneous, ocular, neurological and oral changes. Oral features in the form of early development of squamous cell carcinoma, usually at the lower lip and tip of the tongue may be seen. The disorder is associated more commonly in populations where marriage of close blood relatives is common. Treatment of the disorder includes avoidance of Ultra violet radiation, topical application of 5- fluorouracil to treat actinic keratoses, and regular evaluation by an ophthalmologist, dermatologist, and neurologist. Genetic counseling is an important aspect as an increased incidence of consanguineous marriages has been reported with this disorder. Here, we report an interesting case of xeroderma pigmentosum in an 18 year old male patient who presented with characteristic desquamation of gingiva, fissured tongue and geographic tongue.
1. Introduction
Xeroderma pigmentosum serves as the prototype heritable disease with increased sensitivity to cellular injury [1-4]. Xeroderma pigmentosum is a rare, genetically heterogeneous, autosomal recessive disorder characterized by photosensitivity, cutaneous pigmentary changes, premature skin ageing, and the development of various cutaneous and internal malignancies at an early age. The basic defect underlying the clinical manifestations is a nucleotide excision repair (NER) defect leading to a defective repair of DNA damaged by ultra violet (UV) radiation [5]. Xeroderma pigmentosum is categorized in at least eight complementation groups according to the capacity of the body to repair DNA. These groups (i.e. genetic subtypes) are labeled A through G, plus the XP variant: XPV Groups A, C, D, and variant make up to 90% of XP cases. Group A, for example, has the lowest level of DNA repair and the most neurological manifestations [6]. Frequency of xeroderma pigmentosum has been reported from a low of 1:250,000 in the United States to a high of 1:400,000 in Japan [7]. It is more common in children of consanguineous parents [8]. However, a case report by Stephanic Christen-Zaech et al. showed that the disease did occurred in an Uncle and a Nephew [9]. XP is characterized by clinical and cellular hypersensitivity to UV radiation manifesting as intolerance of skin and eyes to light. The skin lesions are comprised of freckles on limbs and face with a dry skin covered with a mixture of mottled, hypopigmented and hyperpigmented, atrophic rounded and oval macules, giving the entire skin a checkered appearance associated with a generalized actinic keratoses, manifesting on black skin as palpable, rough, blackish spots covered with adherent scales [10]. These skin lesions cover both the sun exposed and covered areas. The skin later develops cutaneous malignancies. Ocular changes include photophobia, ocular pigmentary changes, conjunctivitis, corneal keratitis, ulcers, blindness, and malignancies. An interesting case study by Syed A. R. Rizvi et al. showed a rare occurrence of orbital amelanotic melanoma in xeroderma pigmentosum [11]. No consistent routine clinical laboratory abnormality is observed in individuals with XP. The diagnosis of XP can be established with studies performed in specialized laboratories. These studies include cellular hypersensitivity to UV radiation and chromosomal breakage studies, complementation studies, and gene sequencing to identify the specific gene complementation group [7]. Treatment modalities for XP include Isotretinoin prophylaxis, avoidance of light exposure, surgical excision of pre malignant and malignant tumors, resurfacing with skin grafts, dermabrasion, radiation therapy, and Mohs micrographic surgery [12,13].
The patient, in our case report, presented with the history of consanguineous marriage of the parents, characteristic atrophic, freckled skin changes, subnormal intelligence and blurred vision along with the highlighting oral manifestations in the form of desquamative gingivitis, fissured and geographic tongue.
2. Case Report
An 18 year old male patient reported to the department of Oral Medicine and radiology, Faculty of Dentistry, J. M. I. University, New Delhi, with the complaint of discolored skin, all over the body, particularly marked over the face, scalp, upper part of the trunk, and upper limbs since the age of 8 months. Patient’s parents revealed that the lesions were initially seen over the face, more evident on both sides of the nose; then the lesions gradually spread all over the body. Severe itching and burning sensation on sun exposure was also experienced by the patient. The patient was born to a Muslim couple and his parents had history of consanguineous marriage.
