 J. Biomedical Science and Engineering, 2009, 2, 550-558 doi: 10.4236/jbise.2009.27080 Published Online November 2009 (http://www.SciRP.org/journal/jbise/ JBiSE ). Published Online November 2009 in SciRes. http://www.scirp.org/journal/jbise Analysis of correlated mutations, stalk motifs, and phylogenetic relationship of the 2009 influenza A virus neuraminidase sequences Wei Hu Department of Computer Science, Houghton College, Houghton, NY, USA. Email: wei.hu@houghton.edu Received 13 October 2009; revised 27 October 2009; accepted 30 October 2009. ABSTRACT The 2009 H1N1 influenza pandemic has attracted worldwide attention. The new virus first emerged in Mexico in April, 2009 was identified as a unique combination of a triple-reassortant swine influenza A virus, composed of genetic information from pigs, hu- mans, birds, and a Eurasian swine influenza virus. Several recent studies on the 2009 H1N1 virus util- ized small datasets to conduct analysis. With new se- quences available up to date, we were able to extend the previous research in three areas. The first was finding two networks of co-mutations that may po- tentially affect the current flu-drug binding sites on neuraminidase (NA), one of the two surface proteins of flu virus. The second was discovering a special stalk motif, which was dominant in the H5N1 strains in the past, in the 2009 H1N1 strains for the first time. Due to the high virulence of this motif, the second finding is significant in our current research on 2009 H1N1. The third was updating the phylogenetic an- alysis of current NA sequences of 2009 H1N1 and H5N1, which demonstrated that, in clear contrast to previous findings, the N1 sequences in 2009 are di- verse enough to cover different major branches of the phylogenetic tree of those in previous years. As the novel influenza A H1N1 virus continues to spread globally, our results highlighted the importance of performin g timely analysis o n th e 2009 H1N1 v irus. Keywords: Entropy; Co-mutation; Mutation; Mutual Information; Neuraminidase; Phylogenetic Analysis; Random Forest; Stalk Motif; Swine Flu 1. INTRODUCTION There are three types of flu viruses, types A, B, and C. Type A viruses are the most pathogenic to humans. The influenza has on its surface two glycoproteins, hemag- glutinin (HA) and neuraminidase (NA), based on which influenza is classified. There are 16 types of HA proteins and 9 types of NA proteins, which are named H1, H2, H3 and etc. For example, “bird flu” is H5N1 and “swine flu” is H1N1. The HA binds the virus to sialic acid re- ceptors on the host cell surface. The NA protein facili- tates the release of virions to infect other cells by re- moving sialic acid residues from the viral HA during entry and release from cells. The NA protein is a te- tramer of four identical polypeptide chains anchored in the membrane of the virus. Its head domain is globular and supported by a long and thin stalk. The “Spanish” influenza pandemic of 1918–1919 cau- sed about 50 million deaths worldwide and about one- third of the world’s population was infected. One unique feature of the 1918 influenza pandemic was the simulta- neous infection of humans and swine. Recent studies on the 1918 virus revealed that the genes encoding the HA and NA surface proteins of the 1918 virus were derived from an avian-like influenza virus shortly before the start of the pandemic [1]. In April 2009, a novel strain of the influenza A (H1N1) virus was discovered in patients from Mexico and the United States and it spread across the globe via human- to-human contact within a very short time. Because of the seriousness of this new flu virus, the World Health Or- ganization (WHO) has officially declared the H1N1 virus a global pandemic. Genomic analysis of the 2009 influ- enza A (H1N1) virus suggested that it is closely related to common reassortant swine influenza A viruses isolated in North America, Europe, and Asia. Its NA sequences have 94.4% similarity at the nucleotide level with European swine influenza A virus strains from 1992 [2]. Flu drugs such as oseltamivir (Tamiflu®) and zana- mivir (Relenza®) currently in use only target the NA proteins, and disrupt the capability of the virus to escape infected cells and move elsewhere to infect other healthy cells. Clinical reports suggested that the new virus is susceptible to the two drugs [3]. However, a growing concern is that more drug resistant mutants will emerge under the selection pressure of constant drug use. Re-  W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 551 searchers from Rensselaer Polytechnic Institute [4] de- signed a new flu drug by targeting both the HA and NA genes of the virus as