International Journal of Organic Chemistry
Vol.07 No.04(2017), Article ID:81102,9 pages
10.4236/ijoc.2017.74032
Alternative and Eco-Friendly Synthesis of Tetrakis(Aminomethyl)Calix-[4]-Resorcinarene
S. Ali Moussaoui1, Ziad Damaj1, Moheddine Wehbie1,2, Stéphane Pellet Rostaing2, Iyad Karamé1*
1Department of Chemistry, Laboratory of Catalysis, Organometallic and Materiels LCOM, Lebanese University, Faculty of Sciences I, Hadath, Lebanon
2Institut de Chimie Séparative de Marcoule, ICSM, UMR 5257 - CEA/CNRS/UM2/ENSCM Site de Marcoule, Bâtiment 426, F-30207 Bagnols sur Cèse Cedex, France

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 30, 2017; Accepted: December 12, 2017; Published: December 15, 2017
ABSTRACT
A cleaner and eco-friendly method was developed for the preparation of tetrakis(aminomethyl)calix-[4]-resorcinarene via a synthetic pathway of five steps starting from methylresorcinol. This alternative methodology is firstly based on avoiding the use of CH2BrCl, which is a non-eco-friendly substance with high ozone depletion potential, and on replacing it by CH2Cl2 as a readily available reagent with much less dangerous effects. Secondly, this method engages acetone or CH2Cl2 as the solvent of the bromination step in the place of the very toxic CCl4, leading to tetrakis(bromomethyl)calix-[4]-resorcinarene. The brominated intermediate has been reacted with sodium azide in acetone instead of the high-boiling solvent DMSO to produce tetrakis(azidomethyl) calix-[4]-resorcinarene without the need of tedious purification. Lastly, this work reports an efficient hydrogenation method of the versatile azido adduct employing Pt/C (5%) as the catalyst for the preparation of the amino cavitand as an alternative route with high atom economy that can replace the classical methods used currently.
Keywords:
Bridged Calix-[4]-Resorcinarene, Eco-Friendly Synthesis, Catalytic Hydrogenation, Atom Economy, Radical Substitution, Nucleophilic Substitution, CH2Cl2

Graphical Abstract

1. Introduction
Resorcinarenes constitute a very attractive class of macrocyclic compounds. Their unique three-dimensional structures offer almost unlimited derivation abilities through relatively viable procedures at their upper rim, methylene bridges and extra annular-OH groups [1] . Their chemistry is focused mainly on two directions: i) synthesis and application of host-guest complexes by non-covalent interactions and ii) construction of novel supramolecular assemblies like dendrimers, nano-particles, nano-capsules, and metal extractants [2] [3] [4] [5] [6] .
The chemistry of resorcinarene attracted huge interest since few decades. Developing new synthetic procedures to realize these cavitands and calixarene oligomers continue to gain growing importance, especially green microwave- assisted syntheses and solvent-free methods to minimize hazardous effects, solvent elimination, and long purifications [7] . The derivatives of tetra-azidomethyl-calix- [4]resorcinarene, an extremely useful and desirable precursor for further functionalization, has not been extensively investigated to-date. [8] In the present work, we report an improved synthetic route to realize tetra-azidomethyl-calix- [4]resorcinarene IV (Scheme 1) using the easy substitution of the bromine nucleophile and its transformation to tetra-aminomethyl-calix[4]resorcinarene V by hydrogenation. This transformation is a cleaner, atom-economical, and facile alternative to prepare tetrakis-amino cavitand V as compared to the methods previously reported. Tetra-azidomethylcalix[4]resorcinarene IV is synthesized from the tetra-bromomethylcalix[4]resorcinarene III. The bromine functionality is introduced at the upper rim of the cavitand II following a method from literature [9] with a major modification of replacing the very toxic CCl4 by acetone or CH2Cl2.
2. Materials and Methods
2.1. General Information
Chemicals and solvents (analytically pure) were purchased from Sigma-Aldrich and were used without further purification. Reactions were monitored by thin layer chromatography (Silica gel 60 on TLC Al foils, F254). Chromatography was performed on silica gel 60 column. NMR spectra were recorded on Bruker spectrometers (300 and 400 MHz). Chemical shifts (δ) are expressed in ppm and are measured by referring to the peak of TMS (singlet at δ = 0 ppm) and the solvent (residual CHCl3: 7.26 ppm for 1H; 77.16 ppm for 13C of CDCl3) as an internal reference; the abbreviations used are: s = singlet, d = doublet, t = triplet, q = quartet. Fourier transform infrared (FTIR) measurements were performed on Perkin Elmer Spectrum 100 instrument, wavenumber range was measured from 400 cm−1 to 4000 cm−1. ESI-MS was performed on a Flexar SQ 300 MS instrument. NMR spectra of cavitands II, III and IV, IR spectra of III and IV, and mass spectrum of cavitand IV (ESI in positive and in negative modes) are given in the supplementary material [10] .