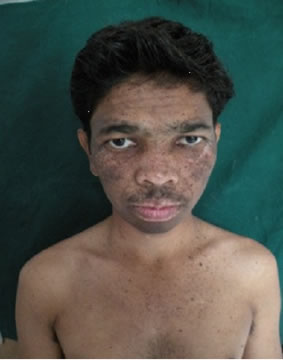
The patient’s sister also had similar discoloration, but less severe, and limited to the face and neck. The medical, surgical and dental history was non contributory. On examination, the patient was irritable and resented light. The patient was of sub normal intelligence and had slurred speech. The skin was pale, dry and atrophic, freckled, scaly and rough. Discrete, multifocal brownblack hyperpigmented macules interspersed with patchy hypopigmentation, particularly abundant over the facial malar area, forehead, ear, eyes, and neck (Figure 1), upper part of the trunk and back (Figures 2(a), (b)), and extensor surfaces of the upper and lower extremities (Figures 2(c), (d)) were seen. The macules varied in size from 1 - 3 mm in diameter, round-ovoid in shape, and slightly rose above the surface. Ocular involvement in the form of blurred vision and difficulty in seeing far away objects (short sightedness) was noticed. Neurological examination was non contributory. The patient was mature for his age. On intra-oral examination, desquamation of gingiva, involving the marginal and attached gingiva was seen in relation to 13 - 23 (Figure 3). There was bleeding on probing. The gingiva was firm and resilient in consistency. Mild fissuring of dorsum of tongue (Figure 4) with multiple depapillated areas surrounded by raised circinate borders on the lateral and anterior border of tongue, suggestive of geographic tongue were also seen (Figure 5). Laboratory investigations (Complete blood count, Renal function tests, Liver function tests, chest X-Ray, ECG etc.) were non contributory. After the informed


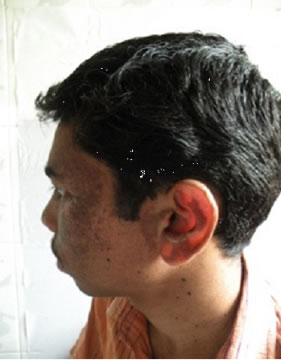

Figure 1. Hyperpigmented macules interspersed with patchy hypopigmentation over face, ear, eyes & forehead.
 (a)
(a)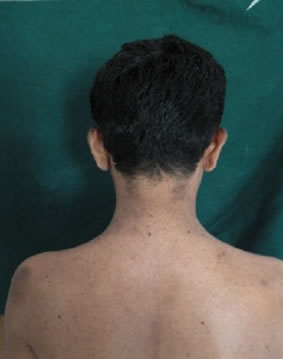 (b)
(b)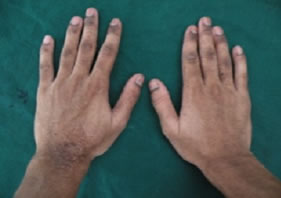 (c)
(c)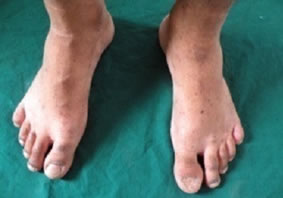 (d)
(d)
Figure 2. Rough, dry, scaly skin with hyperpigmented macules over the truncal area, back, hands & feet.

Figure 3. Area of desquamation involving marginal & attached gingival in relation to 13 - 23.
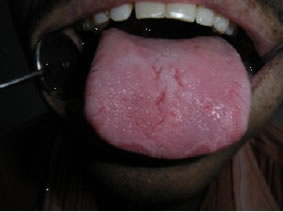
Figure 4. Fissuring over the dorsum of tongue.