an effective way to treat the next mutation of H1N1 swine flu. Two recent studies [5,6] provided insights into the in- teractions of flu drugs with NA of the 2009 H1N1 virus. One study [6] developed a 3D structure of 2009 H1N1 NA and compared it with the crystal structure of 2006 H5N1 NA and the structure of 1918 H1N1 NA. It found that the hydrophobic Try347 in H5N1 NA does not ma- tch with the hydrophilic carboxyl group of oseltamivir as in the case of H1N1 NA, which explains in part the rea- son why the H5N1 avian influenza virus is drug-resistant to oseltamivir. Another study [5] found that the NA sequences of 2009 H1N1 are phylogenetically more closely related to European H1N1 swine flu and H5N1 avian flu rather than to the H1N1 counterparts in the America. It also investigated the sequence variations of 2009 H1N1 NA, using three sequences of NA, A/H1N1 /California/04/ 2009, A/H5N1/Vietnam/2004, and A/H1N1/Brevig Mis- sion/1/18 (the 1918 Spanish flu). With multiple sequence alignment, they found that among the 387 residues of the NA domain, the 2009 H1N1 NA differs from the other two strains in 21 positions. The novel mutations of NA are mainly located at the protein surface and not near the binding pocket for currently used NA inhibitors. It is na- tural to explore whether there are any potential mutation sites that may interfere with the active sites in the near future. In light of the possible emerging of new mutations of 2009 H1N1 that could lead to serious drug resistance, it is imperative to study the potential mutations and co- mutations of the current sequences of 2009 H1N1, be- cause mutations tend to function in concert to achieve some biological purposes. In this study, we employed entropy and mutual information theory [7,8] to investi- gate this issue. Because the NA stalk supports the head domain, its length can influence the function of NA. A special NA stalk motif with a 20-amino acid deletion in the 49th to 68th positions of the stalk region was first identified in H5N1 in 2000. There was a gradual increase of this spe- cial NA stalk motif in H5N1 isolates from 2000 to 2007, and it was in all 173 H5N1 human isolates from 2004 to 2007. The H5N1 virus carrying this special NA stalk motif has the highest virulence and pathogenicity in chicken and mice [9]. This finding prompted us to search for similar stalk motifs in the current 2009 H1N1 virus strains. In summary, the goal of our study is to conduct a timely analysis of mutations, co-mutations, stalk motifs, and phylogenetic relationship of the 2009 H1N1 NA sequences available up to date. Such information can be valuable in further efforts to improve drug design and flu treatment. 2. MATERIALS AND METHODS 2.1. Sequence Data Published NA sequences of 7251 influenza A virus were downloaded from the Influenza Virus Resource (http:// www.ncbi/nlm.nih.giv/genomes/FLU/FLU.html) of the National Center for Biotechnology Information (NCBI) on Sept 13, 2009. We were mainly interested in the se- quences in 2009, but also needed the sequences in 2008 and 2007 to provide comparison in the study of stalk motifs. There were 283 sequences of H1N1 and H5N1 and 52 of H3N2 in 2009. All the sequences used in the study were aligned with MAFFT [10]. 2.2. Entropy and Mutual Information In information theory [7,8], entropy is a measure of the uncertainty associated with a random variable. Let x be a discrete random variable that has a set of possible values {a1, a2, a3,…,an} with probabilities {p1, p2, p3,…,pn} where p(x=ai)= pi. The entropy H of x is () log ii i xp p The mutual information of two random variables is a quantity that measures the mutual dependence of the two variables or the average amount of information that x conveys about y, which can be defined as: (, )()()(, ) xyHx Hy Hxy where H(x) is the entropy of x, and H(x,y) is the joint entropy of x and y. I(x,y)=0 if and only if x and y are independent random variables. In current study, each of the n columns in a multiple sequence alignment of a set of NA sequences of N resi- dues is considered as a discrete random variable xi (1 ≤i ≤N) that takes on one of the 20 (n=20) amino acid types with some probability. H(xi) has its minimum value 0 if all the residues at position i are the same, and achieves its maximum if all the 20 amino acid types appear with equal probability at position i, which can be verified by the Lagrange multiplier technique. A position of high entropy means that the amino acids are often varied at this position. While H(xi) measures the genetic diversity at position i in our current study, I(xi, yj) measures the correlation between residue substitutions at positions i and j. Entropy and