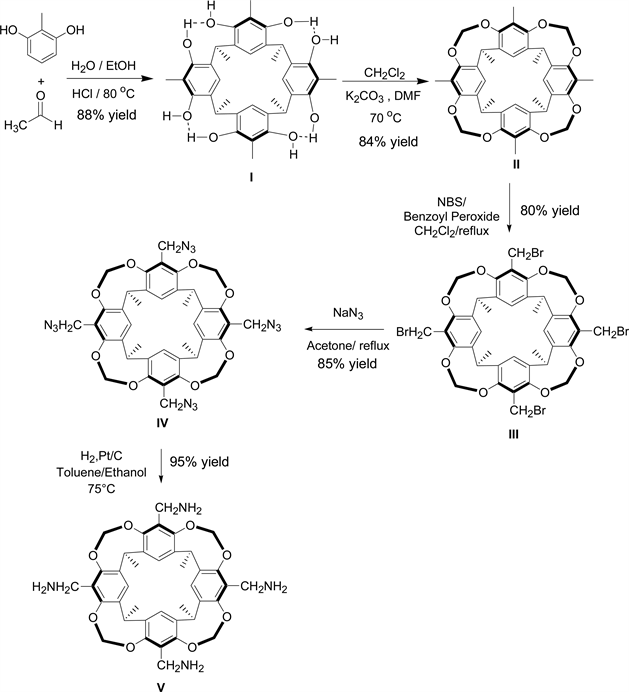
Scheme 1. Synthetic route of tetramethyl calix-[4]-resorcinarene derivatives.
2.2. Synthetic Procedures
1) Synthesis of cavitand I. This compound was prepared as described by Cram et al. [11] and was identified by comparison of its 1H NMR spectrum with the one reported in literature, yield 88%, color: dark yellow, 1H NMR (400 MHz, DMSO) δ [ppm]: 1.70 (d, 12H, CH3CH), 2.00 (s, 12H, ArCH3), 4.46 (q.4H, CH3CH), 7.41 (s, 4H, ArH), 8.70 (s, 8H, ArOH). MS: m/z: 543 (M-H).
2) Synthesis of cavitand II. 6.0 g of cavitand I (9.87 mmol) was dissolved in 150 mL DMF. Then, 28.8 g of K2CO3 (0.20 mol) and 50 ml of CH2Cl2 were added in several portions to the previous mixture and this was agitated for 24 hours at 70˚C. The mixture was then cooled down to room temperature and filtered over Celite. The collected solvent was evaporated using a rotary evaporator, and the solid residue was purified over silica gel column chromatography using ethyl acetate/cyclohexane (30/70) as eluent. The cavitand II was obtained as a white powder (yield 84%). Final product was characterized by comparison of its 1H NMR spectrum with the one reported in literature [11] . 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.73 (d, 12H, CH3CH), 2.00 (s, 12H, ArCH3), 4.29 (d, 4H, inner of OCH2O), 5.20 (q, 4H, CH3CH), 5.92 (d, 4H, outer of OCH2O), 7.14 (s, 4H, ArH).
3) Synthesis of cavitand III. 3.0 g of Cavitand II (4.62 mmol) was dissolved in 200 mL of CH2Cl2. Then, 3.22 g of NBS (18.51 mmol) and a small quantity of benzoyl peroxide (15 mg, 0.06 mmol) were subsequently added to the solution. The mixture (clear orange color) was agitated and refluxed (40˚C) for 4 hours. The precipitation of succinimide was observed. The mixture was cooled to room temperature and the precipitate was eliminated by filtration over Celite. The volatiles were evaporated under vacuum giving withe solid. The crude was purified over silica gel column chromatography using ethyl acetate /cyclohexane (30/70) as eluent. The cavitand III was obtained as an off-white (cream) powder. The final product was characterized by comparison of its 1H NMR spectrum with spectrum already reported in literature [9] . 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.75 (d, 12H, J = 7.4 Hz, CHCH3), 4.42 (s, 8H, CH2Br), 4.57 (d, 4H, J = 7.0 Hz, inner OCH2O), 5.02 (q, 4H, J = 7.4 Hz, CH), 6.04 (d, 4H, J = 7.0 Hz, outer OCH2O), 7.26 (s, 4H, ArH).
