Journal of Materials Science and Chemical Engineering
Vol.06 No.07(2018), Article ID:85790,8 pages
10.4236/msce.2018.67005
Orbital Phase Theory and the Diastereoselectivity of Some Organic Reactions*
Yuji Naruse
Department of Chemistry and Biomolecular Science, Gifu University, Yanagido, Gifu, Japan




Received: January 19, 2018; Accepted: July 1, 2018; Published: July 4, 2018
ABSTRACT
The orbital phase refers to the relationship between orbitals that originates from their wave character. We show here that the orbital phase essentially determines the diastereoselectivity of the following three organic reactions. 1) Torquoselectivity of the electrocyclicring-opening reaction of 3-substituted cyclobutenes; 2) Contradictory torquoselectivity of the retro-Nazarov reaction; 3) Diastereoselectivity in electrophilic addition to substituted ethylenes.
Keywords:
Orbital Phase Theory, Diastereoselectivity, Orbital Phase Continuity, Bond Model Analysis

1. Introduction
Many theories have been proposed to explain the diastereoselectivity of organic reactions [1] [2] [3] [4]. However, they have been limited to qualitative discussions since most are derived from various experimental results. Furthermore, there are often exclusions, although no explanation is offered for why there should be exclusion. On the other hand, there are two major theories regarding reactivity from the perspectives of theoretical and quantum chemistry: the frontier molecular orbital (FMO) theory by Fukui [5] and the theory of the conservation of orbital symmetry by Woodward and Hoffmann (W.-H. rule) [6]. The former emphasizes that reactions proceed due to the delocalization between two molecules. More overlap and a smaller HOMO-LUMO gap between the molecules result in greater stabilization, which lowers the energy of the transition state and accelerates the reaction. The latter approach focuses on the symmetry of the molecular orbital. However, it is mainly applicable to reactions with symmetrical substrates, and thus has a limited application.
Another theory is the orbital phase theory developed by Inagaki [7] [8], which focuses on the wave aspect of the orbital, especially the phase among the orbitals. Since it depends on the more fundamental nature of the wave of the orbital, this theory applies not only to molecules in the ground state, but also to those intransition states.
Here, we explain some organic reactions from the perspective of orbital phase theory. One of the most important points is the cyclic orbital interaction [9]. When a series of orbitals interact with each other to result in ring closure, the electron(s) delocalize(s) among them to produce stabilization when the orbitals satisfy the requirements for phase continuity (Figure 1): i) donating orbitals are out-of-phase; ii) donating and accepting orbitals are in-phase; and iii) accepting orbitals are in-phase. For the interaction among more than three orbitals, two additional requirements must be satisfied: iv) ring closure of the interacting orbitals is monocyclic, so that the orbitals interact only with adjacent orbitals, and not with those at remote locations; and v) the interacting orbitals are divided into two parts―a donor and an acceptor―and not into four, six, or so on.
Note that the overlap between the orbitals is always less than 1, i.e., |Sij| < 1, and a lower number of corresponding orbitals is often preferred. With these additional requirements, cyclic orbital interaction satisfies the requirements for producing stabilization.

Figure 1. Requirements for orbital phase continuity.
2. Torquoselectivity of the Electrocyclic Ring-Opening Reaction of 3-Substituted Cyclobutenes
Here we take a phenomenological approach to explain the torquoselectivity of the electrocyclicring-opening reaction of 3-substituted cyclobutenes to eliminate or minimize any bias. The phenomenological model involves finding regularity in a series of data, which preferably would be a series of experimental results. However, when it would be difficult to obtain reliable results, a simulation may offer a reasonable alternative. Thus, we performed theoretical calculations for substrates with a variety of substituents and searched for some characteristic features among the data. For the torquoselectivity of ring-opening reactions, we chose 3-substituted cyclobutenes as model compounds (Scheme 1) [10] [11] [12] [13]. The results of the theoretical calculations are shown in Table 1 [14].
These results show some characteristic features: 1) the torquoselectivities switch from inward to outward with elements toward the right in the periodic table; 2) the outward rotation has a maximal value of ca. 14.8 kcal/mol in the case of R = NH2, OH, F, Cl. Characteristic (a) suggests that the electronegativity should be considerably related to the selectivity, since electronegativity increases as we move to the right in the periodic table [15] [16] [17]. Electronegativity is closely related to the orbital energies of σC-R bonds. However, N in an amino
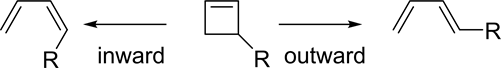
Scheme 1. Torquoselectivity of 3-substituted cyclobutenes.