mutual information were applied to se- quence analysis extensively. Mutual information was employed to identify groups of covariant mutation po- sitions in the sequences of HIV-1 protease and to dis- tinguish the correlated residue substitutions resulting from neutral mutations and those induced by multi- drug resistance [11]. Based on entropy a simple infor- mational index was proposed in [12] to characterize the patterns of synonymous codon usage bias. In another SciRes Copyright © 2009 JBiSE  W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 552 study, sequence data of 1032 complete genomes of in- fluenza A virus (H3N2) during 1968-2006 were used to construct networks of genomic co-occurrence to de- scribe H3N2 virus evolutionary patterns and dynamics. It suggested that amino acid substitutions correspond- ing to nucleotide co-changes cluster preferentially in known antigenic regions of HA [13]. Further, mutual information was used to construct site transition net- work based on 4064 HA1 of A/H3N1 sequences from 1968 to 2008, which was able to model the evolution- ary path of the influenza virus and to predict seven possible HA mutations for the next antigenic drift in the 2009–2010 season [14]. Recently, entropy and mutual information were also applied to indentify critical posi- tions and co-mutated positions on HA for predicting the antigenic variants [15]. 2.3. Mutual Information Evaluation In order to assess the significance of our mutual infor- mation values of residue pairs of NA, it is necessary to show that these values are significantly higher than those based on random sequences. For each residue position of NA, we randomly permuted the amino acids from dif- ferent sequences at that position and calculated the mu- tual information of these random sequences. This pro- cedure was repeated 1000 times. The P value was calcu- lated as the percentage of the mutual information values of the permuted sequences that were higher than those of the sequences of NA. 2.4. Random Forest Clustering Random Forest, proposed by Leo Breiman in 1999 [16], is an ensemble classifier based on many decision trees. The structure of a single tree could be easily altered by a small perturbation of data. Random Forest overcomes this problem by averaging across different decision trees. For many data sets, Random Forest produces a highly accurate classifier for supervised learning, com- parable to Support Vector Machine, the state of the art machine-learning algorithm. It computes proximities between cases and this technique can be extended to unlabeled data, leading to unsupervised clustering. In [17] random forest clustering was applied to renal cell carcinoma. To view the clusters formed by Random Forest, mul- tidimensional scaling [18] was utilized to project high- dimensional data down into a low-dimensional space while preserving the distances between them. First the proximities between cases i and j form a symmetric and positive definite matrix {prox(i,j)}. Then a second posi- tive definite and symmetric matrix {cv(i,j)} is con- structed using the entries of {prox(i,j)}. Random Forest extracts a few largest eigenvalues of the cv matrix and their corresponding eigenvectors. The values of () ()eivi are referred to as the ith scaling coordinate, where e(i) and v(i) re the ith eigenvalue and eigenvector of matrix cv [19]. In this study, the first and second scaling coordinates were utilized to visualize the data. 2.5. Important Sites in NA The N1 active site is a shallow pocket constructed from conserved residues, some of which contact the substrate directly and participate in catalysis, while others provide a structural framework [12]. According to the numbering in [5], these residue positions of N1 are 118, 119, 151, 152, 156, 179, 180, 223, 225, 228, 247, 277, 278, 293, 295, 368, and 402. The antigenic sites of N1 are residues 83–143, 156–190, 252–303, 330, 332, 340–345, 368, 370, 387–395, 431–435, and 448–468. 3. RESULTS 3.1. Mutations and Co-mutations The NA molecule is a homotetramer consisting of four identical polypeptide chains, each of about 470 amino acids. The exact number varies depending on the strain of the virus. The enzymatic domain of the NA is sup- ported from the virus envelope by a polypeptide stalk of variable length. The major molecular determinants that are known to influence the functional activities of the NA protein are the enzyme active site, the stalk length, the sialic acid binding site, and potential glycosylation sites. In this study, entropy analysis was applied to locate the positions that have elevated likelihood to develop mutations and those that already had mutated. The top 31 positions