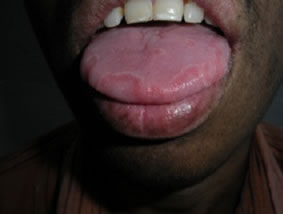
Figure 5. Depapillated areas with whitish circinate borders, suggestive of geographic tongue.
consent to the patient, incisional biopsy was made, both from the skin lesions and the desquamated gingiva. Skin biopsy confirmed the diagnosis of XP. Histopathology of the desquamated area showed parakeratinized stratified squamous epithelium of variable thickness, dense connective tissue stroma with moderate inflammatory cells and areas of vascularity and hemorrhage, features suggestive of chronic desquamative gingivitis (Figure 6). The patient was subjected to thorough oral prophylaxis and oral hygiene instructions. Thereafter, the patient was prescribed topical application of low potency steroids (triamcinolone acetonide) two times daily for 30 days. The patient was reviewed every week for the first one month. The lesions had subsided with topical steroids within 2 weeks of starting the treatment (Figure 7). The patient was asked to stop the topical application and reinforcement of oral hygiene instructions was given.
Since the lesions can recur, the patient was under observation for 3 months and the lesions showed no signs of recurrence.
3. Discussion
Xeroderma pigmentosum (XP) is a rare autosomal-recessive, progressive, degenerative disease associated with sun sensitivity, cutaneous pigmentation, neoplasia, abnormal DNA repair, and, in some patients, neurologic degeneration [14]. About 20% to 30% of patients with XP develop severe progressive neurologic deterioration
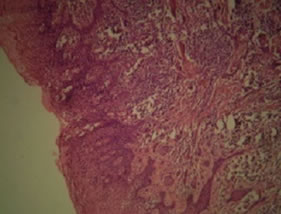
Figure 6. Histopathology suggestive of chronic desquamative gingivitis.
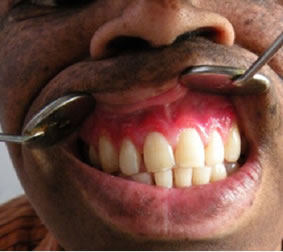
Figure 7. Remission of lesions post operatively.
characterized by loss of reflexes, sensorineural hearing loss, ataxia, dysphagia, and decreasing cognition [2,15].
3.1. Historical Background
In 1870, Moritz Kaposi first used the term “xeroderma” to characterize the dry, dyspigmented skin that is the first permanent cutaneous change observed in patients with this disease [16]. Xeroderma pigmentosum was was first described in 1874 by Hebra and Kaposi [7]. 12 years later Kaposi added “pigmentosum” [17]. The first XP case with neurological signs was described by Dr. Albert Neisser [18] and in 1932, DeSanctis and Cacchione [19] helped coin the term “DeSanctis-Cacchione syndrome” to apply to cases of XP with severe neurological deficiency. In 1968, James Cleaver [5] conducted a study which demonstrated that fibroblasts from patients with XP were unable to perform nucleotide excision repair (NER) of DNA.
3.2. Etio-Pathogenesis
UV radiation-exposure to UV light induces DNA lesions [20]. The majority of DNA damage is caused by UVB light, which leads to the most common forms of DNA damage, cyclobutane pyrimidine dimers (CPDs) and 6-pyrimidine-4-pyrimidone photoproducts [21]. Recently, UVA light-induced DNA damage has also been linked to identical mutations [20]. Individuals with XP are unable to remove these lesions, and studies have strongly correlated these errors with early onset of tumorigenesis [21]. It is noteworthy that subjects with XP have a 10 to 20-fold increase in various internal neoplasms that have no UV aetiology compared to the general population [22], suggesting that the repair of endogenous oxidative DNA damage may also be dysregulated in some subjects with XP [23]. Normally, DNA repair is achieved through nucleotide excision repair (NER) or postreplication repair (PRR) [24,25].
3.2.1. Consanguinity
Consanguinity in the parents of the patients has been implicated as an etiologic factor. This has been reported to varying degrees of up to 92.8% in XP patients in Libya [26]. It has also been reported in studies from Egypt, Pakistan, and Nigeria among some of the countries that have a high incidence of XP [10,27]. F. Handa et al. described a series of four cases and consanguinity had not been observed in those parents of the patients [28]. The patient in the present case was born to parents who had a history of consanguineous marriage. There is, however, a varying degree of severity and the rate of progression of the disease even among siblings [10,26, 27]. In our case, the patient’s sister was affected to a less severe extent, and the pigmentary changes were restricted only to the face and neck.