Table 1. Calculated activation enthalpies for 3-substituted cyclobutenes (kcal/mol, B3LYP/6-31G(d)) and electronegativity (EN).
ΔΔE‡ = ΔE‡ (inward) − ΔE‡ (outward).
group, O in a hydroxyl group, Fin afluoro group and Cl in achloro group are more electronegative than H. Thus, a C-H bond should be more electron-donating than C-N, C-O, C-F and C-Cl bonds, which should affect characteristic (b).
We consider the relationship between the activation enthalpies and the orbital energies of σC-R bonds. Initially, we used the bond energies of σCH3-R [18], which are shown in Figure 2.
The relationship clearly shows that the σ-bond of CH3-R affects the torquoselectivity.
With our orbital phase theory, the cyclic orbital interaction of σC-C-π*C=C-σC-R- and σ*C-C-πC=C-σC-R-satisfies the phase continuity requirements (Figure 3) [10]. They produce considerable stabilization of the transition state, thus controlling the torquoselectivity. An increase in theelectron-donating character of σC-R results in enhanced delocalization, to produce stabilization of the transition state, leading to inward rotation of substituentR (blue line in Figure 2). When the σC-H orbital is more electron-donating than σC-R, i.e., the orbital energy of σC-H is higher than that of σC-R, the σC-H bond controls the torquoselectivity of the outward rotation of R with a maximal difference of 14.8 kcal/mol (red line in Figure 2). Thus, our orbital phase theory should be able to properly explain the torquoselectivities, since it can describe the characteristic features of a series of calculation results.

Figure 2. Dependence of activation enthalpy on the σ-bond energy of CH3-R.

Figure 3. Cyclic orbital interaction in the electrocyclicring-opening reaction of 3-substituted cyclobutenes including the σC-R orbital.
3. Contradictory Torqueselectivity in the Retro-Nazarov Reaction
Harmata reported the torquoselectivity of the retro-Nazarov reaction [19] [20]. Their calculations showed some interesting results. For amino-hydroxyl-substituted substrates, cis-substituted 1 shows outward rotation of the amino group (Figure 4). In contrast, geminally-substituted 3 shows inward rotation of the amino group in our calculations. These reactions proceed in a conrotatory manner. If the substituent effect controls the torquoselectivity, it should result in the same torquoselectivity.
We show that this contradictory torqueselectivity is under the control of geminal bond participation [21]. The cationic nature of this reaction prevents the participation of the vacant orbitals on the substituent(s) due to electron-deficiency of the π2* orbital of the allyl cation moiety. Thus, ingeminal bond participation, only the cyclic orbital interaction among σC-C-π2*allyl-σC-R/geminal-is operative (Figure 5). The electron-donating character is in the order σC-H > σC-N > σC-O, since the order of the electronegativity is in the order H(2.1) < N(3.1) < O(3.5). The σC-H orbital geminal to the amino group is more electron-donating than that geminal to the hydroxyl group due to the inductive effect. Thus, the σC-H orbital geminal to the amino group is the most electron-donating (Figure 6), and thus it should control the torqueselectivity. Inward rotation of σC-H geminal to the amino group results in outward rotation of the amino group, and the hydroxyl group rotates inward since electrocyclicring-opening should occur in a conrotatory manner.