4) Synthesis of cavitand IV. Into a flask containing 80 ml of acetone, 2.0 g of cavitand III (2.40 mmol) and 0.9 g of sodium azide NaN3 (15 mmol) were subsequently added. The mixture was agitated and refluxed for 3 h. After that, the mixture was filtered over Celite and the collected solution was evaporated using a rotary evaporator. The residue was purified over silica gel column chromatography with ethyl acetate/cyclohexane (65/35) as eluent. The cavitand IV was obtained as white powder (yield 85%). The final product was characterized by 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.75 (d, 12H, J = 7.4 Hz, CHCH3), 4.3 (s, 8H, CH2N3), 4.42 (d, 4H, J = 7.0 Hz, inner OCH2O), 5.0 (q, 4H, J = 7.4 Hz, CH3CH), 6.0 (d, 4H, J = 7.0 Hz, outer OCH2O), 7.3 (s, 4H, ArH). X. 13C-NMR (CDCl3, 100 MHz), δ [ppm]: 153.326 (Ar-Cq), 139.058 (Ar-Cq), 122.19 (Ar-Cq), 120.277 (Ar-H), 99.677 (O-CH2-O), 45.078 (Ar-CH2-N3), 31.211 (Ar-CH-Ar), 16.072 (CH3). MS, ESI, in positive mode: m/z = 835 = [M + Na]+, m/z = 853 = [M + MeCN]+; in negative mode m/z = 847 = [M-H + 2H2O]+ and m/z = 874 = [M+ + 4H2O]+. FTIR: ν = 2100 cm−1 (band of N3).
5) Synthesis of cavitand V. 0.5 g of cavitand IV was dissolved in a toluene/ ethanol mixture (5 mL, 8 mL) followed by the addition of 0.3 g of Pt/C (5%). The mixture was agitated and refluxed for 24 h at 75˚C under one atmosphere of hydrogen pressure (or under 5 bars at 25˚C). The mixture was then filtered over Celite and the collected solvent was evaporated using a rotary evaporator. The cavitand V was obtained as white powder (yield 95%). The final product was characterized by comparison of its 1H NMR spectrum with literature [12] [13] . 1H NMR (400 MHz, CDCl3) δ [ppm]: 7.1 (s, 4H; ArH), 5.8 (d, J = 7.2 Hz, 4H; outer OCHO), 4.9 (q, J = 7.35 Hz, 4H; CH3CH), 4.3 (d, J = 7.2 Hz, 4H; inner OCHO), 3.5 (s, 8H; ArCH2NH2), 1.7 (d, J = 7.35 Hz, 12H; CH3CH). 13C-NMR (CDCl3, 100 MHz), δ [ppm]: 152.8 (Ar-Cq), 138 (Ar-Cq), 129 (Ar-Cq), 118 (Ar-H), 99.4 (O-CH2-O), 37 (Ar-CH2-NH2), 31 (Ar-CH-Ar), 15 (CH3). ESI (in positive ionisation mode): m/z = 709 for [M + H]+, 727 for [M + H + H2O]+, 765 [M + K + H2O]+, FTIR: ν = 3370 cm−1 (band of N-H).
3. Discussion
3.1. Synthesis of Cavitands I, II
The cavitand I was prepared as described by Cram et al., methylresorcinol and acetaldehyde were mixed in stoichiometric ratio in a proportional mixture of water and ethanol, in the presence of HCl as the catalyst. The resulting mixture was then refluxed for 16 h, and cavitand I was obtained with a high yield (88%) similar to that mentioned in the literature [11] .
