 Open Journal of Gastroenterology, 2011, 1, 13-22 doi:10.4236/ojgas.2011.12003 Published Online November 2011 (http://www.SciRP.org/journal/ojgas/ OJGas ). Published Online November 2011 in SciRes. http://www.scirp.org/journal/OJGas Role of constitutive nitric oxide synthase in regulation of Helicobacter pylori-induced gastric mucosal cyclooxygenase-2 activation through S-nitrosylation: mechanism of ghrelin action Bronislaw L. Slomiany, Amalia Slomiany University of Medicine and Dentistry of New Jersey, Newark, USA. Email: slomiabr@umdnj.edu Received 18 July 2011; revised 21 September 2011; accepted 3 October 2011. ABSTRACT Gastric mucosal inflammatory responses to H. pylori lipopolysaccharide (LPS), are characterized by the excessive NO and prostaglandin (PGE2) generation due to the disturbances in nitric oxide synthase (NOS) and cyclooxygenase (COX) systems. Here, we report that the LPS-induced enhancement in gastric muco- sal inducible (i) iNOS) activity and up-regulation in PGE2 production was associated with the suppression in Akt kinase activity and the impairment in consti- tutive (c) cNOS activation. The stimulatory effect of the LPS on PGE2 production, furthermore, was sus- ceptible to suppression by COX-2 inhibitor, NS-398, and iNOS inhibitor, 1400 W. Further, we show that the countering effect of peptide hormone, ghrelin, on the LPS-induced changes was reflected in up-regu- lation in Akt activity and the increase in cNOS acti- vation through phosphorylation, and accompanied by the suppression in iNOS expression and the reduction in COX-2 activity associated with the loss in COX-2 protein S-nitrosylation. Moreover, the effect of ghre- lin on the LPS-induced COX-2 S-nitrosylation was subject to repression by Akt inhibition. Our findings demonstrate that induction in iNOS with H. pylori in- fection leads to COX-2 activation through S-nitro- sylation and up-regulation in PGE2 generation, and that ghrelin counters these untoward consequences of the LPS through Akt-mediated up-regulation in cNO- S activation required for the iNOS gene repression. Keywords: H. pylori; Gastric Mucosa; iNOS Induction; COX-2 Activation; S-Nitrosylation; Ghrelin 1. INTRODUCTION Helicobacter pylori, a Gram-negative bacterium colo- nizing the gastric mucosa, is recognized as a primary cause of gastric disease, and its cell-wall lipopolysac- charide (LPS) has been identified as a potent endotoxin capable of eliciting mucosal inflammatory changes that characterize gastritis and duodenal ulcer [1-3]. Indeed, the gastric mucosal inflammatory responses to H. pylori infection in humans as well as well as those characteriz- ing mucosal inflammatory changes in the animal model of H. pylori LPS-induced gastritis are manifested by a marked increase in epithelial cell apoptosis and proin- flammatory cytokine production, and the excessive nitric oxide (NO) and prostaglandin (PGE2) generation [4-7]. A growing body of evidence, moreover, points to the disturbances in NO generated by nitric oxide synthase (NOS) isozyme system, and the formation of PGE2 syn- thesized from arachidonic acid (AA) by the action of cyclooxygenase (COX) system, as the events defining the extent of gastric mucosal inflammatory involvement in response to H. pylori colonization [8-12]. The physiological and pathophysiological implica- tions of NO and PGE2 depend on the type of isozyme system involved in their generation, their subcellular targeting and the local concentrations [12-16]. Of the three NOS isozymes responsible for NO generation, the two expressed constitutively (cNOS) are calcium/calmo- dulindependent and provide precise pulses of NO for a fine modulation of the cellular processes [14,16]. The third, inducible isoform (iNOS) is calcium/calmodu- lin-independent, and its activation by proinflammatory cytokines and bacterial LPS provides the high output of NO that is importance to host defense against microbial invasion. However, its massive and sustained production is also associated with transcriptional disturbances and the induction of apoptosis [14-18]. The cyclooxygenase system consists of two COX isozymes, the constitutive isoform or COX-1, responsible for maintaining normal  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 14 physiological prostaglandin production, and the induc- ible form or COX-2, which accounts for up-regulation in PGE2 production in response to inflammatory stimulus [8,9,12]. Moreover, a large volume of data supports the existence of functional and signaling cross-talk between the products of NOS and COX systems [13,14,19-22]. Indeed, the stimulation of NO production through iNOS induction or the exogenous NO donors leads to COX enzymes activation and the increase in PGE2 gen- eration, while a decrease in PGE2 formation has been observed in the presence of pharmacological inhibitors of NOS system [20-23]. Furthermore, the COX-2 activa- tion for the increase in PGE2 synthesis has been linked to the enzyme protein S-nitrosylation via NO derived through LPS-elicited induction in iNOS [20]. There are also indications for the role of cNOS in the iNOS-de- pendent COX-2 activation [21,24]. Interestingly, the disturbances in NO and PGE2 production associated with H. pylori colonization are reflected in the massive up- regulation in iNOS activity and the suppression of cNOS activation [4,5,25,26]. Moreover, we have recently sh- own that Akt-mediated cNOS activation through phos- phorylation at Ser1179 plays an essential role in the mod- ulatory influence of peptide hormone, ghrelin, on the extent of gastric mucosal inflammatory responses to H. pylori LPS [26]. As gastric ghrelin is recognized as an important regu- lator of NOS and COX enzyme systems, and implicated in the control of local inflammations, gastroprotection, and modulation of the mucosal inflammatory responses to bacterial infection [18,27-31], in this study we inves- tigated the nature of inflammatory changes induced in gastric mucosal cells by H. pylori LPS and the mecha- nism of ghrelin modulatory influence on the cross-talk between the NOS and COX systems. Our data revealed that the LPS-elicited induction in iNOS leads to COX-2 activation through S-nitrosylation and up-regulation in PGE2 generation, and that ghrelin counters these unto- ward consequences of the LPS through up-regulation in cNOS phosphorylation and the suppression of iNOS gene induction. 