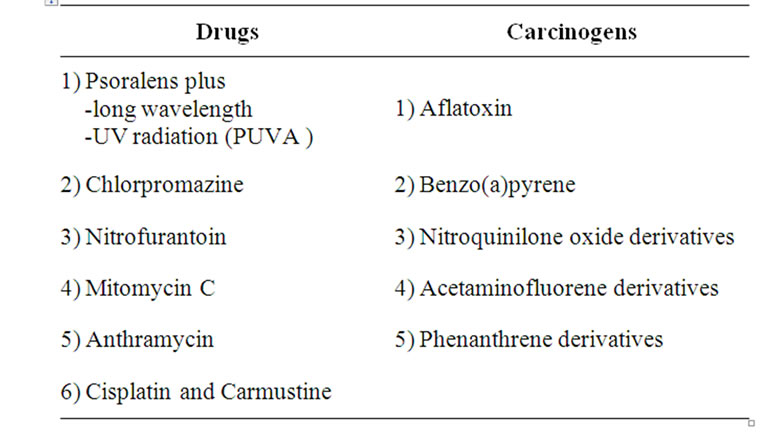
3.2.2. Drugs and Chemicals
A number of DNA-damaging agents other than UV radiation have been found to yield hypersensitive responses with XP cell [4,29].
3.3. Clinical Manifestations
3.3.1. Skin
Approximately half of the patients with XP have a history of acute sunburn reaction on minimal UV exposure. The other patients apparently tan normally without excessive burning. The median age of onset of cutaneous symptoms is between 1 and 2 years [2]. In our patient, the symptoms appeared at the age of 8 months. The disease usually progresses through 3 stages.
3.3.1.1. First Stage
The skin is healthy at birth. The first stage occurs around 6 months after birth.
• Diffuse erythema, scaling, and freckle like areas of increased pigmentation characterize this stage.
• These findings are seen over light-exposed areas, appearing initially on the face. With progression of the disease, the skin changes appear on the lower legs, the neck, and even the trunk in extreme cases.
• While these features tend to diminish during the winter months with decreased sun exposure, as time passes, these findings become permanent.
3.3.1.2. Second Stage
Continued sun exposure will lead to the second stage, which is characterized by poikiloderma.
• Poikiloderma consists of skin atrophy, telangiectasias, and mottled hyperpigmentation and hypopigmentation, giving rise to an appearance similar to that of chronic radiodermatitis.
3.3.1.3. Third Stage
The third stage is the development of solar keratoses and skin cancers [30]. Physical findings in our case were suggestive of second stage of the disease.
3.3.2. Cancer
Patients with XP under 20 years of age have a greater than 1000-fold increased risk of cutaneous basal cell or squamous cell carcinoma or melanoma [2,31]. The median age of onset of non-melanoma skin cancer reported in patients with XP was 8 years, in comparison to 60 years in the general population [2,31]. Multiple primary cutaneous neoplasms, including melanomas, commonly occur. Review of the world literature has revealed a substantial number of cases of oral cavity neoplasms, particularly squamous cell carcinoma of the tip of tongue—a presumed sun-exposed location [2,31].
3.3.3. Ocular
Between 40% and 80% of patients with XP have ocular abnormalities caused by UV induced DNA alteration to epithelial cells of the conjunctiva, the cornea, and the eyelid [32,33]. They also have photophobia, conjunctivitis, keratitis that may lead to corneal opacification, hyperpigmentation of the eyelids, loss of eyelashes, and malignancies including SCC, basal cell carcinoma and melanoma [15,22]. In our patient, there was blurring of vision along with difficulty in seeing far away objects.
3.3.4. Nervous System
Approximately 30% of individuals with XP may present with neurological abnormalities including isolated hyporeflexia, progressive mental retardation, sensorineural deafness, spasticity, or seizures [34,35]. Neurological abnormalities are only seen in individuals with defects in complementation groups XPA, XPB, XPD, and XPG [34]. The most common neurological abnormality is a loss of high-frequency hearing while the most severe neurological deficits are seen in DeSanctis-Cacchione syndrome [34,36]. In the present case, the patient was irritable and of sub normal intelligence.
