 Advances in Biological Chemistry, 2011, 1, 109-118 doi:10.4236/abc.2011.13013 Published Online November 2011 (http://www.SciRP.org/journal/abc/ ABC ). Published Online November 2011 in SciRes. http://www.scirp.org/journal/ABC The interaction between Fe65 and Tip60 is regulated by S-nitrosylation on 440 cystein residue of Fe65 Eun Jeoung Lee1, Sung Hwa Shin2, Sunghee Hyun1, Jaesun Chun3, Sang Sun Kang2,4* 1Department of Pre-Medicine, Eulji University School of Medicine, Daejeon, Republic of Korea; 2Department of Biology Education, Chungbuk National University, Cheongju, Republic of Korea; 3Department of Biology Education, Korea National University of Education, Cheongwon, Republic of Korea; 4Biotechnology Research Institute, Chungbuk National University, Cheongju, Republic of Korea. Email: *jin95324@cbu.ac.kr Received 27 August 2011; revised 30 September 2011; accepted 9 October 2011. ABSTRACT The S-Nitrosylation of protein thiol groups by NO is a widely recognized protein modification. The treat- ment of cells with NOBF4 induces the S-nitrosylation of FE65. In this study, we present evidence showing that FE65 modified by NO (Nitric Oxide) via S-ni- trosylation induces functional changes in the protein that inhibits the HAT activity of Tip60. The results of mutational analysis of FE65 demonstrated further that the cysteine residue of FE65 (Cys440) is critical to the process of S-nitrosylation. The mutation of the cysteine residue which completely ablated the S-ni- trosylation of FE65 also lost its inhibitory effects on Tip60 HAT activity. Thus, our findings show, for the first time, that the novel regulation mechanism of Tip60 activity may operate via FE65 binding, which is enhanced by S-nitrosylation on the FE65 Cys440 residue. This study describes the interaction between FE65 and Tip60, which is en hanced by a posttrans la- tional modification of FE65 (through S-nitrosylation) by NO, promoting the association of the FE65-Tip60 protein complex and inhibiting both the HAT activity of Tip60 and cell death. Keywords: S-Nitrosylation; NO (Nitric Oxide); FE65; HAT Activity; Tip60 1. INTRODUCTION FE65 functions as an adaptor protein within the cell, the identification of ligand proteins for the PID1/PID2 do- main or the WW domain may provide us with some clues as to its biological roles [1-3]. A few proteins ha- ve been identified as ligand proteins to the PID1 domain of FE65. FE65 interacts with the transcription factor (CP2/ LSF/LBP1), Tip60 (Histone acetyltransferase; HA-T), Teashirt, and calsyntenin and low-density lipoprotein re- ceptor-related protein through its PI-D1 domain [4,5]. Further, because the protein is regulated by posttransla- tional modifications, including protein S-nitrosylation, glycosylation, or nitrosylation, it is also crucial to deter- mine the manner in which the modification affects bind- ing with each of its ligand proteins [2,3]. NO is a gaseous signal mediator that evidences a nu- mber of important biological effects [6]. NO has been shown, in many instances, to exert its effects via protein S-nitrosylation, in which the binding of NO to Cys resi- dues regulates protein function [7,8]. S-nitrosylation re- gulates protein function via covalent attachment me- chanisms that control the addition or removal of NO from a cysteine thiol. It has been previously reported that the targets of S- nitrosylation modification, among others, include bovine serum albumin, tissue-type plasminogen activator, XIAP, the N-methyl-D-aspartate receptor, oncogenic p21ras, TR- PC1, and transcriptional activators [9-15]. After analysis of the S-nitrosylation proteins harboring more than one Cys residue, a specific consensus motif for