Cavitand I was transformed into cavitand II cone structure by introducing methylene bridges between the oxygen atoms in ortho positions to the methyl groups of the methylresorcinol. Cram’s group has described the methylene insertion at the upper rim to form the bridged cavitand II by reacting the relaxed-open cavitand I with an excess amount of bromochloromethane in basic medium in DMA. They have demonstrated that a higher yield is obtained with CH2BrCl as compared to using CH2I2 and CH2Br2 as methylene sources. Pellet Rostaing et al. [14] have realized the same insertion in basic medium in DMF instead of DMA. The key step of our methodology concerns the use of dichloromethane (DCM). Cavitand I was mixed with a large excess amount of CH2Cl2 in K2CO3/DMF mixture to afford cavitand II in a very good yield (84%); much better than that obtained by Cram’s group. The first use of CH2Cl2 as methylene bridges precursors as well as in the protection of aromatic diol, has two advantages: it is a very good replacer of Bromochloromethane which destroys the ozone layer and its production has been banned since 1st January 2002, at the Eleventh Meeting of the Parties of Montreal Protocol on the substances that deplete the ozone layer. The second advantage is that the dichloromethane is less expensive and available in large quantities. In our protocol, DCM was used efficiently and its volatility (bp = 40˚C) was overcome by adding several portions during the progress of the refluxing process (70˚C) to establish the desired product.
3.2. Synthesis of Cavitands III
The rigidified-bridged cavitand II was subjected to a bromination reaction using N-bromosuccinimide (NBS) as a source of bromide in dichloromethane (DCM) or in acetone to obtain cavitand III. The use of acetone as a solvent for this reaction has been described by Bourgeois and Evans for the bromination on aromatic of resorcinarene derived from resorcinol [15] .
The bromination takes place through a free radical mechanism in the presence of traces of benzoyl peroxide as initiator. The most electron-rich methyl groups on the upper rim between two donor alkoxy functions are the most reactive sites towards the radical reaction since the generated radicals are stabilized by the neighboring electron donating groups. The obtained yield (80%) was better than the one attained in the literature (60%) [9] . Cavitand III was collected as a pure cream powder. It is noteworthy to mention that only traces of benzoyl peroxide must be added as initiator since its excess prevents the reaction from proceeding to completion and conduces to the formation of benzoyl bromide as a side product, as detected by proton NMR (See Supp. Materials.).
3.3. Synthesis of the Cavitand IV
Alkyl halides are usually very reactive toward nucleophilic substitution. Bromide is a very good leaving group, and is a strong nucleophile. Thus, a substitution reaction on cavitand III occurs easily.
To achieve the synthesis of cavitand IV, we used sodium azide NaN3. This substitution needs a solvent with a high dielectric constant in order to dissociate NaN3. We have recently realized in our laboratory the azide substitution of chlorinated products using NaN3 in acetone. Therefore, we have adopted this method for the preparation of cavitand IV. These conditions proved to be successful for this bromide substitution reaction establishing the desired azido cavitand with no side products or impurities. A simple filtration over Celite followed by evaporation of the solvent affords the product with a very good yield (85%) and purity. Hence, the purification of this step is avoided in this synthetic route. It is important to note the versatility of the azido cavitand IV for further functionalization via click chemistry highlighting the importance of straightforward synthesis for this type of cavitands. For instance, Hooley’s group has published the synthesis of a compound similar to cavitand IV as precursor to advanced cavitands substituted with tri-azole groups which have been used as iron-coordinate water-soluble catalysts for C-H Oxidation [16] . Their method of synthesis consisted on using THF as a solvent mixed with a solution of NaN3 in DMSO. This method allows obtaining the corresponding compound in good yield but it is more demanding than our described method. Furthermore, acetone is much less toxic, less odorous, and more volatile than THF and DMSO; and hence, its elimination by evaporation is much easier.
3.4. Synthesis of the Cavitand V
The traditional routes to introduce amino groups into the aromatic scaffolds of calixarene chemistry are i) nitration of cavitands followed by reduction of the nitro group with common reducing agents [17] [18] [19] , or ii) formation of azo compound followed by reduction with Na2S2O4 [20] [21] . An amino resorcinarene similar to cavitand V, is recently used by Sheela et al. (2011) as a stabilizer for the dispersion of nanodiamonds in organic solvents for the purpose of growing diamond films [12] . This group uses synthetic procedures already described in the literature to prepare the tetra-amino cavitand in two steps from tetra-brominated cavitand [13] [22] . Their described method enables the transformation of the tetra-brominated cavitand into tetrakis (phthalimidomethyl), and then the phthalimido group is hydrolyzed by hydrazine hydrate under reflux in ethanol/THF. In fact, we have realized the reduction reaction of the tetraazide adduct into its corresponding tetra-amine product with molecular hydrogen. The hydrogenation reaction was catalyzed by Platinum on charcoal Pt/C in toluene/ethanol at 75˚C under one atmosphere of hydrogen, to afford to product V in 95% yield. The developed Pt/C catalytic hydrogenation is an advantageous alternative to the classical methods used to synthesize the tetrakis(aminomethyl)- calix-[4]-resorcinare; It is a simpler, cleaner, atom-economical, and higher yielding method with no side products or impurities.