with high entropy are displayed in Figure 1. All except four, 248, 339, 340, and 454, were mutational positions discovered in [5]. These exceptional positions are of interest because they have the potential to mutate and position 248 is close to one active site 247. Notably, there were two clusters of mutations, one near position 286 and another near position 386 (Figure 1). For the sake of overview and comparison, we also plotted in Figure 1 the distinct entropy distributions of the N1 and N2 sequences deposited at the NCBI web site during 2009 so far. The sequences of N1 and N2 varied the most in the neighborhood of the stalk region between positions 36 and 76. In addition, the N1 sequences in H1N1 and H5N1 are experiencing a much greater ge- netic change than the N2 sequences in H3N2 this year as illustrated by their entropy (Figure 1). Next, we seek to probe the potential mutations that may affect the drug binding sites from a greater distance. We calculated the mutual information of each possible residue pairs from 469 residues of NA, a total of 109746 pairs. The top 44 pairs (top 0.04% of all pairs) were se- lected, all with a P value of zero. Because we were in- terested in the mutations in the NA domain, only those pairs in that region were chosen, which gave us 11 pairs: SciRes Copyright © 2009 JBiSE 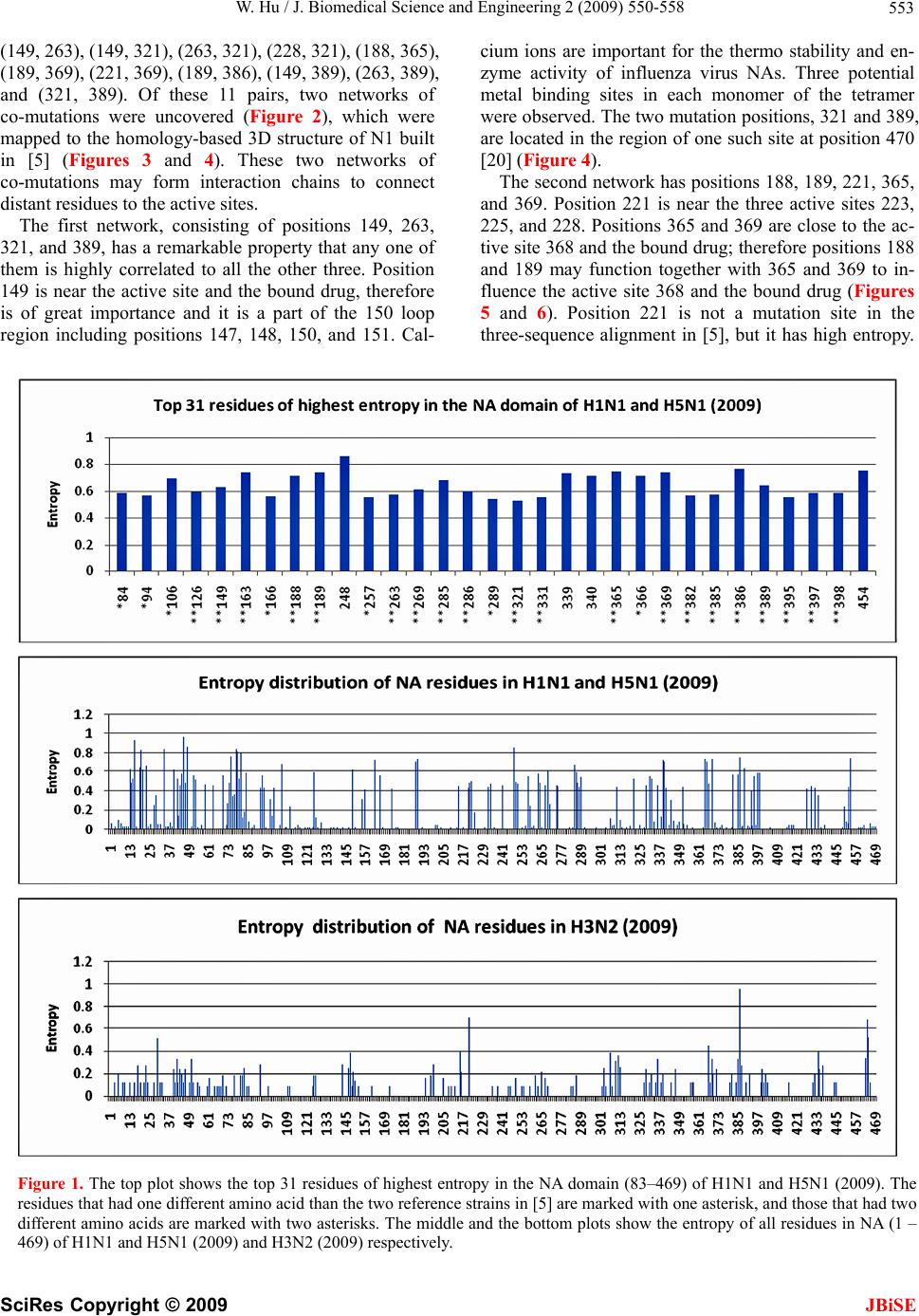 W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 SciRes Copyright © 2009 553 (149, 263), (149, 321), (263, 321), (228, 321), (188, 365), (189, 369), (221, 369), (189, 386), (149, 389), (263, 389), and (321, 389). Of these 11 pairs, two networks of co-mutations were uncovered (Figure 2), which were mapped to the homology-based 3D structure of N1 built in [5] (Figures 3 and 4). These two networks of co-mutations may form interaction chains to connect distant residues to the active sites. The first network, consisting of positions 149, 263, 321, and 389, has a remarkable property that any one of them is highly correlated to all the other three. Position 149 is near the active site and the bound drug, therefore is of great importance and it is a part of the 150 loop region including positions 147, 148, 150, and 151. Cal- cium ions are important for the thermo stability and en- zyme activity of influenza virus NAs. Three potential metal binding sites in each monomer of the tetramer were observed. The two mutation positions, 321 and 389, are located in the region of one such site at position 470 [20] (Figure 4). The second network has positions 188, 189, 221, 365, and 369. Position 221 is near the three active sites 223, 225, and 228. Positions 365 and 369 are close to the ac- tive site 368 and the bound drug; therefore positions 188 and 189 may function together with 365 and 369 to in- fluence the active site 368 and the bound drug (Figures 5 and 6). Position 221 is not a mutation site in the three-sequence alignment in [5], but it has high entropy. Figure 1. The top plot shows the top 31 residues of highest entropy in the NA domain (83–469) of H1N1 and H5N1 (2009). The residues that had one different amino acid than the two reference strains in [5] are marked with one asterisk, and those that had two different amino acids are marked with two asterisks. The middle and the bottom plots show the entropy of all residues in NA (1 – 469) of H1N1 and H5N1 (2009) and H3N2 (2009) respectively. JBiSE 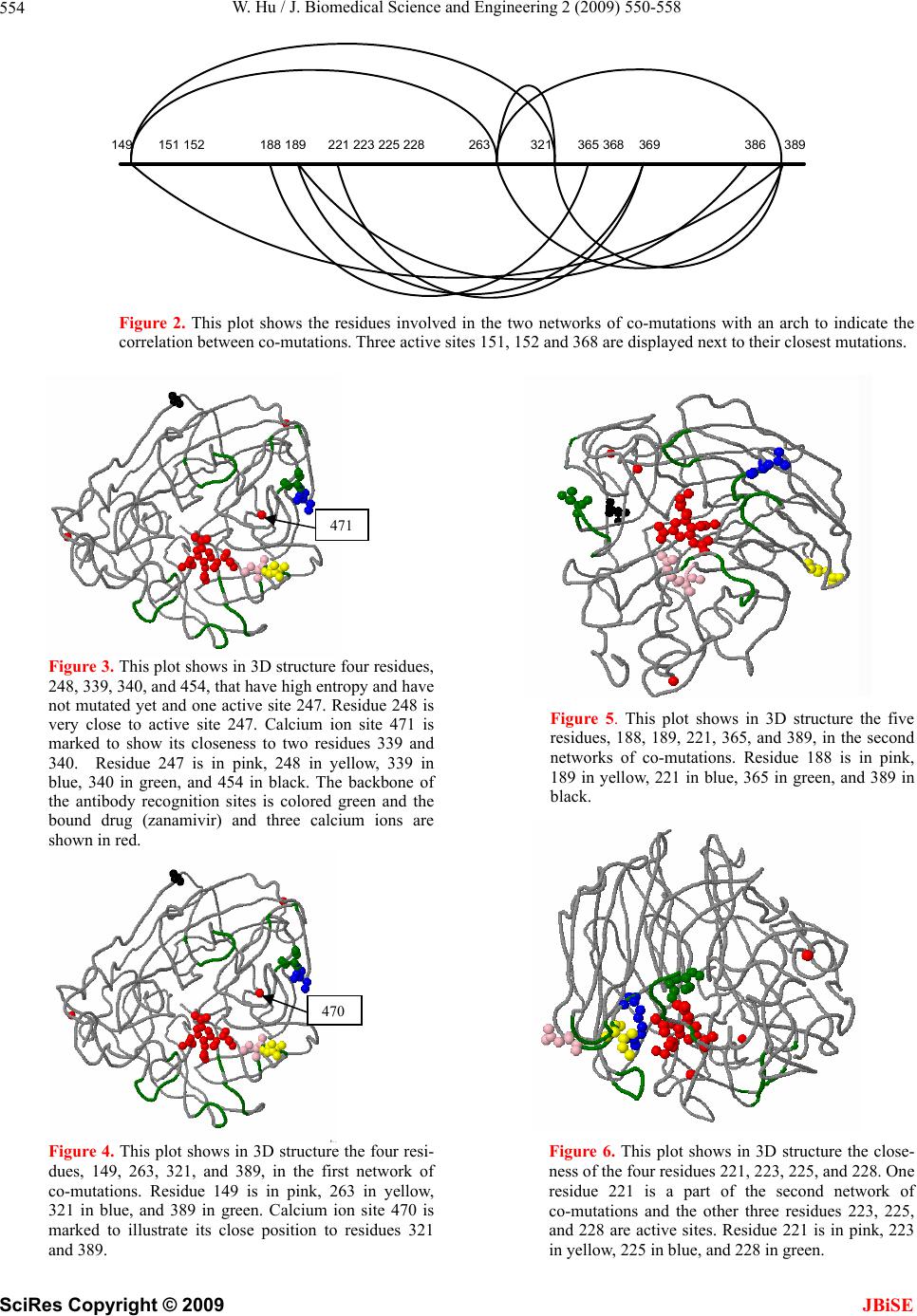 W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 554 149 151 152 188 189 221 223 225 228 263 321 365 368 369 386 389 Figure 2. This plot shows the residues involved in the two networks of co-mutations with an arch to indicate the correlation between co-mutations. Three active sites 151, 152 and 368 are displayed next to their closest mutations. Figure 3. This plot shows in 3D structure four residues, 248, 339, 340, and 454, that have high entropy and have not mutated yet and one active site 247. Residue 248 is very close to active site 247. Calcium ion site 471 is marked to show its closeness to two residues 339 and 340. Residue 247 is in pink, 248 in yellow, 339 in blue, 340 in green, and 454 in black. The backbone of the antibody recognition sites is colored green and the bound drug (zanamivir) and three calcium ions are shown in red. Figure 4. This plot shows in 3D structure the four resi- dues, 149, 263, 321, and 389, in the first network of co-mutations. Residue 149 is in pink, 263 in yellow, 321 in blue, and 389 in green. Calcium ion site 470 is marked to illustrate its close position to residues 321 and 389. 471 Figure 5. This plot shows in 3D structure the five residues, 188, 189, 221, 365, and 389, in the second networks of co-mutations. Residue 188 is in pink, 189 in yellow, 221 in blue, 365 in green, and 389 in black. 