S-nitro-syla- tion (Lys /Arg/Hi /Asp/Glu) Cys (Asp/Glu) (sites –1, 0, and +1, respectively) is determined [7,8,16]. After reco- gnizing the putative S-nitrosylation Cys residue in FE65 with the consensus sequence information, we evaluated the S-nitrosylation of FE65 and determined its biological significance [1,17]. The results of our sitedirected mu- genesis analysis (C440A) and in vitro Snitrosylation as- y revealed that S-nitrosylation occurs on FE65 and the 440 cysteine residue (439Arg/Cys/Glu441) of FE65 as its spefic modulation site. Additionally, we noted that FE65 netively modulates Tip60 HAT activity via protein-rotein interactions, which are enhanced by S-nitrosylation. He- in, we reported for the first time that the S-nitrosylation of FE65 appears to perform a crucial function in the  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 110 regulation of Tip60 activity via the modulation of their protein interactions. 2. MATERIALS AND METHODS 2.1. Materials NOBF4 was acquired from Aldrich. Mouse anti-FE65 monoclonal antibody was obtained from Chemikon. Ra- bbit muscle FE65 (80 units/mg) was purchased from Boeer Mannheim. Other chemicals were of the highest grade of purity, and were obtained from Sigma. All chemicals were purchased from Sigma-Aldrich, unless stated otherwise. HEK293T and HEK 293 cells were maintained in DMEM (Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco) at 37˚C with 5% CO2. Transfection was conducted with Lipo- fectamine and Plus Reagent (Invitrogen) in accordance with the manufacturer’s instructions. 2.2. DNA Constructions FE65 and Tip60 were cloned from the SuperScript hu- man brain cDNA library. Full-length FE65 was cloned into pRK5-GFP and HA vectors for the cell culture study and into the pGEX4T-2 vector for the production of re- combinant protein. The truncated fragments of FE65 encoding for amino acids 1-470 (WW and PID1), 1-265 (PID1-2) were generated via PCR, using the full-length FE65 as a template, then cloned into pRK5-GFP vector. The FE65 fragment (1-470 aa C470A) was also cloned into pGEX4T.2 vector for recombinant protein produc- tion. The sequence integrity of all constructs was con- firmed via sequencing. 2.3. Overexpression and Purification of Recombinant Proteins GST, GST-FE65, and GST-FE65 C440A were expressed in Rosetta (DH5α) pLys Escherichia coli (RBC). Over- expression of bacterial cultures in the linear growth phase (0.6 OD) were induced by 1 mM IPTG at 37˚C overnight, and the recombinant proteins were subse- quently purified by GSH-Sepharose (GE Healthcare). GFP-tagged recombinant FE65 was generated in accor- dance with the methods developed by Stennicke and Sa- lvesen, and was purified using Ni-NTA Sepharose (GE Healthcare). Concentrations of the recombinant proteins were quantified via SDS/PAGE, using BSA as a stan- dard. 2.4. In Vitro S-Nitrosylation Assay The biotin switch assay was conducted according to the method described by Jaffrey and Snyder, with some mo- difications. Nitrosylated cell lysates or recombinant pro- teins in HENT buffer (250 mM Hepes, 1 mM EDTA, 0.1 mM Neocupoine, 1% Triton X-100) were incubated for 20 min with 10 mM methyl methanethiosulfonate (MM- TS) (Thermo Scientific) at 50˚C, and then excess MM- TS was removed via three passages through a G25 Se- phadex spin column. The samples were then incubated with 5 mM ascorbate and 0.4 mM biotin-HPDP (Thermo Scientific) for 1 h at room temperature with rotation. Unreacted biotin-HPDP was then removed with a G25 Sephadex spin column and the biotinylated samples were then incubated for 1 h with 50 μL of Neutravidin- agarose (Thermo Scientific). The pellets were then washed 5 times with neutralization buffer [20 mM Hepes (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5% Triton X- 100] with 0.6 M NaCl and eluted with an SDS sample buffer and subjected to Western blot analysis. 