2. MATERIALS AND METHODS 2.1. Gastric Mucosal Cell Incubation The mucosal cells, collected by scraping the mucosa of freshly dissected rat stomachs with a blunt spatula, were suspended in five volumes of ice-cold Dulbecco’s modi- fied (Gibco) Eagle’s minimal essential medium (DM- EM), supplemented with fungizone (50 µg/ml), penicil- lin (50 U/ml), streptomycin (50 µg/ml), and 10% fetal calf serum, and gently dispersed by trituration with a syringe, and settled by centrifugation [5]. Following rinsing, the cells were resuspended in the medium to a concentration of 2 × 107 cell/ml, transferred in 1 ml ali- quots to DMEM in culture dishes and incubated under 95% O2 - 5% CO2 atmosphere at 37˚C for 16 h in the presence of 0 ng/ml - 200 ng/ml of H. pylori LPS [18]. In the experiments evaluating the effect of ghrelin (rat, Sigma), cNOS inhibitor, L-NAME, iNOS inhibitor, 1400 W, Akt inhibitor, SH-5 (Calbiochem), COX-1 inhibitor, SC-560, COX-2 inhibitor, NS-398 and ascorbate (Sig- ma), the cells were first preincubated for 30 min with the indicated dose of the agent or vehicle before the addition of the LPS. The viability of cell preparations before and during the experimentation, assessed by Trypan blue dye exclusion assay [11], was greater than 97%. 2.2. Helicobacter pylori Lipopolysaccharide H. pylori used for LPS preparation was cultured from clinical isolates obtained from ATCC No. 4350 [2,5]. The bacterium was homogenized with liquid phenol- chloroform-petroleum ether, centrifuged, and the LPS contained in the sup ernatant was precipitated with water, washed with 80% phenol solution and dried with ether. The dry residue was dissolved in a small volume of wa- ter at 45˚C, centrifuged at 100,000 × g for 4 h, and the resulting LPS sediment subjected to lyophilization. 2.3. PGE2 and NO Quantification The aliquots of cell suspension from the control and various experimental conditions were centrifuged at 1500 × g for 5 min and the conditioned medium super- natant collected. PGE2 assays were carried out using an enzyme-linked immunoassay (Cayman) and 100 µl aliquots of the spent medium supernatant, according to the manufacturer’s instructions. The amount of PGE2 released into culture medium was determined by meas- uring the absorbance at 405 nm [30]. To assess NO production in gastric mucosal cells, we measured the stable NO metabolite, nitrite, accumulation in the cul- ture medium using Griess reaction [32]. A 100 µl of spent culture medium was incubated for 10 min at room temperature with 0.1 ml of Griess reagent and the absorbance was measured at 570 nm. 2.4. COX-2 Activity Assay For measurements of COX-2 activity, the gastric mu- cosal cells from the control and various experimental conditions were settled by centrifugation, rinsed with phosphate-buffered saline, and homogenized in 0.3 ml cold sample buffer containing 0.1 M Tris-HCl, pH 7.8, and 1 mM EDTA, centrifuged at 12,000 × g for 10 min at 4˚C, and the supernatant collected. The COX-2 ac- tivity in 40 µl aliquots of the resulting supernatant was measured using COX activity assay kit in the absence and the presence of COX-1 inhibition (SC-560), ac- cording to the manufacturer’s (Cayman) instruction. The absorbance was read at 590 nm. C opyright © 2011 SciRes. OJGas  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 15 2.5. cNOS and iNOS Activity Assay Nitric oxide synthase activities of cNOS and iNOS enzymes in the gastric mucosa l cells were measured by monitoring the conversion of L-[3H] arginine to L-[3H] citrulline using NOS-detect kit (Stratagene). The cells from the control and experimental treatments were homogenized in a sample buffer containing either 10 mM EDTA (for Ca2+-independent iNOS) or 6 mM CaCl2 (for Ca2+-depenedent cNOS), and centrifuged. The aliquots of the resulting supernatant were incu- bated for 30 min at 25˚C in the presence of 50 µCi/ml of L-[3H] arginine, 10 mM NAPDH, 5 µM tetrahydro- biopterin, and 50 mM Tis-HCl buffer, pH 7.4, in a final volume of 250 µl. Following addition of stop buffer and Dowex-50 W (Na+) resin, the mixtures were tran- sferred to spin cups, centrifuged and the formed L-[3H] citrulline contained in the flow through was quantified by scintillation counting. 2.6. Akt Activity Assay The kinase activity of Akt in gastric mucosal cells was measured with the Akt Activity Kit (Calbiochem) by quantifying phosphorylation of a biotinylated peptide substrate (GRPRTSSFAEG). The cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1% deoxycholate, 2 mM EDTA, 1 mM sodium orthovanadate, 1 mM PAF, and 1 mM NaF), containing protease inhibitor cocktail (Sigma), at 4˚C for 30 min, centrifuged at 14,000 × g for 15 min, and immunoprecipitated with anti-Akt antibody for 1 h at 4˚C. Protein A/G agarose beads were then added for an additional 1 h, and the immune complex was recovered by centrifugation and thoroughly washed with lysis buffer [26]. The agarose beads were then sus- pended for 30 min at room temperature in the kinase assay buffer, centrifuged, and the supernatants used for the Akt activity assay by following the manufacturer’s instruction. 