4 Figure 6. This plot shows in 3D structure the close- ness of the four residues 221, 223, 225, and 228. One residue 221 is a part of the second network of co-mutations and the other three residues 223, 225, and 228 are active sites. Residue 221 is in pink, 223 in yellow, 225 in blue, and 228 in green. SciRes Copyright © 2009 JBiSE  W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 555 This is a position of interest, because it is close to the active sites 223, 225 and 228 and co-mutates with a cluster of mutation positions 365, 366, and 369. The latter cluster also encloses another active site 368 (Fig- ures 2, 5 and 6). 3.2. Stalk Motifs In this study, our intention was to discover the NA stalk motif patterns of 2009 H1N1and H5N1, which is the focus of this work, and those of 2008 and 2007 to place our findings in the right historical context. As noted in the previous section, the N1 strains in 2009 are experi- encing a rapid genetic variation in the stalk region com- pared with other regions of N1, which could be reflected in the different stalk motifs appearing this year. We extracted the sub-sequences consisting of posi- tions from 36 to 79 in the NA sequences and discarded those that contained no amino acids. All the different motifs found are displayed in Tab le 1. In 2007, all NA stalk motifs in H1N1 and H5N1 had three different types referred to as types 1, 2, and 3. In 2008, type 3 stalk mo- tif disappeared and type 4 appeared. In 2009, types 1, 2 and 4 persisted. In all these three years, types 1 and 2 persisted. Type 2, referred to as the special stalk motif in [9], was in H5N1 in all three years and type 1 was in H1N1 in all three years. The important change in 2009 was that type 1, a com- mon stalk motif in H1N1, was more prevalent in H5N1 and type 2, a common stalk motif in H5N1, was in H1N1 for the first time, a dramatic exchange of the two different motifs between these two subtypes of flu vi- ruses. The special stalk motif was dominant in H5N1 in 2007 and 2008, but it was no longer the case in 2009. The new type 4 was more evident in H5N1 in 2009. These new patterns or exchange of different patterns of stalk motifs reminded us again of the fast evolutionary nature of the flu virus and the need for timely analysis of its data. The occurrence of the special stalk motif in 2009 H1N1, which may bring increased virulence to the current swine flu epidemic, is worthy of further attention and surveillance. These alterations in the stalk region of NA in H1N1 and H5N1 could also be reflected in the phylogenetic analysis conducted in the next section. 3.3. Phylogenetic Analysis Even though the phylogenetic tree of a small number of NA sequences was constructed before [5]. With new H1N1 NA sequences being deposited at the NCBI web site regularly, it is constructive to perform phylogenetic analysis on these new sequences. For easy comparison, software MEGA [21] was used to reproduce the phy- logenetic tree in Figure 3 in [5] with the same NA se- quences, which had eight sequences of 2009 H1N1 (numbered from 1 to 8) available at the time of the re- search (as of April 29th) in [5] and 44 different represen- tative sequences of H1N1 or H5N1(numbered from 9 to 52) in previous years (left plot of Figure 7). We em- ployed Random Forest to cluster these sequences to get a different view of their phylogenetic relationship (Figure 8), where a number is used to represent a sequence due to the limited space in that plot. The association of these numbers with their sequences can be found in Figure 7, where a number is printed before each flu subtype such as “2 H1N1 California 09 2009” and “6 H1N1 Texas 05 2009”. Random Forest clustering revealed that all eight 2009 H1N1 sequences, which formed their own single cluster, were similar to others only in the second scaling coordinate, but not in the first. We reasoned this was because all eight sequences were from the same country and the structures of these clusters might be different if the plentiful sequences deposited recently at the NCBI web site were used. We took note of some minor but interesting differ- ences between the phylogenetic tree in the left plot of Figure 7 and Random Forest-based clusters of the same sequences in the left plot of Figure 8. In Figure 8, the eight sequences 1, 2, 3, …, 8 were clustered in one clus- ter away from all the other sequences. This cluster was close to two groups of sequence numbers in the second scaling coordinate. The first group consisted of sequences Table 1. Different NA stalk motifs 36th ---------------------------- Stalk