3.3.5. Oral Cavity
Leukoplakia, erythroplakia and SCC of the tip of the tongue, actinic cheilitis and SCC of the lips are associated with XP. The precancerous and cancerous lesions of the tip of the tongue, sites seldom affected in the normal population group, are presumed to be induced by UV radiation. This is not a convincing explanation but it is the only one offered [22,37,38]. In the general population, SCC most frequently affects the posterolateral and ventral surfaces of the tongue and floor of the mouth of elderly users of tobacco and alcohol, and runs an aggressive course. By contrast XP associated SCC affects the tip of the tongue of persons younger than 20 years of age and runs a slowly progressive course [37].
Chronic desquamative gingivitis was first described by Tomes and Tomes [39] in 1894. However, it was not until the 1930s that Prinz [40] and Merrit [41] first proposed the term of chronic diffuse desquamative gingivitis and first attempted to define the disease process. DG is a fairly common disorder in which the gingivas are desquamated. Chronic soreness is common and can be worse with the intake of spicy foods [42]. The clinical appearance is of gingival erythema and loss of stippling, extending apically from the gingival margins to the alveolar mucosa. The desquamation may vary from mild, almost insignificant small patches to widespread erythema [43] with a glazed appearance. DG is now recognized to be mainly a manifestation of a number of disorders ranging from vesiculobullous diseases to adverse reactions to a variety of chemical or allergens [44,45]. Other disorders manifesting as DG include dermatitis herpetiformis, linear IgA disease, chronic ulcerative stomatitis, pemphigus, erythema multiforme, pyostomatitis vegetans, epidermolysis bullosa acquisita, and Kindler syndrome; similar appearance may be seen in reactions to various mouthwashes, chewing gums, medications, and dental materials and in candidosis, lupus erythematosus, plasma cell gingivitis, Crohn’s disease, sarcoidosis, some leukaemias, and even some factitious lesions [45].
Fissured tongue is a condition frequently seen in the general population that is characterized by grooves that vary in depth and are noted along the dorsal and lateral aspects of the tongue. Although a definitive etiology is unknown, a polygenic mode of inheritance is suspected because the condition is seen clustering in families who are affected. Patients are usually asymptomatic, and the condition is initially noted on routine intraoral examination as an incidental finding. Fissured tongue is also seen in Melkersson-Rosenthal syndrome [46] and Down syndrome and in frequent association with benign migratory glossitis (geographic tongue). Fissured tongue and geographic tongue have been reported in association with chronic granulomatous disease [47].
In the present case, the patient presented with desquamative gingivitis in relation to 13 - 23 along with developmental anomalies of tongue, like geographic tongue and fissured tongue. To our knowledge, this is the first case in literature of Xeroderma pigmentosum, associated with desquamation of gingiva and developmental tongue lesions.
4. Treatment
There is no cure for XP. The DNA damage is cumulative and irreversible [6]. Persons with XP must avoid exposure to any sources of UV light including sunlight, fluorescent, halogen and mercury-vapour lights [37], and must wear protective clothing and UV-absorbing eye glasses, and must use high protection factor sunscreens [22]. Regarding medical care, oral retinoids have been shown to decrease the incidence of skin cancer in patients with XP. Chemical therapy with 5-fluorouracil may be useful for actinic keratoses. A new approach to photoprotection is to repair DNA damage after UV exposure. This can be accomplished by delivery of a DNA repair enzyme into the skin by means of specially engineered liposomes. Surgical care includes complete excision of the malignancies associated with XP [7]. Regular surveillance for and treatment of all neoplasms is very important [6]. Regular visits to the dermatologist might be necessary for the purposes of patient education as well as early detection and treatment of any malignancy [7]. Ophthalmologic and neurologic consultations are recommended for XP. It must be remembered that persons with XP who are properly protected from sun-light may suffer consequential vitamin D deficiency, and they should routinely take vitamin D supplements [37].