2.7. COX-2 Protein S-Nitrosylation Assay A biotin switch procedure was employed to assess COX-2 protein S-nitrosylation [33,34]. The gastric mu- cosal cells were treated with iNOS inhibitor, 1400 W (40 µM) or ghrelin (0.5 µg/ml), or Akt inhibitor, SH-5 (30 µM) + ghrelin (0.5 µg/ml), and incubated for 16 h in the presence of 100 ng/ml of H. pylori LPS. Following cen- trifugation at 500 × g for 5 min, the recovered cells were lysed in 0.2 ml of HEN lysis buffer (250 mM HEPES, 1 mM EDTA, 0.1 mM neocuprin, pH 7.7), and the unni- trosylated thiol groups were blocked with S-methyl me- thanethiosulfonate reagent at 50˚C for 20 min [34]. The proteins were precipitated with acetone, resuspended in 0.2 ml of HEN buffer containing 1% SDS, and subjected to targeted nitrothiol group reduction with sodium as- corbate (100 mM). The free thiols were then labeled with biotin and the biotinylated proteins were recovered on streptavidin beads. The formed streptavidin bead- protein complex was washed with neutralization buffer, and the bound proteins were dissociated from strepta- vidin beads with 50 µl of elution buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, pH 7.7) containing 1% 2-mercaptoethanol [34]. The obtained proteins were then analyzed by Western blotting. 2.8. Immunoblotting Analysis The mucosal cells from the control and experimental treatments were collected by centrifugation and resus- pended for 30 min in ice-cold lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 % glycerol, 1% Tri- ton X-100, 2 mM EDTA, 1 mM sodium orthovanadate, 4 mM sodium pyrophosphate, 1 mM PMSF, and 1 mM NaF), containing 1 µg/ml leupeptin and 1 µg/ml pep- statin [18]. Following brief sonication, the lysates were centrifuged at 12,000 g for 10 min, and the supernatants were normalized with respect to protein concentration using BCA protein assay kit (Pierce). The samples, in- cluding those subjected to biotin switch procedure, were then resusp ended in load ing buffer, boiled for 5 min, and subjected to SDS-PAGE using 40 µg protein/lane. The separated proteins were transferred onto nitrocellulose membranes, blocked for 1 h with 5% skim milk in Tris-buffered Tween (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and probed with specific poly- clonal rabbit antibodies directed against COX-1, COX-2, and iNOS (Calbiochem). The phosphorylated cNOS (pcNOS) was analyzed using specific antibody (Calbio- chem) directed against phospho-cNOS (mouse anti- eNOS, pSer11 79), and following stripping, probed with antibody against to tal cNOS. 2.9. Data Analysis All experiments were carried out using duplicate sam- pling, and the results are expressed as means ± SD. Analysis of variance (ANOVA) and nonparametric Kruskal-Wallis tests were u sed to determine significance. Any difference detected was evaluated by means of post hoc Bonferroni test, and the significance level was set at P < 0.05. 3. RESULTS To further ascertain the nature of gastric mucosal in- flammatory responses to H. pylori and to reveal the modulatory role ghrelin, we investigated the interaction between the systems responsible for NO generation and prostaglandin production. Using rat gastric mucosal cells exposed to H. pylori key virulence factor, LPS, we C opyright © 2011 SciRes. OJGas 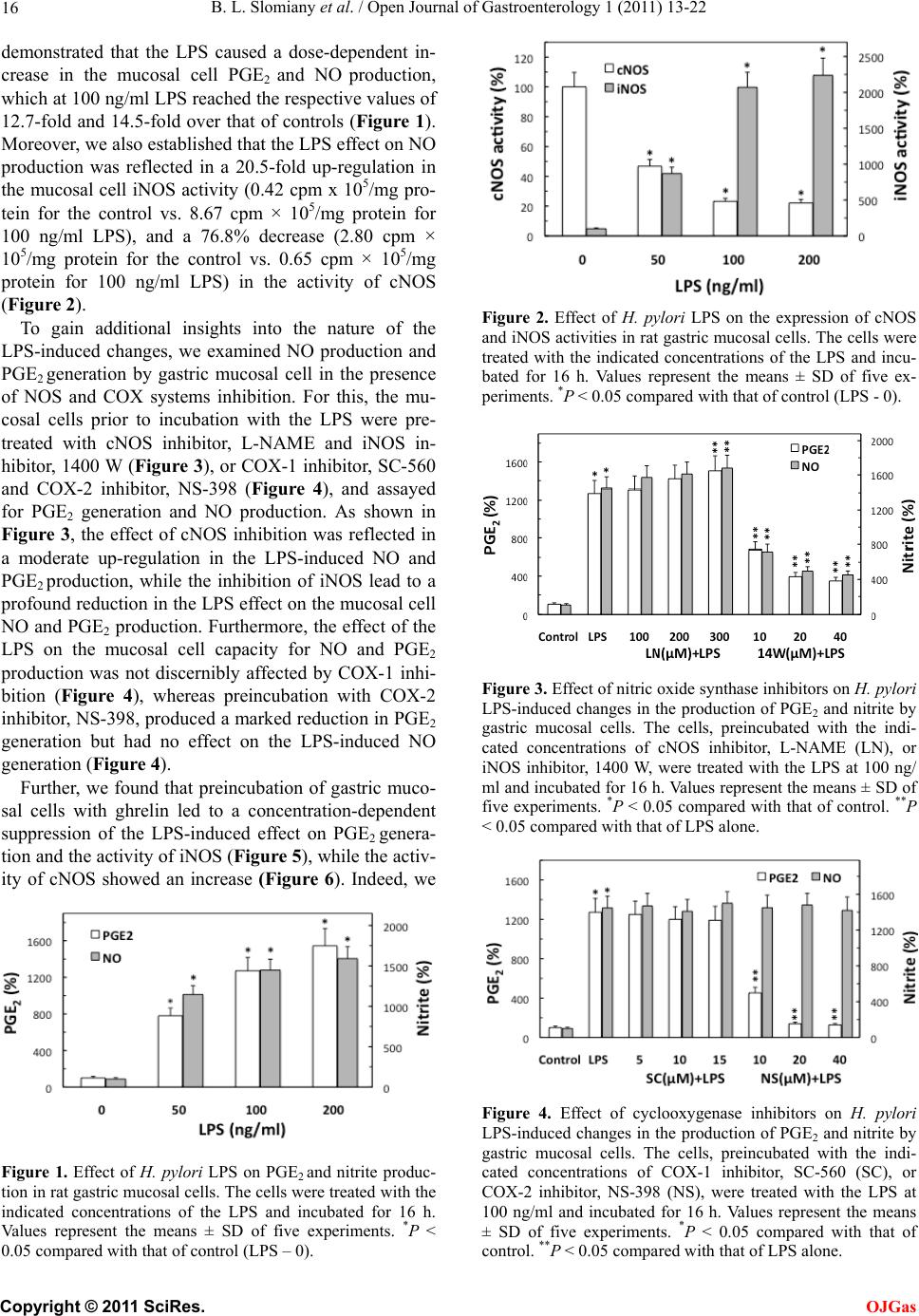 B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 16 demonstrated that the LPS caused a dose-dependent in- crease in the mucosal cell PGE2 and NO production, which at 100 ng/ml LPS reached the respective values of 12.7-fold and 14.5-fold over that of controls (Figure 1). Moreover, we also established that the LPS effect on NO production was reflected in a 20.5-fold up-regulation in the mucosal cell iNOS activity (0.42 cpm x 105/mg pro- tein for the control vs. 8.67 cpm × 105/mg protein for 100 ng/ml LPS), and a 76.8% decrease (2.80 cpm × 105/mg protein for the control vs. 0.65 cpm × 105/mg protein for 100 ng/ml LPS) in the activity of cNOS (Figure 2). To gain additional insights into the nature of the LPS-induced changes, we examined NO production and PGE2 generation by gastric mucosal cell in the presence of NOS and COX systems inhibition. For this, the mu- cosal cells prior to incubation with the LPS were pre- treated with cNOS inhibitor, L-NAME and iNOS in- hibitor, 1400 W (Figure 3), or COX-1 inhibitor, SC-560 and COX-2 inhibitor, NS-398 (Figure 4), and assayed for PGE2 generation and NO production. As shown in Figure 3, the effect of cNOS inhibition was reflected in a moderate up-regulation in the LPS-induced NO and PGE2 production, while the inhibition of iNOS lead to a profound reduction in the LPS effect on the mucosal cell NO and PGE2 production. Furthermore, the effect of the LPS on the mucosal cell capacity for NO and PGE2 production was not discernibly affected by COX-1 inhi- bition (Figure 4), whereas preincubation with COX-2 inhibitor, NS-398, produced a marked reduction in PGE2 generation but had no effect on the LPS-induced NO generation (Figur e 4). Further, we found that preincubation of gastric muco- sal cells with ghrelin led to a concentration-dependent suppression of the LPS-induced effect on PGE2 genera- tion and the activity o f iNOS (Figure 5), while the activ- ity of cNOS showed an increase (Figure 6). Indeed, we Figure 1. Effect of H. pylori LPS on PGE2 and nitrite produc- tion in rat gastric mucosal cells. The cells were treated with the indicated concentrations of the LPS and incubated for 16 h. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control (LPS – 0). Figure 2. Effect of H. pylori LPS on the expression of cNOS and iNOS activities in rat gastric mucosal cells. The cells were treated with the indicated concentrations of the LPS and incu- bated for 16 h. Values represent the means ± SD of five ex- periments. *P < 0.05 compared with that of control (LPS - 0). Figure 3. Effect of nitric oxide synthase inhibitors on H. pylori LPS-induced changes in the production of PGE2 and nitrite by gastric mucosal cells. The cells, preincubated with the indi- cated concentrations of cNOS inhibitor, L-NAME (LN), or iNOS inhibitor, 1400 W, were treated with the LPS at 100 ng/ ml and incubated for 16 h. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. Figure 4. Effect of cyclooxygenase inhibitors on H. pylori LPS-induced changes in the production of PGE2 and nitrite by gastric mucosal cells. The cells, preincubated with the indi- cated concentrations of COX-1 inhibitor, SC-560 (SC), or COX-2 inhibitor, NS-398 (NS), were treated with the LPS at 100 ng/ml and incubated for 16 h. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. C opyright © 2011 SciRes. OJGas 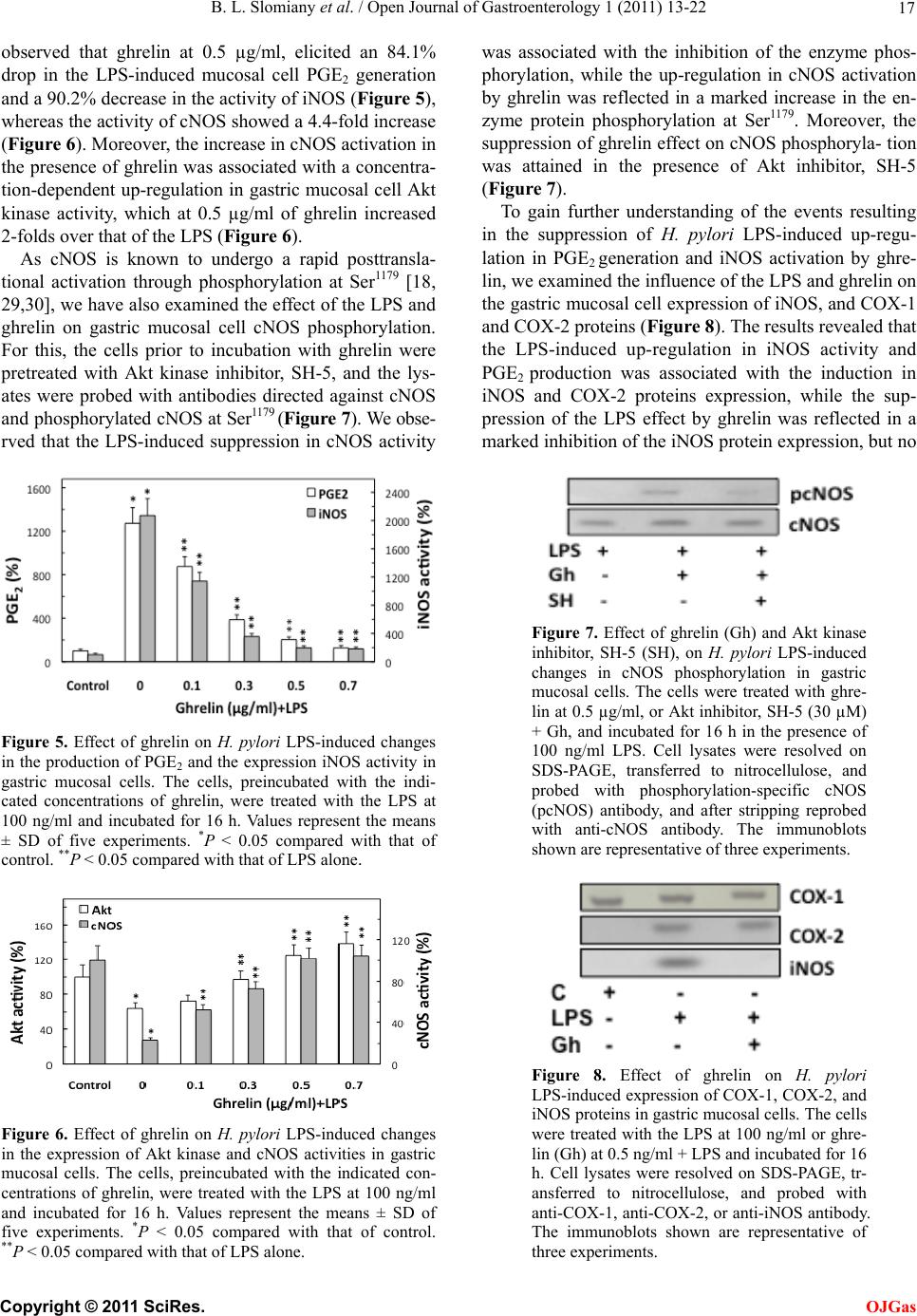 B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 17 observed that ghrelin at 0.5 µg/ml, elicited an 84.1% drop in the LPS-induced mucosal cell PGE2 generation and a 90.2% decrease in the activity of iNOS (Figure 5), whereas the activity of cNOS showed a 4.4-f old incr ease (Figure 6). Moreover, the increase in cNOS activation in the presence of ghrelin was associated with a concentra- tion-dependent up -regulation in gastric mucosal cell Akt kinase activity, which at 0.5 µg/ml of ghrelin increased 2-folds over that of the LPS (Figure 6). As cNOS is known to undergo a rapid posttransla- tional activation through phosphorylation at Ser1179 [18, 29,30], we have also examined the effect of the LPS and ghrelin on gastric mucosal cell cNOS phosphorylation. For this, the cells prior to incubation with ghrelin were pretreated with Akt kinase inhibitor, SH-5, and the lys- ates were probed with antibodies directed against cNOS and phosphor ylated cNOS at Ser1179 (Figure 7). We obse- rved that the LPS-induced suppression in cNOS activity Figure 5. Effect of ghrelin on H. pylori LPS-induced changes in the production of PGE2 and the expression iNOS activity in gastric mucosal cells. The cells, preincubated with the indi- cated concentrations of ghrelin, were treated with the LPS at 100 ng/ml and incubated for 16 h. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. Figure 6. Effect of ghrelin on H. pylori LPS-induced changes in the expression of Akt kinase and cNOS activities in gastric mucosal cells. The cells, preincubated with the indicated con- centrations of ghrelin, were treated with the LPS at 100 ng/ml and incubated for 16 h. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. was associated with the inhibition of the enzyme phos- phorylation, while the up-regulation in cNOS activation by ghrelin was reflected in a marked increase in the en- zyme protein phosphorylation at Ser1179. Moreover, the suppression of ghrelin effect on cNOS phosphoryla- tion was attained in the presence of Akt inhibitor, SH-5 (Figure 7). To gain further understanding of the events resulting in the suppression of H. pylori LPS-induced up-regu- lation in PGE2 generation and iNOS activation by ghre- lin, we examined the influence of the LPS and ghrelin on the gastric mucosal cell expression of iNOS, and COX-1 and COX-2 prot ei ns (Figure 8). The results revealed that the LPS-induced up-regulation in iNOS activity and PGE2 production was associated with the induction in iNOS and COX-2 proteins expression, while the sup- pression of the LPS effect by ghrelin was reflected in a marked inhibition of the iNOS protein expression, but no Figure 7. Effect of ghrelin (Gh) and Akt kinase inhibitor, SH-5 (SH), on H. pylori LPS-induced changes in cNOS phosphorylation in gastric mucosal cells. The cells were treated with ghre- lin at 0.5 µg/ml, or Akt inhibitor, SH-5 (30 µM) + Gh, and incubated for 16 h in the presence of 100 ng/ml LPS. Cell lysates were resolved on SDS-PAGE, transferred to nitrocellulose, and probed with phosphorylation-specific cNOS (pcNOS) antibody, and after stripping reprobed with anti-cNOS antibody. The immunoblots shown are representative of three experiments. Figure 8. Effect of ghrelin on H. pylori LPS-induced expression of COX-1, COX-2, and iNOS proteins in gastric mucosal cells. The cells were treated with the LPS at 100 ng/ml or ghre- lin (Gh) at 0.5 ng/ml + LPS and incubated for 16 h. Cell lysates were resolved on SDS-PAGE, tr- ansferred to nitrocellulose, and probed with anti-COX-1, anti-COX-2, or anti-iNOS antibody. The immunoblots shown are representative of three experiments. C opyright © 2011 SciRes. OJGas  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 18 COX-2 protein expression was discerned. We have also apparent diminution in observed that the COX-1 protein expression remained essentially unaffected by the inclu- sion of the LPS or ghrelin (Figure 8). Thus, the enzy- matic activity of the LPS-induced COX-2 protein for up-regulation in PGE2 production shows an apparent dependence on NO generated by the iNOS system. Therefore, to ascertain the requirement of H. pylori LPS-induced up-regulation in COX-2 activ ation for NO, the mucosal cells prior to incubation with ghrelin were pretreated with iNOS inhib itor, 1400 W, or Akt inhibito r, SH-5, or nitrosothiol reducing agent, ascorbate, and as- sayed for COX-2 activity. We found that the LPS-in- duced up-regulation