4. Conclusion
A new approach to synthesize the tetrakis(aminomethyl)calix-[4]-resorcinarene was developed, according to an improved synthetic pathway. The synthesis procedures and steps allow us to obtain the final product in good five step global yield (48%). The successful use of acetone as the solvent for the bromination of cavitands with NBS is confirmed. Bromochloromethane, which is banned worldwide due to its high potential of destroying the ozone layer, and the toxic CCl4 are replaced by dichloromethane during the preparation of intermediates II and III. An atom-economical hydrogenation procedure is described to establish the amino-cavitands with a set of advantages over the classical methods. The presented improvements furnish a higher yielding synthetic route to the amino cavitand with less tedious purifications and fewer hazards for the environment.
Acknowledgements
We gratefully acknowledge the financial support for this project by the Lebanese University, CEDRE program, CNRSL (the National Council for Scientific Research, Lebanon).
Cite this paper
Moussaoui, S.A., Damaj, Z., Wehbie, M., Pellet Rostaing, S. and Karamé, I. (2017) Alternative and Eco- Friendly Synthesis of Tetrakis(Aminomethyl) Calix-[4]-Resorcinarene. International Journal of Organic Chemistry, 7, 403-411. https://doi.org/10.4236/ijoc.2017.74032
References
- 1. Timmerman, P., Verboom, W. and Reinhoudt, D.N. (1996) Resorcinarenes. Tetrahedron, 52, 2663-2704. https://doi.org/10.1016/0040-4020(95)00984-1
- 2. Boas, U.P. and Heegaard, M.H. (2004) Dendrimers in Drug Research. Chemical Society Reviews, 33, 43-63. https://doi.org/10.1039/b309043b
- 3. Antesberger, J., Cave, G.W.V., Ferrarelli, M.C., Heaven, M.W., Raston, C.I. and Atwood, J.L. (2005) Solvent-Free, Direct Synthesis of Supramolecular Nano-Capsule. Chemical Communications, 7, 892-894. https://doi.org/10.1039/b412251h
- 4. Gualbert, J., Shahgaldian, P., Lazar, A. and Coleman, A. (2004) Solid Lipid Nanoparticles (SLNs): Preparation and Properties of CalixResorcinarene Derived Systems. Journal of Inclusion Phenomena and Macrocyclic Chemistry, 48, 37-44. https://doi.org/10.1023/B:JIPH.0000016598.29935.6e
- 5. Jain, V.K., Pillai, S.G., Pandya, R.A., Agrawal, Y.K. and Shrivastav, P.S. (2005) Selective Extraction, Preconcentration and Transport Studies of Thorium(IV) Using Octa-Functionalized CalixResorcinarene-Hydroxamic Acid. Analytical Sciences, 21, 129-136. https://doi.org/10.2116/analsci.21.129
- 6. Jain, V.K., Pillai, S.G., Pandya, R.A., Agrawal, Y.K. and Shrivastav, P.S. (2005) Molecular Octopus: Octa Functionalized CalixResorcinarene-Hydroxamic Acid [C4RAHA] for Selective Extraction, Separation and Preconcentration of U(VI). Talanta, 65, 466-475. https://doi.org/10.1016/j.talanta.2004.06.033
- 7. Sadarjino, R.E. and Racamawati, R. (2017) Green Synthesis of Oligomer Calixarenes. Chapter 4, In: Karamé, I. and Srour, H., Eds., Green Chemical Processing and Synthesis, Intech. Publisher, Liss, CC BY 3.0 Zagreb-Croatia, 71-90. Open Access, Print ISBN 978-953-51-3259-2.
- 8. Gramage-Doria, R., Armspach, D. and Matt, D. (2013) Metallated Cavitands (Calixarenes, Resorcinarenes, Cyclodextrins) with Internal Coordination Sites. Coordination Chemistry Reviews, 257, 776-816. https://doi.org/10.1016/j.ccr.2012.10.006
- 9. Sorrell, T.N. and Pigge, F.C. (1993) A Convenient Synthesis of Functionalized Cavitands via Free-Radical Bromination. The Journal of Organic Chemistry, 58, 784-785. https://doi.org/10.1021/jo00055a044
- 10. Supplementary Material Is Given in an Independent File “Supplementary Material” and Available upon Request Directly from Corresponding Author.