region -------------------------------79th Subtype Year Number of strains shared this motif Motif type number HS IQTGSQNHTGICNQRIITYENSTWVNHTYVNINNTNVVAG KD H1N1 2007 196 1 HS IQTGNQHQAEP----------------------------------------ISNTNFLTE KA H5N1 2007 189 2 -- ---------NQNQVEP---------------------------------------ISNTNFLTE KA H5N1 2007 1 3 HS IQIGSQGYPETCNQSVITYENNTWVNQTYINISNTNLIGG QA H5N1 2007 2 1 HS IQTGSQNNTGICNQRIITYENSTWVNHTYVNINNTNVVAG ED H1N1 2008 147 1 -- ------------------------------------------------NHTYVNINNTNVVAG ED H1N1 2008 1 4 HS IQTGNQCQAEP----------------------------------------ISNTKFLTE KA H5N1 2008 70 2 HS IQLGNQNQIETCNQSVITYENNTWVNQTYVNISNTNFAAG QS H1N1 2009 157 1 HS IQTGNQCQDEP----------------------------------------ISNTKFLTE KA H1N1 2009 21 2 ------------------------------SVITYENNTWVNQTYVNISNTNFAAG QS H1N1 2009 39 4 HS INTGNQHQAEP----------------------------------------ISNANFLTE KA H5N1 2009 1 2 HS IQLGNQNQIETCNQSVITYENNTWVNQTYVNISNTNFAAG QS H5N1 2009 20 1 SciRes Copyright © 2009 JBiSE  W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 556 2 H1N1 Cali fornia 0 9 2009 gi |227 831806|g 6 H1N1 Texas 05 2 009 gi |22 7831797 |gb|A CP 5 H1N1 Texas 04 2 009 gi |22 7831824 |gb|A CP 1 H1N1 Cali fornia 0 4 2009 gi |227 809834|g 3 H1N1 Cali fornia 05 2009 gi |22 78317 68|g 4 H1N1 Cali fornia 06 2009 gi |22 80177 56|g 7 H1N1 NewYork 20 20 09 gi |2279 77174|gb |A 8 H1N1 Ohi o 07 2009 g i|2 27977 158|gb |A CP 4 11 H1N1 Thailand 271 2005 gi|117935805|g 13 H1N1 swine Chachoengsao NIAH587 2005 9 H1N1 s wi ne B el g iu m 74 85 gi |2006 8225|e 10 H1N1 s win e S cotla nd WVL1 7 1999 gi |225 14 H1N1 s win e Zhejian g 1 2007 gi |210 0766 15 H1N1 swine Italy 1509-6 97 gi|2006821 16 H1N1 s wine Hungary 19774 2006 gi |2249 12 H1N1 swi ne F i n i s tere 3616 84 gi |20068 19 H6N1 grayteal A ust ralia 1 1979 gi|115 17 H7N1 ost ric h Ital y 984 00 gi|12396720 20 H1N1 duc k E asternChi na 15 2 2003 gi |16 28 H5N1 HongK ong 156 97 gi |28336 59|gb|A A 34 H6N1 partridge S hant ou 5028 2004 gi|1 25 H5N1 duc k S ha nt ou 1930 20 01 gi |16912 4 33 H5N1 Chick e n HongKong Y U5 62 01 gi |288 22 H5N1 egret HongK o ng 757 . 2 03 gi|5 6548 32 H5N1 Mus c ovyduc k V iet nam 39 2007 gi|1 23 H5N1 Chick en P aul auRam p ang B PP V11 200 29 H5N1 Vi etNam HN3124 2 20 07 g i|16256900 30 H5N1 duck E as t ernChina 150 2003 gi|16 37 H5N1 Thailand NKNP 2005 gi|116295106| 24 H5N1 Thailand 1K AN-1 2004 gi |46578136 36 H5N1 c hi ck e n Thailan d ICRC-V586 20 08 21 H6N1 c hi cken Taiwan S P1 00 g i|8 724711 18 H3N1 mall ardduc k Mi nnesota 1979 gi|14 35 H11N1 ruddy t u rns tone De lawa re 2589 87 27 H7N1 FP V Rostock 1934 gi |58577|emb|CA 31 H1N1 Brevig Mi ss i on 1 18 gi |85721 69|g 42 H1N1 swine Ohio 75004 04 gi|188572594 51 H1N1 s wine A l berta 5662 6 03 gi |8262 28 43 H1N1 turkey KS 488 0 1980 gi |189 312997 40 H1N1 c hi ck e n NY 21665-7 3 1998 gi|1 938 46 H1N1 s wine S hang hai 3 2005 gi |2244828 48 H3N1 sw i ne IN PU542 04 gi |757565 70|gb 49 H1N1 s win e Ontari o 111 12 04 gi |8262 29 26 H1N1 Swine Iowa 30 gi |85 721 85|gb |AA F 7 38 H1N1 W i l son-S m i th 33 gi |89782161 |gb|A 41 H3N1 NYMCX-161AP uertoRic o 8 1934-Wi s c 39 H1N1 W ei ss 43 g i|8 572187|g b|A A F 77 045. 47 H1N1 Fort M on m outh 1 47 gi |217 17612|gb 44 H1N1 swi ne Tianjin 01 20 04 g i |151 3355 45 H1N1 Len i ngra d 54 1 gi|325336|gb |AA A4 50 H1N1 NewCaledoni a 20 19 99 gi |15882 753 52 H1N1 Taiwan 117 96 gi |597 97382|gb |A A X 99 26 13 8 17 79 100 88 58 74 51 46 99 72 86 48 67 100 89 77 99 99 35 38 41 86 97 25 6 22 35 94 96 53 41 100 47 19 17 38 36 96 44 44 13 30 28 0.02 24 H5N1 Thailand 1KA N-1 2004 gi|46578136 36 H5N1 chick en Thailand ICRC-V586 2008 37 H5N1 Thailand NKNP 2005 gi|116295106| 30 H5N1 duck E asternChina 150 2003 gi|16 6 H1N1 Crete 2307 2009 gi|254688553|gb|A 29 H5N1 Viet Nam HN31242 2007 gi|16256900 23 H5N1 Chick en PaulauRampang B P PV 11 200 5 H1N1 Stockholm 35 2009 gi|251833568|gb 32 H5N1 Muscovyduck Vietnam 39 2007 gi|1 7 H1N1 swine Hong Kong NS 29 2009 gi|2396 22 H5N1 egret HongK ong 757.2 03 gi |56548 25 H5N1 duck S hant ou 1930 2001 gi|169124 33 H5N1 Chic ken HongK ong YU562 01 gi|288 28 H5N1 HongKong 156 97 gi|2833659|gb|AA 34 H6N1 partri dge S hant ou 5028 2004 gi|1 17 H7N1 ost rich Italy 984 00 gi|12396720 20 H1N1 duck E ast ernChina 152 2003 gi|16 19 H6N1 grayt eal A us t rali a 1 1979 gi|115 12 H1N1 swine Fi ni st ere 3616 84 gi|20068 16 H1N1 swine Hungary 19774 2006 gi|2249 4 H1N1 Dublin 11 2009 gi|253828472|gb|AC 14 H1N1 swine Zhejiang 1 2007 gi|2100766 15 H1N1 swine Italy 1509-6 97 gi|2006821 9 H1N1 swine B el gium 74 85 gi|20068225|e 10 H1N1 swine S cot land WVL17 1999 gi|225 3 H5N1 grey heron Hong Kong 779 2009 gi| 11 H1N1 