in COX-2 activity was subject to suppression not only by the pretreatment with ghrelin, but also sho wed su scep tibility to iNOS inh ibito r, 1400 W, whereas Akt inhibitor, SH-5, had no effect on the extent of the LPS-induced COX-2 activation (Figure 9). Moreover, while the iNOS inhibitor, 1400 W, produced amplification in the inhibitory effect of ghrelin on COX-2 activity, the Ak t inhibitor, SH-5, caused the sup- pression in ghrelin effectiveness. Further, we found that the LPS-induced COX-2 activation displayed suscepti- bility to suppression by nitrosothiol reducing agent, as- corbate, which also produced enhancement in the effect of ghrelin on COX-2 activity (Figure 9). Finally, we also examined the dependence of COX-2 S-nitrosylation on the LPS-induced iNOS activation by the biotin switch method [33,34]. The gastric mucosal cells were incubated with H. pylori LPS or ghrelin + LPS in the absence and presence of Akt inhibitor, SH-5, or iNOS inhibitor, 1400 W + LPS, and t he lysates following the biotin switch procedure were examined for COX-2 protein S-nitrosylation (Figure 10). Western blot analy- sis revealed that COX-2 in the cells exposed to the LPS alone showed a marked increase in the protein S-nitro- sylation, whereas the prein cubation with iNOS inhibitor, 1400 W, resulted in the loss of the LPS-induced COX-2 S-nitrosylation. A pronounced decrease in the LPS-in- duced COX-2 S-nitrosylation was also attained in the presence of ghrelin. Moreover, this effect of ghrelin on COX-2 S-nitrosylation was subject to suppression by the inclusion of Akt inhibitor, SH-5. These data suggest that induction in iNOS elicited by H. pylori LPS leads to COX-2 activation through S-nitrosylation that results in an excessive PGE2 generation, and that ghrelin counters the detrimental consequences of iNOS induction by way of cNOS-dependent suppression of iNOS gene induc- tion. 4. DISCUSSION H. pylori lipopolysaccharide is recognized as a potent endotoxin capable of eliciting mucosal inflammatory Figure 9. Effect of iNOS inhibitor, 1400 W, Akt inhibitor, SH-5, and ascorbate on the ghrelin (Gh)-induced changes in COX-2 activity in gastric mucosal cell exposed to H. pylori LPS. The cells, preincubated with 40 µM 1400 W (14 W), 30 µM SH-5 (SH), or 300 µM ascorbate (As), were treated with Gh at 0.5 µg/ml and incubated for 16h in the presence of 100 ng/ml LPS. Values represent the means ± SD of five experi- ments. *P < 0.05 compared with t hat of control. **P < 0.05 com- pared with that of LPS alone. ***P < 0.05 compared with that of Gh LPS . Figure 10. Effect of ghrelin (Gh) on H. pylori LPS-induced COX-2 S-nitrosylation. The gastric mucosal cells were treated with 40 µM of iNOS inhibitor, 1400 W (14 W), or Gh (0.5 µg/ml), or 30 µM Akt inhibitor, SH-5 (SH) + Gh, and incu- bated for 16 h in the presence of 100 ng/ml L PS. A portion of the cell lysates was processed by biotin switch procedure for protein S-nitrosylation and, along with the reminder of the lysates, re- solved on SDS-PAGE, transferred to nitrocellu- lose and probed with anti-COX-2 antibody. The immunoblots shown are representative of three experiments. responses akin to those that characterize gastritis and duodenal u lcer [1-3]. Indeed, H. pylori LPS, like LPS of other Gram-negative bacteria, is known to trigger a wide variety of transcriptional factors, including NF-B and AP-1, that exert transcrip tional control over such impor- tant mediators of inflammation as iNOS and COX-2 systems, which along with the constitutively expressed isozyme forms, cNOS and COX-1, are responsible for NO and PGE2 production [4-6,20-22]. Moreover, a growing volume of data consistently points towards the existence of a functional and signaling relationship be- tween the products of NOS and COX enzyme systems [13,14,19,20]. In particular, there are strong indications for the involvement of iNOS-derived NO in COX-2 ac- tivation through S-nitrosylation for the increase in PGE2 C opyright © 2011 SciRes. OJGas  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 19 generation [20-22]. Furthermore, the gastric mucosal inflammatory responses to H. pylori as well as its LPS are reflected in the suppression in cNOS activation [5,7,25,26]. Hence, in this study presented herein, we investigated the nature of interaction between the systems responsible for NO generation and PGE2 production. Using rat gastric mucosal cells exposed to H. pylori LPS, we demonstrated that the LPS-induced enhance- ment in iNOS activity and up-regulation in PGE2 pro- duction was accompanied by the suppression in Akt ki- nase activity and the impairment in cNOS activation through phosphorylation. The stimulatory effect of the LPS on PGE2 production was susceptible to suppression by COX-2 inhibitor, NS-398, as well as the inhibitor of iNOS, 1400 W. However, the LPS-induced up-regulatio n in the mucosal cell NO generation was not affected by the inhibitors of COX-1 and COX-2 systems. These lat- ter findings are thus in keeping with the literature data as to the role of iNOS in the regulation of COX-2 activa- tion for the increase in PGE2 production [2 0 -23]. Further, we found that preincubation of the mucosal cells with gastric hormone, ghrelin, recognized for its modulatory influence on the inflammatory responses to bacterial infection [18,27,29-31,35], exerted countering effect on the LPS-induced suppression in Akt activity and lead to the increase in cNOS activation through phosphorylation at Ser1179. We also observed that these effects of ghrelin were accompanied by the suppression in iNOS protein expression and the reduction in COX-2 activity. Moreover, ghrelin-induced