Figure 4. Calculated torquoselectivities of retro-Nazarov reactions (B3LYP/6-31G(d)).

Figure 5. Cyclic orbital interaction in TS of the retro-Nazarov reaction.
4. Diastereoselectivity in Electrophilic Addition to Substituted Ethylenes
Stork and Kishi reported that electrophiles often approach to the C=C double bond in a directionanti to the most electron-donating σ-bond at the vicinal position in electrophilic addition [22] [23]. This selectivity is often described in terms of antiperiplanar interaction between cationic three-membered species and the σ-electron-donating substituent at the vicinal position. From the perspective of FMO theory, however, it is still unclear why the electrophile, an acceptor, attacks in a direction so as to maintain the largest overlap with the HOMO as a result of the interaction between πC=C and σC-D (Figure 7). There are no explanations for why the HOMO shows larger expansion anti to the σ electron-donating substituent D with interaction between πC=C and σC-D, or why the cationic electrophile-π-complex is stabilized with the vicinal donating σ-bond at the anti position.
We show here that cyclic orbital interaction determines the selectivity in the electrophilic addition reaction [24]. The cyclic orbital interaction among πC=C-σC=R/vicinal-σC-C/geminal-σC=C-ϕ*E+-is phase-continuous, so that it produces considerable stabilization of the TS to enhance the reactivity and selectivity (Figure 8).

Figure 6. σ-orbitalenergies (Fii elements) of the bond orbitals of protonated 1 (RHF/6-31G(d)//B3LYP/6-31G(d)).

Figure 7. FMO perspective of the diastereoselectivity in electrophilic addition.