Thailand 271 2005 gi|117935805|g 13 H1N1 swine Chachoengs ao NIAH587 2005 21 H6N1 chick en Taiwan SP 1 00 gi|8724711 18 H3N1 mallardduck M i nnesot a 1979 gi|14 35 H11N1 ruddyturns t one Delaware 2589 87 27 H7N1 FPV Ros toc k 1934 gi|58577|emb|CA 31 H1N1 Brevig Miss i on 1 18 gi|8572169|g 8 H1N1 swine Guangdong 2 2009 gi|2555298 46 H1N1 swine S hanghai 3 2005 gi|2244828 40 H1N1 chick en NY 21665-73 1998 gi|1938 43 H1N1 turkey K S 4880 1980 gi|189312997 42 H1N1 swine Ohio 75004 04 gi|188572594 51 H1N1 swine A l bert a 56626 03 gi|826228 48 H3N1 swine IN PU542 04 gi|75756570|gb 49 H1N1 swine Ont ario 11112 04 gi|826229 26 H1N1 Swine Iowa 30 gi|8572185|gb|AAF 7 1 H1N1 Was hi ngt on 01 2009 gi |243031412|g 2 H1N1 North Caroli na 02 2009 gi|2559606 50 H1N1 NewCaledonia 20 1999 gi|15882753 52 H1N1 Taiwan 117 96 gi|59797382|gb|AAX 38 H1N1 Wil son-S m ith 33 gi|89782161|gb|A 41 H3N1 NYMCX-161APuert oRic o 8 1934-Wi s c 39 H1N1 Weiss 43 gi|8572187|gb|AAF 77045. 47 H1N1 FortMonm out h 1 47 gi|21717612|gb 44 H1N1 swine Tianjin 01 2004 gi|1513355 45 H1N1 Leningrad 54 1 gi|325336|gb|AAA 4 67 92 100 99 90 31 26 14 26 87 100 52 76 42 55 100 100 86 64 99 45 99 34 27 4 10 35 88 43 82 98 95 52 41 99 56 27 29 45 34 95 92 96 34 41 19 21 39 15 0.02 Figure 7. Left plot: a reproduced phylogenetic tree of the NA protein sequences of the N1 subtype family in [5]. Right plot: A phy- logenetic tree of the same sequences used in the left plot except that the first eight sequences were replaced with eight new represen- tative sequences from 283 N1 sequences in 2009. 13, 14, 16, 10, and 11, and the second had sequences 38, 35, and 31. The phylogenetic tree displayed the close relationship between the first group and the group of sequences 1, 2,…., 8, but did not do so for the second group. The first group had H1N1 swine Chachoengsao 2005 (13), H1N1 swine Zhejiang 2007 (14), H1N1 swine Hungary 2006 (16), H1N1 swine Scotland 1999 (10), and H1N1 Thailand 2005 (11). The second group had H1N1 Wilson Smith 33 (38), H11N1 Delaware 1987 (35), and H1N1 Brevig Mission 1918 (31). These two groups were all H1N1 subtype and the second group inherited from the ancient strains in the first group, and that is why they were similar and clustered together by Random Forest. To get a new view of the clusters of the NA sequences of 2009 H1N1 and H5N1 available up to date, eight new representative sequences were selected from 283 se- quences in the same year with cd-hit [22] to replace the eight NA sequences, numbered from 1 to 8, used in the left plot of Figures 7 and 8. The new sequences were H1N1 Washington 2009 (1), H1N1 North Carolina 2009 (2), H5N1 Hong Kong 2009 (3), H1N1 Dublin 2009 (4), H1N1 Stockholm 2009 (5), H1N1 Crete 2009 (6), H1N1 swine Hong Kong 2009 (7), and H1N1 swine Guang- SciRes Copyright © 2009 JBiSE 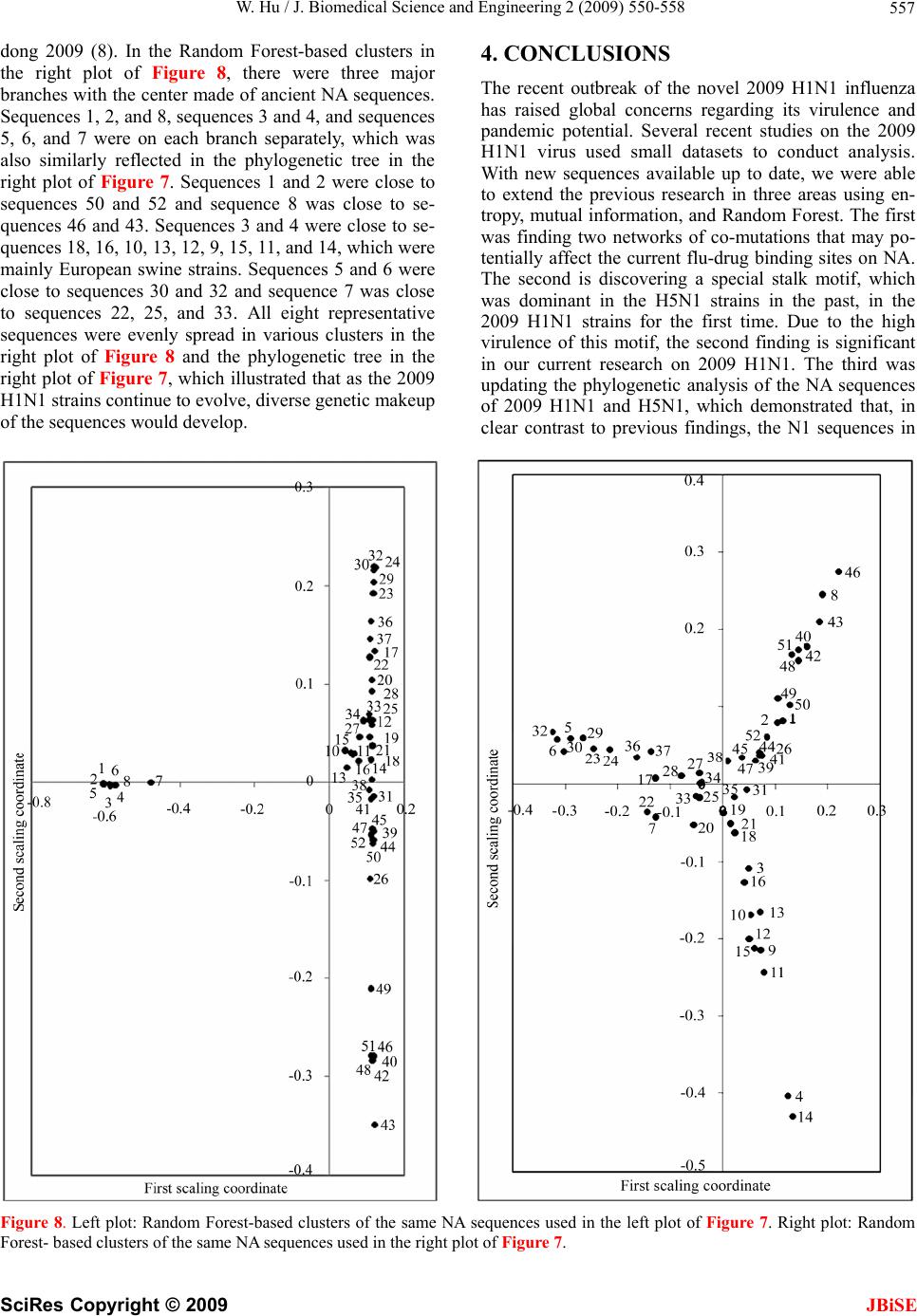 W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 557 dong 2009 (8). In the Random Forest-based clusters in the right plot of Figure 8, there were three major branches with the center made of ancient NA sequences. Sequences 1, 2, and 8, sequences 3 and 4, and sequences 5, 6, and 7 were on each branch separately, which was also similarly reflected in the phylogenetic tree in the right plot of Figure 7. Sequences 1 and 2 were close to sequences 50 and 52 and sequence 8 was close to se- quences 46 and 43. Sequences 3 and 4 were close to se- quences 18, 16, 10, 13, 12, 9, 15, 11, and 14, which were mainly European swine strains. Sequences 5 and 6 were close to sequences 30 and 32 and sequence 7 was close to sequences 22, 25, and 33. All eight representative sequences were evenly spread in various clusters in the right plot of Figure 8 and the phylogenetic tree in the right plot of Figure 7, which illustrated that as the 2009 H1N1 strains continue to evolve, diverse genetic makeup of the sequences would develop. 4. CONCLUSIONS The recent outbreak of the novel 2009 H1N1 influenza has raised global concerns regarding its virulence and pandemic potential. Several recent studies on the 2009 H1N1 virus used small datasets to conduct analysis. With new sequences available up to date, we were able to extend the previous research in three areas using en- tropy, mutual information, and Random Forest. The first was finding two networks of co-mutations that may po- tentially affect the current flu-drug binding sites on NA. The second is discovering a special stalk motif, which was dominant in the H5N1 strains in the past, in the 2009 H1N1 strains for the first time. Due to the high virulence of this motif, the second finding is significant in our current research on 2009 H1N1. The third was updating the phylogenetic