up-regulation in cNOS activation was susceptible to suppression by Akt inhibitor, SH-5, which also caused the abrogation in ghrelin-induced reduction in COX-2 activity. From this, we concluded that ghrelin countering effect on H. pylori LPS-induced inflammatory changes occurs with the in- volvement of Akt-mediated cNOS activation through phosphorylation, and that Akt kinase plays an essential role in the action of ghrelin on COX-2 activity. Indeed, Akt kinase occupies a central role in the receptor (GHS- R)-mediated responses to ghrelin stimulation [26,36], and the involvement of signaling through Akt for a rapid up-regulation in cNOS activity through the enzyme pro- tein phosphorylation at Ser1179 is well documented [29, 30,37]. Our further examination of the influence of ghrelin on H. pylori LPS-induced up-regulation in iNOS activity and PGE2 generation revealed that while the expression of COX-1 protein remained essentially unaffected by the LPS or ghrelin, the effect of the LPS was associated with the induction in iNOS and COX-2 proteins. The coun- tering effect of ghrelin, moreover, was reflected in a marked inhibition of the iNOS protein expression, but no apparent diminution in COX-2 protein was discerned. Thus, in concordance with our earlier results, it is ap- parent that whilst the countering effect of ghrelin on the LPS-induced enhancement in iNOS activity is associated with the inhibition of iNOS gene expression at the tran- scriptional level [38], the suppression of the LPS-in- duced COX-2 activity by ghrelin does not involve the inhibition in the enzyme protein expression. However, the enzymatic activity of the LPS-induced COX-2 pro- tein for up-regulation in PGE2 production shows an ap- parent dependence on NO generated by the iNOS system. Indeed, the stimulation of NO production through iNOS induction has been reported to lead to COX-2 protein modification through S-nitrosylation that results in the enzyme activation and the increase in PGE2 generation [20-22]. Hence, to reveal further insights into the mechanism of ghrelin suppression of the LPS-induced gastric mu- cosal inflammatory disturbances, we examined the COX-2 activity and its protein S-nitrosylation in the presence of the inhibitors of iNOS and Akt, as well as nitrosothiols reducing agent, ascorbate. We found that the countering effect of ghrelin on the LPS-induced up-regulation in COX-2 activity was further amplified in the presence of iNOS inhibitor, 1400 W, while the Akt inhibitor, SH-5, caused the suppression in ghrelin effec- tiveness. Moreover, the LPS-induced COX-2 activation displayed susceptibility to suppression by ascorbate, which also produced an amplification in ghrelin effect on the mucosal COX-2 activity. These data, together with the demonstrated susceptibility of the LPS-induced COX-2 activation to inhibition by iNOS inhibitor, 1400 W, as well as known vulnerability of S-nitrosylated pro- teins to reduction by ascorbate [20,21,26,33,34], sug- gest that the countering effect of ghrelin on H. pylori LPS-induced changes in COX-2 activation are intimately linked to the events of Akt activation and a rapid up- regulation in cNOS activity through Akt-med iated cNOS protein phosphorylation. Consequently, our results point to the involvement of cNOS in controlling the extent of iNOS-dependent COX-2 activation. The supporting evidence as to the involvement of Akt-mediated cNOS activation in the modulatory action of ghrelin against H. pylori LPS-induced gastric mucosal consequences of iNOS induction and up-regulation in COX-2 activation comes from the results of biotin switch assay. We found that the mucosal cells exposed to incubation with H. pylori LPS showed a marked incr e as e in COX-2 protein S-nitrosylation, while the suppression of the LPS-induced iNOS activity with a specific inhibi- tor, 1400 W, caused the loss in COX-2 S-nitrosylation. Further, we observed a pronounced drop in the LPS- induced COX-2 S-nitrosylation in the presence of ghre- lin, the effect of which was subject to the repression by Akt inhibitor, SH-5. These findings, together with the results of COX-2 activity assays, suggest that induction C opyright © 2011 SciRes. OJGas  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 20 in gastric mucosal iNOS expression elicited by H. pylori LPS leads to COX-2 activation through S-nitrosylation that results in an excessive PGE2 generation. It is also apparent that ghrelin counters these untoward conse- quences of H. pylori via Akt-mediated up-regulation in cNOS activation that is required for the suppression of iNOS gene induction. While the intricate details of the role of cNOS in the regulation of iNOS g ene induction remain ob scure, there are indications that cNOS is capable of affecting tran- scriptional factor NF-B activation and therefore to in- fluence the extent of promoter activity and iNOS gene transcription [12,21,24]. More interestingly, the litera- ture data suggest that S-n itrosylation of an inhibitor pro- tein, IB kinase complex (IKK) interferes with ubiquit- inylation and proteasomal degradation of IB, thus pre- venting the nuclear translocation of NF-B, and result- ing in its inability to promote target gene transcription [39-41]. Furthermore, we have shown that the LPS-in- duced suppression in cNOS activity was associated with the induction in iNOS protein expression, while the countering effect of ghrelin, like that NF-B inhibitor, PPM-18, was reflected in a marked inhibition of the iNOS protein expression [38]. Moreover, the effect of ghrelin was intimately linked to Akt-mediated cNOS activation through phosphorylation [26]. Taken togeth er, these