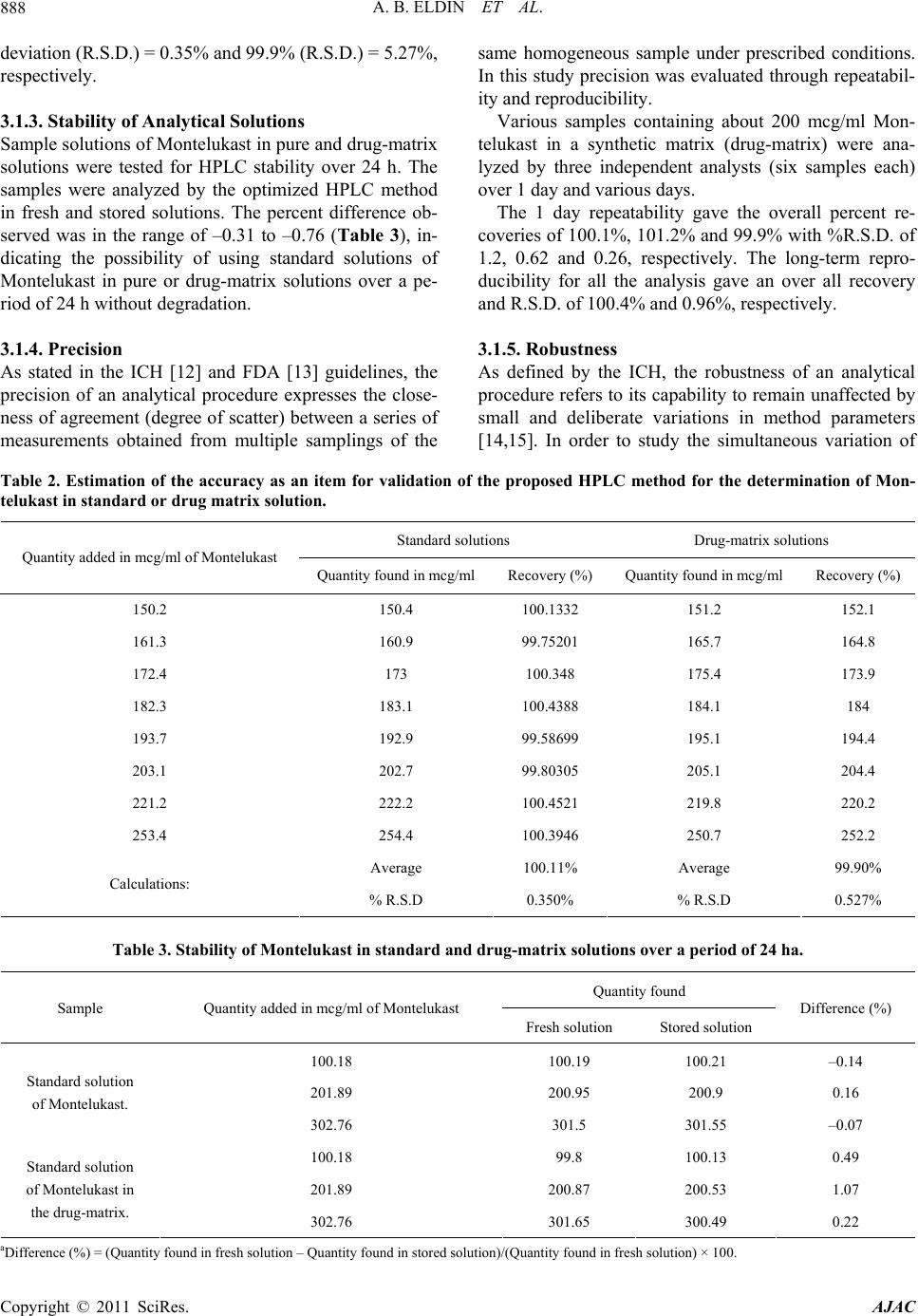
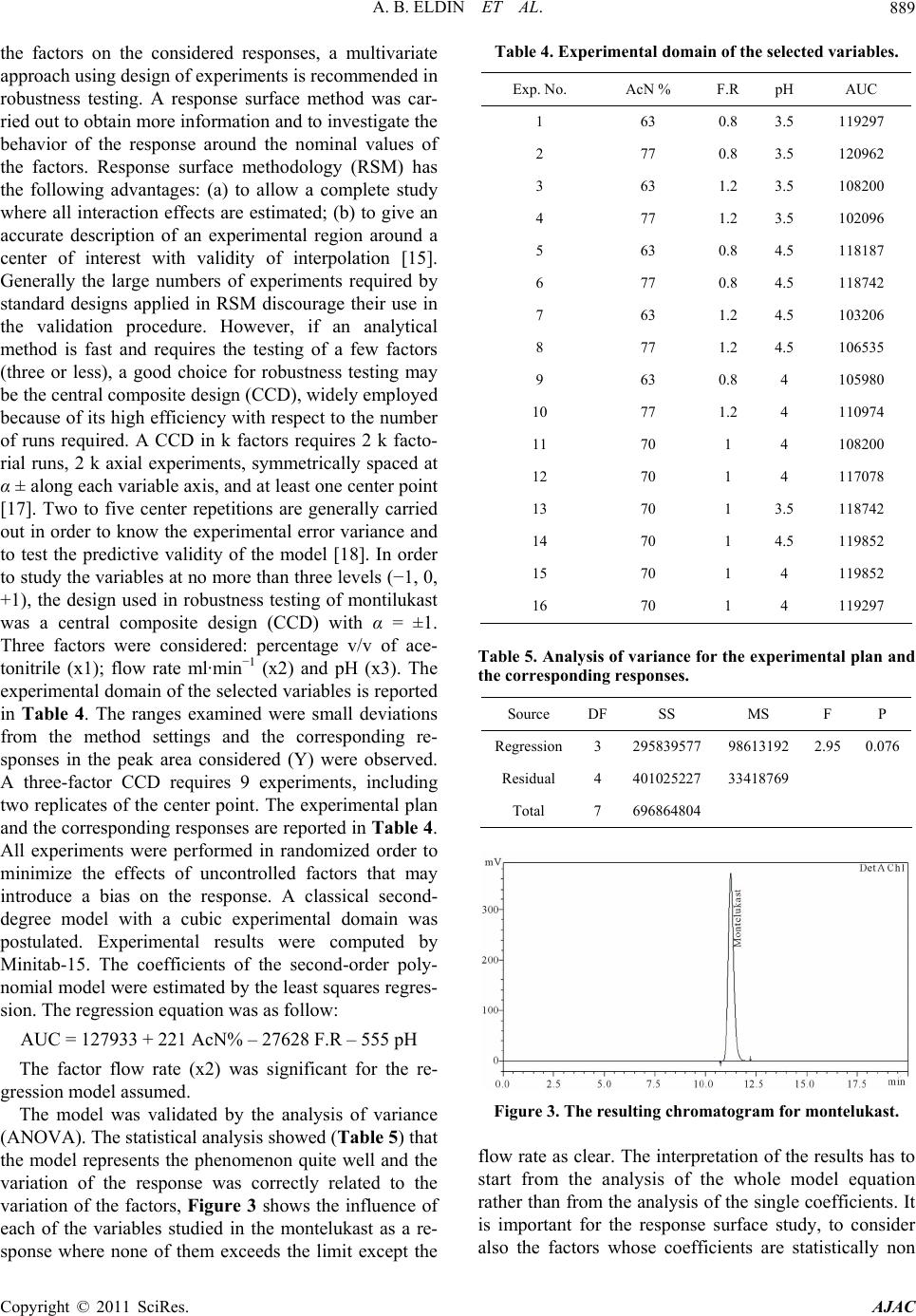
A. B. ELDIN ET AL.
Copyright © 2011 SciRes. AJAC
891
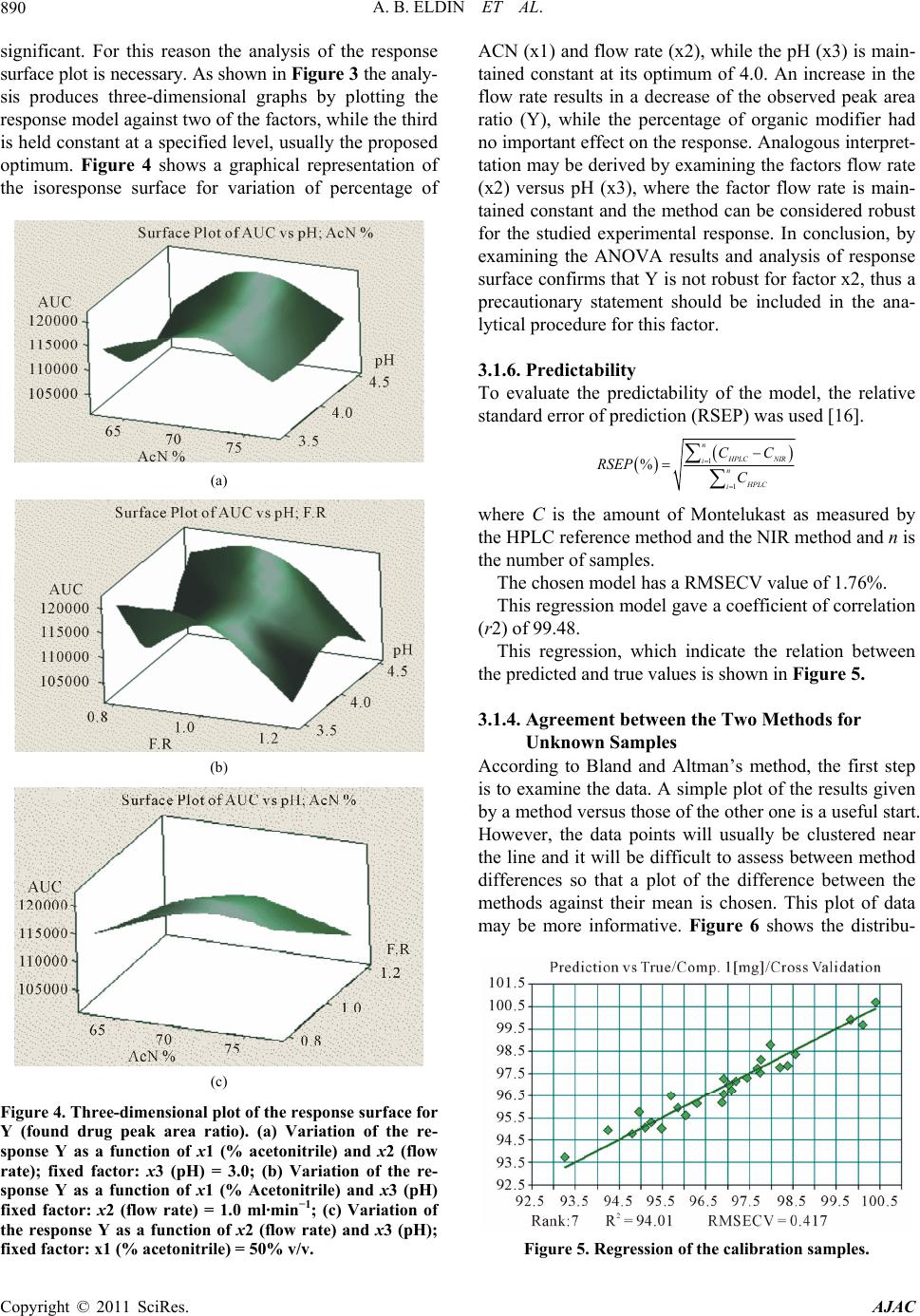
Difference vs True / Montelukast % / Cross Validation
-0.8
-0.6
-0.4
-0.2
0
0.2
0.4
0.6
0.8
051015202530
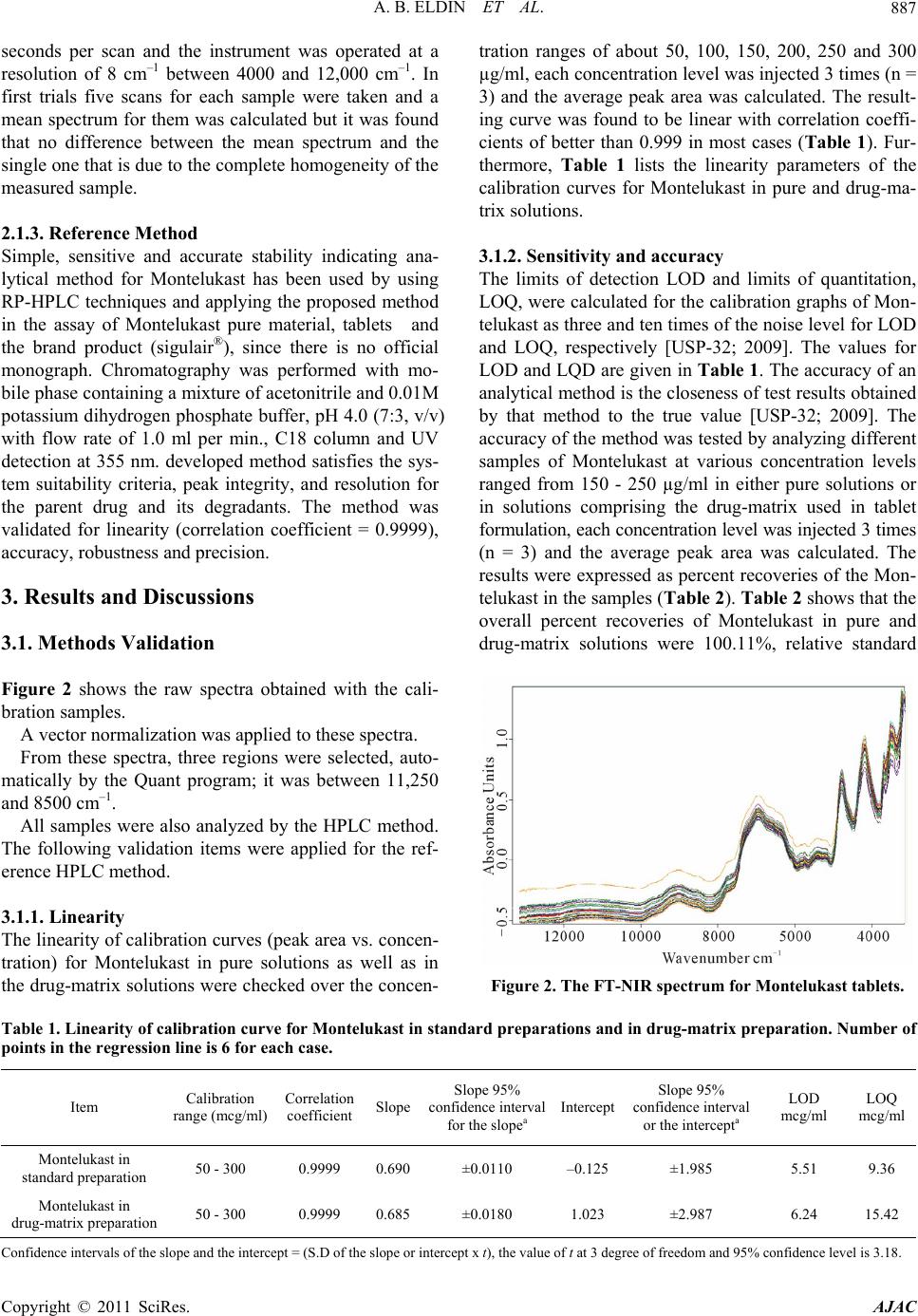
Figure 6. Distribution of the differences against their mean.
tion of the differences against their mean.
4. Conclusions
NIR spectroscopy has been shown to be a viable alterna-
tive to HPLC with UV detection for the assay of Monte-
lukast tablets, and it takes only few minutes to analyse a
batch once the calibration model has been set up. The
proposed model is easy to use and give accurate results.
It is a non-destructive method and thus lends itself very
well for on-line/at-line production control purposes.
Compared to the conventional technique, the NIR
spectroscopy method is faster, non-destructive, and gives
less variability. It has been shown that NIRspectroscopy
can replace safely the UV-vis spectrophotometry.
5. Acknowledgements
The authors thank Sigma Pharmaceutical Corp., Egypt
for technical support.
6. References
[1] FDA, “PAT—A Framework for Innovative Pharmaceu-
tical Manufacturing and Quality Assurance,” 2004.
http://fda.gov/cder
[2] E. W. Ciurczak and J. K. Drennen, “Practical Spectros-
copy Series: Pharmaceutical d Medical Applications of
Near-Infrared Spectroscopy,” Marcel Dekker, New York,
2002.