2.5. Interaction Between FE65 and Tip60 HEK293T lysates with or without GFP-FE65 expression were treated for 15 min with GSH (500 μM), NOBF4 (500 μM), or DTT (1 mM) at 37˚C as indicated. The lysates were then passed once through a G25 Sephadex spin column and the recombinant FE65 (8 μg) was added to the lysates, and the co-IP protocol was con- ducted as described. Tip60 was detected with Tip60 an- tibody (Sigma). 2.6. Cell Death Analysis HEK293T cells were transfected with 0.25 μg of GFP- FE65 or control vector. Thirty hours after transfection, the cells were pretreated for 6 h with 100 μM NO donor NOBF4 (Calbiochem). The cells were then treated with the selected drugs and under the indicated conditions: 50 ng/mL TNF-α and 0.1 μg/mL actinomycin D for 24 h; 50 μM rotenone for 16 h; 2 mM dopamine for 24 h; 3 μM MG132 for 24 h. In the FE65-induced cell death assay, in addition to GFP-FE65 C470A, or (1-470 aa) and con- trol vector, the cells were also cotransfected with GFP as a control. Cell death was analyzed 60 h after transfection via a Trypan blue exclusion assay. 2.7. Tip60 Binding Assay Tip60 (1 µg/assay) and up to 200 µM individual NO donor were incubated for 5 min in 50 mM triethylam- monium buffer (pH 7.5) in a total volume of 50 µl at 37˚C. The samples were diluted into 950 µl of 50 mM triethylammonium buffer (pH 7.5) containing 50 µM arsenate, 2.4 mM glutathione, and 100 µg/ml of gly- ceraldehyde-3-phosphate at 37˚C. The enzymatic reduc- tion of NAD+ to NADH was initiated via the addition of 250 µM NAD+. FE65 activity was monitored by re- cording fluorescence emissions above 430 nm after ex- citation at 313 and 366 nm, respectively. Samples with- out NO donors were used as controls. C opyright © 2011 SciRes. ABC 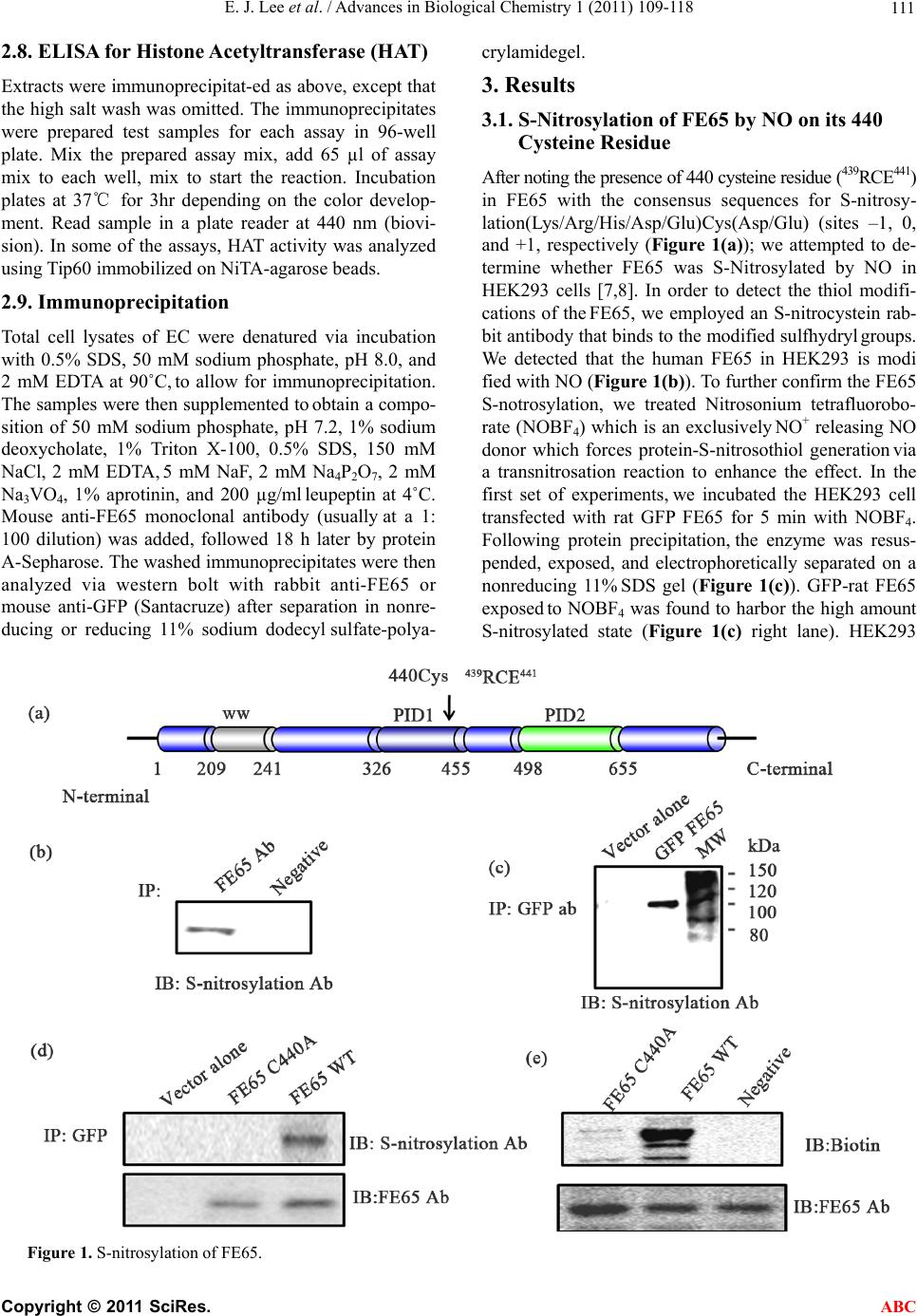 E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 Copyright © 2011 SciRes. 111 2.8. ELISA for Histone Acetyltransferase (HAT) crylamidegel. 3. Results Extracts were immunoprecipitat-ed as above, except that the high salt wash was omitted. The immunoprecipitates were prepared test samples for each assay in 96-well plate. Mix the prepared assay mix, add 65 µl of assay mix to each well, mix to start the reaction. Incubation plates at 37℃ for 3hr depending on the color develop- ment. Read sample in a plate reader at 440 nm (biovi- sion). In some of the assays, HAT activity was analyzed using Tip60 immobilized on NiTA-agarose beads. 3.1. S-Nitrosylation of FE65 by NO on its 440 Cysteine Residue After noting the presence of 440 cysteine residue (439RCE441) in FE65 with the consensus sequences for S-nitrosy- lation(Lys/Arg/His/Asp/Glu)Cys(Asp/Glu) (sites –1, 0, and +1, respectively (Figure 1(a)); we attempted to de- termine whether FE65 was S-Nitrosylated by NO in HEK293 cells [7,8]. In order to detect the thiol modifi- cations of the FE65, we employed an S-nitrocystein rab- bit antibody that binds to the modified sulfhydryl groups. We detected that the human FE65 in HEK293 is modi fied with NO (Figure 1(b)). To further confirm the FE65 S-notrosylation, we treated Nitrosonium tetrafluorobo- rate (NOBF4) which is an exclusively NO+ releasing NO donor which forces protein-S-nitrosothiol generation via a transnitrosation reaction to enhance the effect. In the first set of experiments, we incubated the HEK293 cell transfected with rat GFP FE65 for 5 min with NOBF4. Following protein precipitation, the enzyme was resus- pended, exposed, and electrophoretically separated on a nonreducing 11% SDS gel (Figure 