observations lead us to postulate that the initial phase gastric mucosal inflammatory re- sponses elicited by H. pylori involves the suppression in Akt kinase-dependent cNOS activation that leads to ab- rogation of cNOS-mediated IKK S-nitrosylation and the increase in proteasomal degradation of IB, thus allow- ing NF-B translocation to the nucleus to promote iNOS gene transcription. The induction in iNOS expression, in turn, leads to iNOS-mediated COX-2 activation through S-nitrosylation that results in an excessive PGE2 genera- tion. We also postulate that ghrelin counters these unto- ward consequences of H. pylori LPS via up-regulation in Akt-dependent cNOS activation through phosphoryla- tion that results in up-regulation in cNOS-mediated IKK coplex S-nitrosylation which interferes with proteasomal degradation of IB, thereby preventing the nuclear translocation of NF-B and the induction in iNOS gene transcription. Hence, the repression of iNOS expression by ghrelin triggers the loss in COX-2 activation through iNOS-dependent S-nitrosylation and leads to a reduction in PGE2 generati o n. REFERENCES [1] Stolte, M. and Edit, S. (1996) Helicobacter pylori and evolution of gastritis. Scandinavian Journal of Gastroen- terology, 3, 13-16. doi:10.3109/00365529609094508 [2] Piotrowski, J., Piotrowski, E., Skrodzka,D., Slomiany, A. and Slomiany, B.L. (1997) Induction of acute gastritis and epithelial cell apoptosis by Helicobacter pylori lipo- polysaccharide. Scandinavian Journal of Gastroenter- ology, 32, 203-211. doi:10.3109/00365529709000195 [3] de Boer, W.A. (2000) Helicobacter py lori infection: Focus on a “search-and-treat” strategy for ulcer disease. Scandi - navian Journal of Gastroenterology, 35, 4-9. [4] Fu, F., Ramanujan, K.S. and Wong, A., et al. (1999) In- creased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase-2 in Helico- bacter pylori gastritis. Gas troentero logy, 116, 1319-1329. doi:10.1016/S0016-5085(99)70496-8 [5] Slomiany, B.L., Piotrowski, J. and Slomiany, A. (1999) Gastric mucosal inflammatory responses to Helico- bac- ter pylori lipopolysaccharide: Down-regulation of nitric oxide synthase-2 and caspase-3 by sulglycotide. Bioche- mical and Biophysical Research Communications, 261, 15-20. doi:10.1006/bbrc.1999.1003 [6] Gupta, R.A., Polk, D.B., Krishna, U., et al. (2001) Acti- vation of peroxisome proliferator-activated receptor sup- presses nuclear factor kB-mediated apoptosis induced by Helicobacter pylori in gastric epithelial cells. Journal of Biological Chemistry, 276, 31059-31066. doi:10.1074/jbc.M104141200 [7] Slomiany, B.L. and Slomiany, A. (2003) Plate-let-acti- vating factor modulates gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccha-ride. Bi- ochemical and Biophysical Research Communications, 306, 261-266. doi:10.1016/S0006-291X(03)00944-6 [8] Wallace, J.L. and Devchand, P.R. (2005) Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal de- fense. British Journal of Pharmacology, 145, 275-282. doi:10.1038/sj.bjp.0706201 [9] Gyires, K. (2005) Gastric mucosal protection: From prostaglandins to gene-therapy. Current Medicinal Che- mistry, 12, 203-215. [10] Reider, G., Hofmann, J.A., Hatz, R.A., Stolte, M. and Enders, G.A. (2003) Up-regulation of inducible nitric oxide synthase in Helicobacter pylori-associated gastritis may represent an increased risk factor to develop gastric carcinoma of the intestinal type. International Journal of Medical Microbiology, 293, 403-412. doi:10.1078/1438-4221-00280 [11] Slomiany, B.L. and Slomiany, A. (2007) Interference by leptin with Helicobacter pylori lipopolysaccha ride-in- duced cytosolic phospholipase A2 activation in gastric mucosal cells. Journal of Physiology and Pharmacology, 58, 117-113. [12] Tsatsanis, C., Androulidaki, A., Venihaki, M. and Margi- oris, A.N. (2006) Signaling network regulating cyc- looxygenase-2. International Journal of Biochemistry and Cell Biology, 38, 1654-1661. doi:10.1016/j.biocel.2006.03.021 [13] Mollace, V., Muscoli, C., Masini, E., Cuzzocrea, S. and Salvemini, D. (2005) Modulation of prostaglandin bio- synthesis by nitric oxide and nitric oxide donors. Phar- macological Reviews, 57, 217-252. doi:10.1124/pr.57.2.1 [14] Cuzzocrea, S. and Salvemini, D. (2007) Molecular me- chanisms involved in the reciprocal regulation of cyc- looxygenase and nitric oxide synthase enzymes. Kidney International, 71, 290-297. doi:10.1038/sj.ki.5002058 C opyright © 2011 SciRes. OJGas  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 21 [15] Skill, N.J., Theodorakis, N.G., Wang, Y.N., Wu, J.M., Redmond, E.M. and Sitzmann, J.V. (2008) Role of cycl- ooxygenase isoforms in prostacyclin biosynthesis and murine prehepatic portal hypertension. American Journal of Physiology-Gastrointestinal and Liver Physiology, 295, G953-964. doi:10.1152/ajpgi.00013.2008 [16] Korhonen, R., Lahti, A., Kankaanranta, H. and Moilanen, E. (2005) Nitric oxide production and signaling in infla- mmation. Current Drug Targets: Inflammation and Al- lergy, 4, 471-479. doi:10.2174/1568010054526359 [17] Maa, M.C., Chang, M.Y., Chen, Y.T., et al. (2008) Re- quirement of inducible nitric-oxide synthase in lipopo- lysaccharide-mediated Src induction and macrophage migration. Journal of Biological Chemistry, 283, 31408- 31416. doi:10.1074/jbc.M801158200 [18] Slomiany, B.L. and Slomiany, A. (2010) Ghrelin protec- tion against lipopolysaccharide-induced gastric mucosal cell apoptosis involves constitutive nitric oxide synthase- mediated caspase-3 S-nitrosylation. Mediators of In- flammation. doi:1155/2010/280464 [19] Marnett, L.J., Wright, T.L., Crews, B.C., Tannenbaum, S.R. and Morrow, J.D. (2000) Regulation of prostag- ndin