5. Conclusions
Xeroderma pigmentosum is a life threatening disorder, as various malignancies may occur at an early age, so early diagnosis and management may prove fruitful. Genetic counseling implicating the effect of consanguineous marriages should be emphasized.
REFERENCES
- J. H. Robbins, et al., “Xeroderma Pigmentosum: An Inherited Diseases with Sun Sensitivity, Multiple Cutaneous Neoplasms, and Abnormal DNA Repair,” Annals of Internal Medicine, Vol. 80, No. 2, 1974, pp. 221-248.
- K. H. Kraemer, et al., “Xeroderma Pigmentosum: Cutaneous, Ocular and Neurologic Abnormalities in 830 Published Case,” Archives of Dermatology, Vol. 123, No. 2, 1987, pp. 241-250. doi:10.1001/archderm.123.2.241
- H. Takebe, et al., “Genetic and Skin Cancer of Xeroderma Pigmentosum in Japan,” Japanese Journal of Cancer Research, Vol. 78, No. 11, 1987, pp. 1135-1143.
- J. E. Cleaver and K. H. Kraemer, “Xeroderma Pigmentosum and Cockayne Syndrome in the Metabolic and Molecular Basis of Inherited Diseases,” 7th Edition, McGraw-Hill, New York, 1995, p. 4393.
- J. E. Cleaver, “Defective Repair Replication of DNA in Xeroderma Pigmentosum,” Nature, Vol. 218, No. 5142, 1968, pp. 652-656. doi:10.1038/218652a0
- Xeroderma Pigmentosum Society, New York, USA, 2005. http://www.xps.org
- M. G. Horenstein and A. H. Diwan, “Xeroderma Pigmentosum,” 2005. http://www.emedicine.com/derm/topic 462.htm
- K. Yagi, A. el-G. Ali, K. el-D. Abbas, et al., “Carcinoma of the Tongue in a Patient with Xeroderma Pigmentosum,” International Journal of Oral Surgery, Vol. 10, No. 1, 1981, pp. 73-76. doi:10.1016/S0300-9785(81)80011-7
- S. Christen-Zaech, K. Imoto, S. G. Khan, et al., “Unexpected Occurrence of Xeroderma Pigmentosum in an Uncle and Nephew;” Archives of Dermatology, Vol. 145, No. 11, 2009, pp. 1285-1291.
- H. Ahmed and R. Hassan, “Xeroderma Pigmentosum in Three Consecutive Siblings of a Nigerian Family: Observations on Oculo Cutaneous Manifestations in Black African Children,” British Journal of Ophthalmology, Vol. 85, No. 1, 2001, p. 110. doi:10.1136/bjo.85.1.110
- S. R. Rizvi, A. K Amitava, G. Mehdi, R. Sharma and M. S. Alam, “Orbital Amelanotic Melanoma in Xeroderma Pigmentosum: A Rare Association: Presentation at a Meeting,” Indian Journal of Ophthalmology, Vol. 56, No. 5, 2008, pp. 421-423.
- K. Agarwal, A. J. Veliath, S. Mishra, et al., “Xeroderma Pigmentosum: Resurfacing Versus Dermabrasion,” British Journal of Plastic Surgery, Vol. 45, No. 4, 1992, pp. 311-314. doi:10.1016/0007-1226(92)90059-7
- S. Leal-Khouri, G. J. Hruza, L. L. Hruza, et al., “Management of a Young Patient with Xeroderma Pigmentosum,” Pediatric Dermatology, Vol. 11, No. 1, 1994, pp. 72-75. doi:10.1111/j.1525-1470.1994.tb00080.x
- K. H. Kraemer and H. Slor, “Xeroderma Pigmentosum,” Clinics in Dermatology, Vol. 3, No. 1, 1985, pp. 33-69. doi:10.1016/0738-081X(85)90096-3
- K. H. Kraemer, N. J. Patronas, R. Schiffmann, B. P. Brooks, D. Tamura and J. J. DiGiovanna, “Xeroderma Pigmentosum, Trichothiodystrophy and Cockayne Syndrome: A Complex Genotype-Phenotype Relationship,” Neuroscience, Vol. 145, No. 4, 2007, pp. 1388-1396. doi:10.1016/j.neuroscience.2006.12.020
- F. Hebra and M. Kaposi, “On Diseases of the Skin including Exanthemata,” New Sydenham Society, Vol. 61, 1874, pp. 252-258.