1(c)). GFP-rat FE65 exposed to NOBF4 was found to harbor the high amount S-nitrosylated state (Figure 1(c) right lane). HEK293 2.9. Immunoprecipitation Total cell lysates of EC were denatured via incubation with 0.5% SDS, 50 mM sodium phosphate, pH 8.0, and 2 mM EDTA at 90˚C, to allow for immunoprecipitation. The samples were then supplemented to obtain a compo- sition of 50 mM sodium phosphate, pH 7.2, 1% sodium deoxycholate, 1% Triton X-100, 0.5% SDS, 150 mM NaCl, 2 mM EDTA, 5 mM NaF, 2 mM Na4P2O7, 2 mM Na3VO4, 1% aprotinin, and 200 µg/ml leupeptin at 4˚C. Mouse anti-FE65 monoclonal antibody (usually at a 1: 100 dilution) was added, followed 18 h later by protein A-Sepharose. The washed immunoprecipitates were then analyzed via western bolt with rabbit anti-FE65 or mouse anti-GFP (Santacruze) after separation in nonre- ducing or reducing 11% sodium dodecyl sulfate-polya- Figure 1. S-nitrosylation of FE65. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 112 cell transfected with GFP vector alone was used as the negative control (left lane). Thus, this result also indi- cates that FE65 is an S-Nitrosylation protein that is modified by NO. Next, in order to map the potential S-nitrosylation sites of FE65, we constructed the FE65 mutant (C440A) which is predicted with the consensus S-nitrosylation site motif in Figure 1(a) and subjected it to immuno- blotting with an S-nitrocystein rabbit antibody (Figure 1(d)) or the biotin switch assay (Figure 1(e)). As shown in Figure 1(d), GFP FE65 C440A mutant was not de- tected with S-nitrocystein Ab, while GFP FE65 WT was recognized by the antibody (Figure 1(d) upper lane). To monitor the rat GFP FE65 in HEK293 cell, the im- munoblotting was performed with ant-FE65 Ab (Figure 1(d) bottom lane), after protein immune-precipitation with GFP Ab. Next, we isolated GST-FE65 WT or C440A mutant protein form E. coli and subjected them to the biotin switch assay. Consistent with Figure 1(c), GST FE65 WT was recognized in the biotin switch assay, while GST FE65 C440A mutant was not reacted (Figure 1(e)). Therefore, together with the biochemical data, the 440 cysteine residue of (439RCE441) in FE65 is the S- nitrosylation site, as predicted in Figure 1(a) . 3.2. S-Nitrosylation of FE65 Enhanced its Binding to Tip60 A number of physiological functions of FE65 are associ- ated with its PID1 domains, including Tip60, CP2, LSF, Teashirt, and calsyntenin; thus, we suspected that the S- nitrosylation of FE65 PID1 domain could affect its Tip60 binding activity in cells exposed to a variety of cell death stimuli [4,5]. We investigated whether NO modification on FE65 influences its binding to Tip60. With NOBF4 treatment concentration, we observed that FE65 increases its binding to Tip60 (Figure 2(b) left lane). To compare the effect of S-nitrosylation on FE65, FE65 C440A (which is mutated the S-nitrosylated site) was also treated with NOBF4. As shown in Fig. 2B right lane, the NOBF4 treatment FE65 C440A does not influ- ence on its binding to Tip60 in HEK293 cell. To monitor S-nitrosylation of FE65 and FE65 C440A with treatment of NOBF4, the western blot using S-nitrosylation spe- cific antibody was performed (Figure 2(c)). As we ex- pected, FE65 WT with NOBF4 treatment was shown the strong western result (Figure 2(c) second lane). Thus, S- nitrosylation on the Cys 440 residue of FE65 seems to increase its binding affinity to Tip60. Further, we moni Figure 2. NO infusion promoted the binding between FE65 and Tip60 but inhibited the HAT activity of Tip60. C opyright © 2011 SciRes. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 113 tored each HAT activity of the total cell lysates with ELISA (Figure 2(d)). The HAT activity of FE65 WT with NOBF4 treatment was reduced significantly, com- paring FE65 C440A. Therefore, it seems to be that S-nitrosylation on the Cys 440 residue of FE65 reduced it HAT activity (Figure 2(d)). Together, these results suggested that the binding enhancement between FE65 and Tip60 by S-nitrosylation on the Cys 440 residue of FE65 decreases the HAT activity of Tip60. 3.3. The Reduction of Tip60 HAT Activi ty by the Increase of FE65 Expression Activity ELISA assay of HEK293 cells transfected with EGFP FE65 or FE65 C440A, and then monitored the FE65, Tip60, or actin expression by using their own spe- cific antibody. The transfection of EGFP FE65 WT in HEK293 lysate resulted in the reduction of Tip60 HAT activity (Figure 3(a)). By way of contrast, the transfec- tion of EGFP FE65 C440A in HEK293 lysate did not effect on Tip60 HAT activity (Figure 3(b)). A variety of previous studies have suggested that FE65’s anti-Tip60 activity depends on direct physical interactions between FE65 and Tip60 [18-20]. Thus, FE65 C440 residue may result in losses in the anti-Tip60 activity of FE65 by in- terfering with the direct interaction between FE65 and Tip60. Taken together, these results demonstrated that the S-nitrosylation on C440 residue of FE65 increases its binding with Tip60, resulting in the inhibition HAT ac- tivity of Tip60. 3.4. S-Nitrosylation of FE65 Does Not Affect its Subcellular Localization To determine whether NO could affect FE65 subcellular localization and Tip60 interaction, HEK293 cells trans- fected with GFP-FE65 were treated with the NO donor, NOBF4, which indicated that the scaffolding activity was affected by FE65 S-nitrosylation (Figure 4). The trans- fected EGFP-FE65 wt or its FE65 mutant (C440A) (all constructs were shown as green color) were detected by fluorescence microscopy and the FE65 position appeared as red (Figure 4). We noted that the FE65 modification proteins were detected primarily in the nuclear speckle (Figure 4 middle lane). EGFP-FE65 wt was merged with FE65 in the nuclear speckles surrounding the nu- clear pore (Figure 4(a)). EGFP-FE65 wt, which was observed as a fibrous form in the cytoplasm (similar to Figure 1(b)), was not merged (yellow color) with FE65 C440A. Thus, these results also implicated FE65 as one of the FE65 S-nitrosylation proteins. Because FE65 WT was shown to have merged with Tip60 (yellow color) in the nuclear speckles (dots) around the nuclear pore but not in the cytoplasm (Figure 4(a) right lane), the S-ni- trosylation of FE65 appears to be related to its nuclear subcellular localization. Figure 3. The increase of FE65 expression promoted its binding with Tip60 but inhibited the HAT activity of Tip60. C opyright © 2011 SciRes. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 114 (a) (b) Figure 4. The subcellular localization of FE65 by S-nitrosylatio. Similar to the confocal results of FE65 wt, its FE65C- 440A mutant was also detected primarily in the nucleus (Figure 4(b)). However, the FE65 C440A mutant (which evidenced a diffuse, rather than a dot form) did not merge well with Tip60 at the nuclear pore (Figure 4(b) right). These results indicated that S-nitrosylation on the C440 of FE65 is required for its nuclear speckle local- ization around the nuclear pore, consistent with the re- sults shown in Figure 4(a). Further, the results (Figure 4(b)) again verified that the C440A of FE65 is the FE65 S-nitrosylation site, based on the results of confocal mi- croscopic analysis. 