biosynthesis by nitric oxide is revealed by targeted deletion of inducible nitric-oxide synthase. Journal of Biological Chemistry, 275, 13427-13430. doi:10.1074/jbc.275.18.13427 [20] Kim, S.F., Huri, D.A. and Snyder, S.H. (2005) Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science, 310, 1966-1970. doi:10.1126/science.1119407 [21] Ye, Y., Martinez, J.D., Perez-Polo, R.J., Lin, Y., Uretsky, B.F. and Brinbaum, Y. (2008) The role of eNOS, iNOS, and NF-B in upregulation and activation of cyclooxy- genase-2 and infarct size reduction by atorvastatin. American Journal of Physiology-Heart and Cir-culatory Physiology, 295, H343-351. doi:10.1152/ajpheart.01350.2007 [22] Lamon, B.D., Upmacis, R.K., Deeb, R.S., Koyuncu, H. and Hajjar, D.P. (2010) Inducible nitric oxide synthase gene deletion exaggerate MAPK-mediated cyclooxy- genase-2 induction by inflammatory stimuli. American Journal of Physiology-Heart and Circulatory Physiology, 299, H613-623. doi:10.1152/ajpheart.00144.2010 [23] Tian, J., Kim, S.F., Hester, L. and Snyder, S.H. (2008) S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proceedings of the National Academy of Sciences of the USA, 105, 10537-10540. doi:10.1073/pnas.0804852105 [24] Bell, R.M., Smith, C.C. and Yellon, D.M. (2002) Nitric oxide as a mediator of delayed pharmacological (A1 re- ceptor triggered) preconditioning; is eNOS masquerading as iNOS? Cardiovascular Research, 53, 405-413. doi:10.1016/S0008-6363(01)00472-2 [25] Slomiany, B.L. and Slomiany, A. (2010) Role of consti- tutive nitric oxide synthase S-nitrosylation in Helico- bacter pylori-induced gastric mucosal cell apoptosis: Ef- fect of ghrelin. Inflammopharmacology, 18, 233-240. doi:10.1007/s10787-010-0051-7 [26] Slomiany, B.L. and Slomiany, A. (2010) Helicobacter pylori induces disturbances in gastric mucosal Akt acti- vation through inducible nitric oxide synthase-dependent S-nitrosylation: Effect of ghrelin. I S R N G a stro e n t e ro l o g y , doi:10.5402/2011/308727 [27] Kojima, M., Hosoda, H., Date, Y., Nakazato, M. and Kangawa, K. (1999) Ghrelin is a growth-hormone-re leasing acylated peptide from stomach. Nature, 402 , 656- 660. doi:10.1038/45230 [28] Sibilia, V., Pagani, F., Rindi, G., et al. (2008) Central ghrelin gastroprotection involves nitric oxide/prostag- landin cross-talk. British Journal of Pharmacology, 154, 688-697. doi:10.1038/bjp.2008.120 [29] Xu, X., Jhun, B.S., Ha, C.H. and Jin, Z.G. (2008) Mo- lecular mechanisms of ghrelin-mediated endothelial ni- tricoxide synthase activation. Endocrinology, 149, 4183- 4192. doi:10.1210/en.2008-0255 [30] Slomiany, B.L. and Slomiany, A. (2009) Involvement of constitutive nitric oxide synthase in ghrelin-induced cy- tosolic phospholipase A2 activation in gastric mucosal cell protection against ethanol cytotoxicity. Inflammoph- armacology, 17, 245-253. doi:10.1007/s10787-009-0013-0 [31] Chen, Y.T., Tsai, S.H., Sheu, S.Y. and Tsai, L.H. (2010) Ghrelin improves LPS-induced gastrointestinal motility disturbances: Roles of NO and prostaglandin E2. Shock, 33, 205-212. doi:10.1097/SHK.0b013e3181ae841b [32] Green, L.C., Wagner, D.A., Glogowski, J., Skipper, P.L., Wishnok, J.S. and Tannenbaum, S.R. (1982) Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Analytical Biochemistry, 126, 131-138. doi:10.1016/0003-2697(82)90118-X [33] Jaffrey, S.R., Erdjument-Bromage, H., Ferris, D., Tempst, P. and Snyder, S.H. (2001) Protein S-nitrosylation: A physiological signal for neuronal nitric acid. Nature Cell Biology, 3, 193-197. doi:10.1038/35055104 [34] Forrester, M.T., Foster, M.W. and Stamler, J.S. (2007) Assessment and application of the biotin switch tech- nique for examining protein S-nitrosylation under condi- tions of pharmacologically induced oxidative stress. Jo- urnal of Biological Chemistry, 282, 13977-13983. doi:10.1074/jbc.M609684200 [35] Waseem, T., Duxbury, M., Ito, H., Ashley, S.W. and Ro-binson, M.K. (2008) Exogenous ghrelin modulates re-lease of proinflammatory and anti-inflammatory cyto- kines in LPS-stimulated macrophages through distinct signaling pathways. Surgery, 143, 334-342. doi:10.1016/j.surg.2007.09.039 [36] Lodeiro, P., Theodoropoulou, M., Pardo, M., Casanueva, F.F. and Camina, J.P. (2009) c-Src regulates Akt signaling in response to ghrelin via barrestin signaling-independent and dependent mechanism. PLoS One, 4, E4686. doi:10.1371/journal.pone.0004686 [37] Haynes, M.P., Li, L., D. Sinha, D., et al. (2003) Src ki- nase mediates phosphatidylinositol 3-kinase/Akt-depen- dent rapid endothelial nitric-oxide synthase activation by estrogen. Journal of Biological Chemistry, 278, 2118- 2123. doi:10.1074/jbc.M210828200 [38] Slomiany, B.L. and Slomiany, A. (2011) Ghrelin sup- pression of Helicobacter pylori-induced S-nitrosyla- tion-dependent Akt inactivation exerts modulatory influ- ence on gastric mucin synthesis. Inflammopharmacology, 19, 98-97. doi:10.1007/s10787-011-0078-4 [39] Park, S.K., Lin, H.L. and Murphy, S. (1997) Nitric oxide regulates nitric oxide synthase-2 gene expression by in- hibiting NF-B binding to DNA. Biochemical Journal, 322, 609-613. C opyright © 2011 SciRes. OJGas  B. L. Slomiany et al. / Open Journal of Gastroenterology 1 (2011) 13-22 Copyright © 2011 SciRes. 22 OJGas [40] Reynaert, N.L., Ckless, K., Korn, S.H., et al., (2004) Nitric oxide represses inhibitory kB kinase through S-nitrosylation. Proceedings of the National Academy of Sciences of the USA, 101, 8945-8950. doi:10.1073/pnas.0400588101 [41] Hess, D.T., Matsumoto, A., Kim, S.O., Marshall, H.E. and Stamler, J.S. (2005) Protein S-nitrosylation: Purview and parameters. Nature Reviews/Molecular Cell Biology, 6, 150-166. doi:10.1038/nrm1569
|