- 11. Cram, D.J., Karbach, S., Kim, H., Knobler, C.B., Maverick, E.F., Ericson, J.L. and Helgeson, R.C. (1988) Host-Guest Complexation. 46. Cavitands as Open Molecular Vessels Form Solvates. Journal of the American Chemical Society, 110, 2229-2237. https://doi.org/10.1021/ja00215a037
- 12. Sheela, V.P., Xiao, W., Han, S., Zhou, X., Albin, S. and Balasubramanian, R. (2011) Resorcinarene Amine Stabilized Nanodiamond Dispersions in Organic Solvents: Applications in Diamond film Growth. Journal of Materials Chemistry, 21, 6395-6400. https://doi.org/10.1039/c0jm04520a
- 13. Boerrigter, H., Verboom, W. and Reinhoudt, D.N. (1997) Novel Resorcinarene Cavitand-Based CMP(O) Cation Ligands: Synthesis and Extraction Properties. The Journal of Organic Chemistry, 62, 7148-7155. https://doi.org/10.1021/jo9703414
- 14. Pellet-Rostaing, S., Nicod, L., Chitry, F. and Lemaire, M. (1999) Synthesis and Caesium Complexing Properties of Water-Soluble Cavitands. Tetrahedron Letters, 40, 8793-8796. https://doi.org/10.1016/S0040-4039(99)01870-5
- 15. Bourgeois, J.M. and Stoeckli-Evans, H. (2005) Synthesis of New Resorcinarenes under Alkaline Conditions. Helvetica Chimica Acta, 88, 2722-2730. https://doi.org/10.1002/hlca.200590211
- 16. Djernes, K.E., Moshe, O., Mettry, M., Richards, D.D. and Hooley, R.J. (2012) Metal-Coordinated Water Soluble Cavitands Act as C-H Oxidation Catalysts. Organic Letters, 14, 788-791. https://doi.org/10.1021/ol203243j
- 17. Desroches, C., Parola, S., Vocanson, F., Perrin, M., Lamartine, R., Letoffe, J.M. and Bouix, J. (2002) Nitration of ThiacalixArene Using Nitrosium Nitrate Complexes: Synthesis and Characterization of Tetranitro-, Tetraamino-, and Tetra(4-Pyridylimino)TetrahydroxythiacalixArene. New Journal of Chemistry, 26, 651-655. https://doi.org/10.1039/b110609k
- 18. Tabakci, M., Memon, S., Yilmaz, M. and Roundhill, M.D. (2004) Synthesis and Evaluation of Extraction Ability of Calix-Crown-6 Cone Conformer and Its Oligomeric Analogue. Reactive & Functional Polymers, 58, 27-34. https://doi.org/10.1016/j.reactfunctpolym.2003.11.002
- 19. Tunstad, L.M., Tucker, J.A., Dalcanale, E., Weiser, J., Bryant, J.A., Sherman, J.C., Helgeson, R.C., Knobler, C.B. and Cram, D.J. (1989) Host-Guest Complexation. 48. Octol Building Blocks for Cavitands and Carcerands. The Journal of Organic Chemistry, 54, 1305-1312. https://doi.org/10.1021/jo00267a015
- 20. Jain, V.K. and Kanaiya, P.H. (2008) Diazo Reductive: A New Approach to the Synthesis of Novel ‘‘Upper Rim’’ Functionalized ResorcinArene Schiff-Bases. Journal of Inclusion Phenomena and Macrocyclic Chemistry, 62, 111-115. https://doi.org/10.1007/s10847-008-9445-1
- 21. Jayswal, K.P., Patel, J.R., Patel, V.B. and Patel, M.H. (2008) A New Approach towards Synthesis of Some Novel “Upper Rim” Functionalized CalixResorcinarene Schiff-Bases. Acta Chimica Slovenica, 55, 302-307.
- 22. Middel, O., Verboom, W. and Reinhoudt, D.N. (2002) Water-Soluble Cavitands Synthesis, Solubilities and Binding Properties. European Journal of Organic Chemistry, 15, 2587-2597. https://doi.org/10.1002/1099-0690(200208)2002:15<2587::AID-EJOC2587>3.0.CO;2-2