[3] W. Plugge and C. Van der Vlies, “The Use of Near In-
frared Spectroscopy in the Quality Control Laboratory of
the Pharmaceutical Industry,” Journal of Pharmaceutical
and Biomedical Analysis, Vol. 10, No. 10-12, 1992, pp.
797-803. doi:10.1016/0731-7085(91)80083-L
[4] C. I. Gerh¨ausser and K. A. Kovar, “Strategies for
Constructing Near-Infrared Spectral Libraries for the
Identification of Drug Substances,” Applied Spectroscopy,
Vol. 51, No. 10, 1997, pp. 1504-1510.
doi:10.1366/0003702971939000
[5] M. J. Vredenbregt, P. W. J. Caspers, R. Hoogerbrugge
and D. M. Barends, “Choice and Validation of a Near
Infrared Spectroscopic Application for the Identity
Control of Starting Materials.: Practical Experience with
the EU Draft Note for Guidance on the Use of Near
Infrared Spectroscopy by the Pharmaceutical Industry
and the Data to be Forwarded in Part II of the Dossier for
a Marketing Authorization,” European Journal of
Pharmaceutics and Biopharmaceutics, Vol. 56, No. 3,
2003, pp. 489-499. doi:10.1016/S0939-6411(03)00119-X
[6] S. S. Sekulic, H. W. Ward and P. K. Aldridge, “On-Line
Monitoring of Powder Blend Homogeneity by Near-In-
frared Spectroscopy,” Analytical Chemistry, Vol. 68, No.
3, 1996, pp. 509-513. doi:10.1021/ac950964m
[7] P. Merckle and K.-A. Kovar, “Assay of Effervescent
Tablets by Near-Infrared Spectroscopy in Transmittance
and Reflectance Mode: Acetylsalicylic Acid in Mono and
Combination Formulations,” Journal of Pharmaceutical
and Biomedical Analysis, Vol. 17, No. 3, 1998, pp. 365-
374. doi:10.1016/S0731-7085(97)00194-5
[8] R. P. Cogdill, C. A. Anderson and J. K. Drennen, Phar-
maceutical Technology, 2004, pp. 29-34.
[9] J. Sun, Journal of Chemometrics, Vol. 11, 1997, pp.
525-532.
[10] R. J. Barnes, M. S. Dhanoa and S. J. Lister, “Standard
Normal Variate Transformation and De-trending of Near-
Infrared Diffuse Reflectance Spectra,” Applied Spectros-
copy, Vol. 43, No. 5, 1989, pp. 772-777.
doi:10.1366/0003702894202201
[11] T. Fearn, NIR News, Vol. 10, 1999, pp. 10-11.
[12] ICH Q2B, International Conference on Harmonisation,
Validation of Analytical Procedures, Methodology, 2002.
[13] FDA, “Guidance for Industry: Validation of Analytical
Procedures,” Food and Drug Administration, Rockville,
1997.
[14] International Conference on Harmonisation Topic Q2B,
“Validation of Analytical Methods: Methodology,” The
Third International Conference on Harmonization of
Technical Requirements for Registration of Pharmaceu-
ticals for Human Use (ICH) Yokohama-Japan.
[15] Y. V. Heyden, A. Nijhuis, J. Smeyers-Verbeke and B. G.
M. Vandeginste D. L., Journal of Pharmaceutical and
Biomedical Analysis, 2001, p. 24723.
[16] A. Eustaquio, P. Graham, R. D. Jee, A. C. Moffat and A.
D. Trafford, “Quantification of Paracetamol in Intact
Tablets Using Near-Infrared Transmittance Spectros-
copy,” Analyst, Vol. 123, No. 11, 1998, pp. 2303-2306.
doi:10.1039/a804528c
[17] R. Ragonese, M. Mulholland and J. Kalman, “Full and
Fractionated Experimental Designs for Robustness Test-
ing in the High-Performance Liquid Chromatographic
Analysis of Codeine Phosphate, Pseudoephedrine Hy-
drochloride and Chlorpheniramine Maleate in a Pharma-
ceutical Preparation,” Journal of Chromatography A, Vol.
870, No. 1-2, 2000, p. 45.
doi:10.1016/S0021-9673(99)00972-3
[18] G. A. Lewis, D. Mathieu and R. Phan-Tan-Luu, “Phar-
maceutical Experimental Design,” Marcel Dekker, New
York, 1999.