- M. Kaposi, “Xeroderma Pigmentosum [in French],” Ann Dermatol Venereol, Vol. 4, 1883, pp. 29-38.
- Neisser A, “Ueber das “Xeroderma Pigmentosum” (Kaposi): Lioderma Essentialis Cummelanosi et Telangiectasia,” Vierteljahrschr Dermatol Syphil, 1883, pp. 47-62.
- C. De Sanctis and A Cacchione, “Xerodermatic Idiocy,” Riv Sper Freniat, Vol. 56, 1932, pp. 269-292.
- A. Sarasin, “The Molecular Pathways of Ultraviolet-Induced Carcinogenesis,” Mutation Research, Vol. 428, No. 1-2, 1999, pp. 5-10. doi:10.1016/S1383-5742(99)00025-3
- H. van Steeg and K. H. Kraemer, “Xeroderma pigmentosum and the Role of UV-induced DNA Damage in Skin Cancer,” Molecular Medicine Today, Vol. 5, No. 2, 1999, pp. 86-94. doi:10.1016/S1357-4310(98)01394-X
- T. M. Rünger, J. J. DiGiovanna and K. H. Kraemer, “Hereditary Disorders of Genome Instability and DNA Repair,” In: K. Wolff, L. A. Goldsmith, S. I. Katz, et al., Eds., Fitzpatrick’s Dermatology in General Medicine, McGraw-Hill, New York, 2008, pp. 1311-1325.
- Y. Shimizu, I. Shigenori, F. Hanaoka, et al., “Xeroderma Pigmentosum Group C Protein Interacts Physically and Functionally with Thymine DNA Glycosylase,” The EMBO Journal, Vol. 22, 2003, pp. 164-173. doi:10.1093/emboj/cdg016
- J. H. Robbins, “Xeroderma Pigmentosum: Defective DNA Repair Causes Skin Cancer and Neurodegeneration,” Journal of the American Medical Association, Vol. 260, No. 3, 1988, pp. 384-388. doi:10.1001/jama.260.3.384
- A. Gratchev, P. Strein, J. Utikal, et al., “Molecular Genetics of Xeroderma Pigmentosum Variant,” Experimental Dermatology, Vol. 12, No. 5, 2003, pp. 529-536. doi:10.1034/j.1600-0625.2003.00124.x
- M. L. Khatri, M. Bemghazil, M. Shafi, et al., “Xeroderma Pigmentosum in Libya,” International Journal of Dermatology, Vol. 38, No. 7, 1999, pp. 520-524. doi:10.1046/j.1365-4362.1999.00751.x
- H. Masinjila and E. Arnbjornsson, “Two Children with Xeroderma Pigmentosum Developing Two Different Types of Malignancies Simultaneously,” Pediatric Surgery International, Vol. 13, No. 4, 1998, pp. 299-300. doi:10.1007/s003830050324
- F. Handa, R. L. Manchanda and R. Singh, “Xeroderma Pigmentosum,” Indian Journal of Dermatology, Venereology and Leprology, Vol. 34, No. 6, 1968, pp. 231-236.
- E. C. Friedberg, et al., “DNA Repair and Mutagenesis,” ASM Press, Washington DC, 1995.