Figure 8. Effect of cyclic orbital interaction in the TS on the diastereoselectivity of electrophilic addition.
Large extension toward the electrophile results from a combination of σC=C and σC=C orbitals.
Cite this paper
Naruse, Y. (2018) Orbital Phase Theory and the Diastereoselectivity of Some Organic Reactions. Journal of Materials Science and Chemical Engineering, 6, 35-42. https://doi.org/10.4236/msce.2018.67005
References
- 1. Pine, S.H. (1987) Chap. 7 Organic Chemistry. 5th Edition, McGraw-Hill, New York.
- 2. Morrison, R.T. and Boyd, R.N. (1992) Chap. 11. In Organic Chemistry. 6th Edition, Prentice Hall International, Inc., London.
- 3. Kirby, A.J. (1996) Chap. 6. Stereoelectronic Effect. Oxford University Press, Oxford, U.K.
- 4. Fleming, I. (2009) Molecular Orbitals and Organic Chemical Reactions. Wiley, Chichester. https://doi.org/10.1002/9780470684306
- 5. Fukui, K. (1975) Theory of Orientation and Stereoselection. Springer, Berlin Heidelberg New York.
- 6. Woodward, R.B. and Hoffmann, R. (1970) The Conservation of Orbital Symmetry. VerlagChemie International, Deerfield Beach FL.
- 7. Inagaki, S. (2009) Orbitals in Chemistry. Springer, Berlin-Heidelberg.
- 8. Inagaki, S. (2009) Anorbital Phase Theory. Top. Curr. Chem., 289, 83-127. https://doi.org/10.1007/128_2008_40
- 9. Inagaki, S. and Hirabayashi, Y. (1982) Cyclic and Noncyclic Electron Delocalization in Cyclic π-Conjugated Molecules. Relative Delocalizabilities of Donor-Acceptor Disposition Isomers. Inorg. Chem., 21, 1798. https://doi.org/10.1021/ic00135a021
- 10. Yasui, M., Naruse, Y. and Inagaki, S. (2004) Anorbital Phase Theory for the Torquoselectivity of the Ring-Opening Reactions of 3-Substituted Cyclobutenes: Geminal Bond Participation. J. Org. Chem., 69, 7246-7249. https://doi.org/10.1021/jo049081y
- 11. Kirmse, W., Rondan, N.G. and Houk, K.N. (1984) Stereoselective Substituent Effects on Conrotatoryelectrocyclic Reactions of Cyclobutenes. J. Am. Chem. Soc., 106, 7989-7991. https://doi.org/10.1021/ja00337a067
- 12. Rondan, N.G. and Houk, K.N. (1985) Theory of Stereoselection in Conrotatoryelectrocyclic Reactions of Substituted Cyclobutenes. J. Am. Chem. Soc., 107, 2099-2111. https://doi.org/10.1021/ja00293a046
- 13. Honda, K., Lopez, S.A., Houk, K.N. and Mikami, K. (2015) Mono-, Di-, and Trifluoromethyl Substituent Effects on the Torquoselectivities of Cyclobutene and Oxeteneelectrocyclic Ring Openings. J. Org. Chem., 80, 11768-11772. https://doi.org/10.1021/acs.joc.5b01361
- 14. Gaussian 94-Gaussian 09 Gaussian, Inc.
- 15. Pauling, L. (1932) The Nature of the Chemical Bond. IV. The Energy of Single Bonds and the Relative Electronegativity of Atoms. J.Am. Chem. Soc., 54, 3570-3582. https://doi.org/10.1021/ja01348a011
- 16. Mulliken, R.S. (1934) A New Electroaffinity Scale; Together with Data on Valence States and on Valence Ionization Potentials and Electron Affinities. J. Chem.Phys., 2, 782-793. https://doi.org/10.1063/1.1749394
- 17. Mulliken, R.S. (1935) Electronic Structures of Molecules XI. Electroaffinity, Molecular Orbitals and Dipole Moments. J. Chem. Phys., 3, 573-585. https://doi.org/10.1063/1.1749731
- 18. Ma, J., Ding, Y., Hattori, K. and Inagaki, S. (2004) Theoretical Designs of Singlet Localized 1,3-Diradicals. J. Org. Chem., 69, 4245-4255. https://doi.org/10.1021/jo035687w
- 19. Harmata, M. and Lee, D.R. (2002) Theretro-Nazarovreaction. J. Am.Chem. Soc., 124, 14328-14329. https://doi.org/10.1021/ja028729q
- 20. Harmata, M., Schreiner, P.R., Lee, D.R. and Kirchhoefer, P.L. (2004) Combined Computational and Experimental Studies of the Mechanism and Scope of the Retro-Nazarovreaction. J. Am. Chem. Soc., 126, 10954-10957. https://doi.org/10.1021/ja048942h
- 21. Naruse, Y. and Tokunaga, M. (2015) Geminalbond Participation Is Essential for the Contradictory Torquoselectivities in Retro-Nazarovreactions. Tetrahedron Lett., 56, 3813-3815. https://doi.org/10.1016/j.tetlet.2015.04.081
- 22. Stork, G. and Kahn, M. (1983) A Highly Stereoselective Osmium Tetroxide-Catalyzed Hydroxylation of γ-Hydroxyα,β-Unsaturated Esters. Tetrahedron Lett., 24, 3951. https://doi.org/10.1016/S0040-4039(00)88233-7
- 23. Cha, J.K., Christ, W.J. and Kishi, Y. (1984) On Stereochemistry of Osmium Tetraoxide Oxidation of Allylic Alcohol Systems. Empirical Rule. Tetrahedron, 40, 2247-2255. https://doi.org/10.1016/0040-4020(84)80008-3
- 24. Naruse, Y., Hasegawa, Y. and Ikemoto, K. (2016) Orbital Theory for Diastereoselectivity in Electrophilic Addition. Tetrahedron Lett., 57, 2029-2033. https://doi.org/10.1016/j.tetlet.2016.03.077
NOTES
*Presented at COC4, Kunming China, July 2018.