analysis of the NA sequences of 2009 H1N1 and H5N1, which demonstrated that, in clear contrast to previous findings, the N1 sequences in Figure 8. Left plot: Random Forest-based clusters of the same NA sequences used in the left plot of Figure 7. Right plot: Random Forest- based clusters of the same NA sequences used in the right plot of Figure 7. SciRes Copyright © 2009 JBiSE  W. Hu / J. Biomedical Science and Engineering 2 (2009) 550-558 558 2009 are diverse enough to cover different major bran- ches of the phylogenetic tree of those in previous years. As the novel influenza A H1N1 virus continues to spread globally, our results highlighted the value of performing timely analysis on the 2009 H1N1 virus. 5. ACKNOWLEDGMENTS We thank Houghton College for its financial support. REFERENCES [1] Taubenberger, J. K. and Morens, D. M., (2006) 1918 Influenza: the mother of all pandemics, Emerg. Infect. Dis., 12(1), 15–22. [2] Trifonov, V., Khiabanian, H., and Rabadan, R., (2009) Geographic dependence, surveillance, and origins of the 2009 influenza A (H1N1) virus, N. Engl. J. Med., 361, 115–119. [3] Centers for Disease Control and Prevention (CDC), (2009) Update: Drug susceptibility of swine-origin in- fluenza A (H1N1) viruses, MMWR Morb Mortal Wkly Rep 2009, 58, 433–435. [4] Weïwer, M., Chen, C. C., Kemp, M. M., and Linhard, R. J., (2009) Synthesis and biological evaluation of non-hydrolyzable 1,2,3-triazole-linked sialic acid deriva- tives as neuraminidase inhibitors, European Journal of Organic Chemistry, 16, 2587. [5] Maurer-Stroh, S., Ma, J., Lee, R. T. C., Sirota, F. L., and Frank, E., (2009) Mapping the sequence mutations of the 2009 H1N1 influenza A virus neuraminidase relative to drug and antibody binding sites, Biol. Direct., 4, 18. [6] Wang, S. Q., Du, Q. S., Huang, R. B., Zhang, D. W., and Chou, K. C., (2009) Insights from investigating the in- teraction of oseltamivir (Tamiflu) with neuraminidase of the 2009 H1N1 swine flu virus, Biochemical and Bio- physical Research Communications, 386(3), 432–6. [7] Cover, T. A. and Thomas, J. A., (1991) Elements of in- formation theory, John Wiley and Sons, NewYork. [8] MacKay, D., (2003) Information theory, inference, and learning algorithms, Cambridge University Press. [9] Zhou, H. B., Yu, Z. J., Hu, Y., Tu, J. G., Zou, W., Peng, Y. P., Zhu, J. P., Li, Y. T., Zhang, A. D., Yu, Z. N., Ye, Z. P., Chen, H. C., and Jin, M. L., (2009) The special neura- minidase stalk-motif responsible for increased virulence and pathogenesis of H5N1 influenza A virus, PLoS One, 4(7), e6277. [10] Katoh, K., Kuma, K., Toh, H., and Miyata, T., (2005) MAFFT version 5: Improvement in accuracy of multiple sequence alignment, Nucleic. Acids. Res., 33, 511–518. [11] Liu, Y., Eyal, E., and Bahar, I., (2008) Analysis of corre- lated mutations in HIV-1 protease using spectral cluster- ing, Bioinformatics, 24(10), 1243–1250. [12] Colman, P. M., Hoyne, P. A., and Lawrence, M. C., (1993) Sequence and structure alignment of paramyxovirus he- magglutinin-neuraminidase with influenza virus neura- minidase, J. Virol., 67, 2972–2980. [13] Du, X. J., Wang, Z., Wu, A. P., Song, L., Cao, Y., Hang, H. Y., and Jiang, T. J., (2008) Networks of genomic co-occurrence capture characteristics of human influenza A (H3N2) evolution, Genome. Res., 18, 178–187. [14] Xia, Z., Jin, G. L., Zhu J., and Zhou, R. H., (2009) Using a mutual information-based site transition network to map the genetic evolution of influenza A/H3N2 virus, Bioinformatics, 25(18), 2309–2317. [15] Huang, J. W., King, C. C., and Yang, J. M., (2009) Co- evolution positions and rules for antigenic variants of human influenza A/H3N2 viruses, BMC Bioinformatics, 10(Suppl 1), S41. [16] Breiman, L., (2001) Random forests, Machine Learning, 45(1), 5–32. [17] Shi, T., Seligson, D., Belldegrun, A. S., Palotie, A., and Horvath, S., (2005) Tumor classification by tissue mi- croarray profiling: Random forest clustering applied to renal cell carcinoma, Mod. Pathol., 18(4), 547–57. [18] Cox, T. F. and Cox, M. A. A., (2001), Multidimensional scaling, Chapman and Hall. [19] http://www.stat.berkeley.edu/~breiman/RandomForests/. [20] Xu, X. J., Zhu, X. Y., Dwek, R. A., Stevens, J., and Wil- son, I. A., (2008) Structural characterization of the 1918 influenza virus H1N1 neuraminidase, Journal of Virology, 82(21), 10493–10501. [21] Kumar, S., Nei, M., Dudley, J., and Tamura, K., (2008) MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences, Brief Bioinfor- matics, 9, 299–306. [22] Li, W. and Godzik, A., (2006) Cd-hit: A fast program for clustering and comparing large sets of protein or nucleo- tide sequences, Bioinformatics, 22, 1658–1659. SciRes Copyright © 2009 JBiSE
|