- Xeroderma Pigmentosum. New Zealand Dermatological Society, Hamilton, New Zealand, 2005. http://www.dermnetnz.org/systemic/xerodermapigmentosum.html
- K. H. Kraemer, et al., “The Role of Sunlight and DNA Repair in Melanoma and Non Melanoma Skin Cancer: The Xeroderma Pigmentosum Paradigm,” Archives of Dermatology, Vol. 130, No. 8, 1994, pp. 1018-1021. doi:10.1001/archderm.130.8.1018
- L. J. Niedernhofer, “Tissue-Specific Accelerated Aging in Nucleotide Excision Repair Deficiency,” Mechanisms of Ageing and Development, Vol. 129, No. 7-8, 2008, pp. 408-411. doi:10.1016/j.mad.2008.04.010
- B. Gupta, R. Anegundi and C. M. Marya, “Xeroderma Pigmentosa; Review and Case Report,” Journal of Oral Health & Community Dentistry, Vol. 1, No. 2, 2007, pp. 43-55.
- K. H. Kraemer, “Heritable Diseases with Increased Sensitivity to Cellular Injury,” In: I. M. Freedberg, A. Z. Eisen, K. Wolff, et al., Eds., Fitzpatrick’s Dermatology in General Medicine, McGraw-Hill, New York, 2003, pp. 1508- 1521.
- J. L. Hawk and H. W. Lim, “Photodermatoses,” In: J. L. Bolognia, J. L. Jorizzo, R. P. Rapini, et al., Eds., Dermatology, Mosby, New York, 2003, pp. 1365-1383.
- W. C. Lambert, H.-R. Kuo and M. W. Lambert, “Xeroderma Pigmentosum,” In: A. C. Chu, R. L. Edelson, Eds., Malignant Tumors of the Skin, Oxford University Press, New York, 1999, pp. 119-137.
- P. Mahindra, J. J. DiGiovanna, D. Tamura, et al., “Skin Cancers, Blindness, and Anterior Tongue Mass in African Brothers,” Journal of the American Academy of Dermatology, Vol. 59, No. 5, 2008, pp. 881-886. doi:10.1016/j.jaad.2008.06.030
- B. W. Neville, D. D. Damm, C. M. Allen, et al., “Dermatologic Diseases,” In: B. W. Neville, D. D. Damm, C. M. Allen, et al., Eds., Oral and Maxillofacial Pathology, Saunders Elsevier, Missouri, 2009, pp. 747-748.
- J. Tomes, G. Tomes, “Dental Surgery,” 4th Edition, J & A Churchill Ltd., London, 1894.
- H. Prinz, “Chronic Diffuse Desquamative Gingivitis,” Dental Cosmos, Vol. 74, 1932, p. 331.
- A. H. Merrit, “Chronic Desquamative Gingivitis,” Journal of Periodontology, Vol. 4, 1932, pp. 30-34.
- A. Alkan, O. Gunhan, A. Alkan and F. Otan, “A Clinical Study of Oral Mucous Membrane Pemphigoid,” Journal of International Medical Research, Vol. 31, No. 4, 2003, pp. 340-344.
- F. Y. Bozcurt, H. Celenligil, A. Sungur and S. Ruacan, “Gingival Involvement in Mucous Membrane Pemphigoid,” Quintessence International, Vol. 29, No. 7, 1998, pp. 438-441.
- C. Scully and S. R. Porter. “The Clinical Spectrum of Desquamative Gingivitis,” Seminars in Cutaneous Medicine and Surgery, Vol. 16, No. 4, 1997, pp. 308-313.
- R. J. Niesengard and R. A. Levine, “Diagnosis and Management of Desquamative Gingivitis,” Periodontal Insights, Vol. 2, 1995, pp. 4-10.
- E. Kaminagakura and J. Jorge Jr., “Melkersson Rosenthal Syndrome: A Histopathologic Mystery and Dermatologic Challenge, Journal of Cutaneous Pathology, Vol. 38, No. 2, 2011, pp. 241-245.
- N. S. Dar-Odeh, W. A. Hayajneh, O. A. Abu-Hammad, et al., “Orofacial Findings in Chronic Granulomatous Disease: Report of Twelve Patients and Review of the Literature,” BMC Research Notes, Vol. 3, No. 1, 2010, p. 37. doi:10.1186/1756-0500-3-37