3.5. Increase in FE65 Protein Stability Induced by S-Nitrosylation In order to compare the protein stability of FE65WT or C440A in HEK293 cells, DNA were transfected in the cells. Then, the transfected cells were treated with cyclo- hexamide, and the protein stabilities were assessed and compared. The FE65 proteins were chased for the indi- cated time periods. The EGFP FE65 proteins were im- munoprecipitated with a polyclonal anti-GFP antibody and subjected to SDS-PAGE followed by Western blot- ting with a monoclonal FE65 antibody (Figure 5(a)). An equal quantity of cell lysate was subjected to Western blotting using an actin antibody to monitor protein levels. As shown in Figure 4(b), we noted that FE65 WT was more stable than the C440A mutant. In order to eliminate the mutational effect of the C440A mutant on its protein stability, the protein stability of the FE65 C440A mutant was also evaluated using the same procedure. It was ob- served that the protein stability of the FE65 C440A mu- C opyright © 2011 SciRes. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 115 tant was not altered as profoundly as that of the C440A mutant (Figure 5(b)). Thus, these data also demon- strated that the 440 Cys residue of FE65 appears to be required for its protein stability. Taken together, we observed that S-nitrosylation on Cys440 residue of Fe65 increases its binding affinity to Tip60, which affects HAT activity and protein stability of Tip60. 4. Figure Legends 4.1. Figure 1. S-Nitrosylation of FE65. 1) Rat FE65 contains 666 amino acids, and functional domains and key amino acids are indicated. C440 is the PID1, where specific modifications (S-nitrosylation, sul- phonation) occur as the result of NO. FE65 binds to sev- eral proteins with its PID, including Tip60. The putative S-nitrosylation site was noted in human FE65 (Cys853). Cys853 (852KCD855) was replaced with alanine (C440A). Among 14 Cys residues in FE65, the Cys853 residue is immediately followed by the acidic amino residue (Lys/ Arg/His/Asp/Glu) Cys (Asp/Glu) (sites –1, 0, and +1, re- spectively) which is required for S-nitrosylation by nitric acid (NO). The FE65 Cys 853 that is replaced with ala- nine is marked with an arrow. 2) HEK293 cell lysate was immunoprecipitated with FE65 Ab or a normal serum (as a negative control). To detect the thiol modifications of the FE65, the im- munoblotting was performed with an S-nitrocystein rab- bit antibody. 3) HEK293 cells transfected with GFP-tagged FE65 WT with 0.5 mM NOBF4. After immnoprecipitating with S-nitrocystein Ab the precipitant subjected to im- munoblot with an S-nitrocystein rabbit antibody. The un- transfected HEK293 cells were used as a negative con- trol. 4) After transfected GFP-FE65 or C440A mutant in HEK293 cells. The immnoprecipitation with a rabbit anti- GFP Ab was conducted. The immunoprecipitant was immune blotted with an S-nitrocystein rabbit antibody (upper lane), or FE65 antibody (bottom lane). The trans- fected DNA was indicated above, and the immunoblot antibody was shown in left. 5) GST fusion proteins with FE65 fragments contain- ing Cys440A (as shown in Figure 1) were prepared from E. coli, and subjected to a biotin-switch assay. The GST fusion proteins with FE65 fragments harboring C440A were prepared from E. coli and subjected to the biotin- switch assay. These results were replicated at least in triplicate. Figure 5. Protein stability of FE65 or C440A mutant. C opyright © 2011 SciRes. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 116 4.2. Figure 2. NO Infusion Promoted the Binding Between FE65 and Tip60 but Inhibited the HAT Activity of Tip60. HEK293 cells were transiently transfected with GFP- FE65 WT or the C440A plasmid. After 48 h, the cells were lysed, the total proteins were recollected, and im- munoprecipitation was conducted using nickel beads. Western blot assays were subsequently conducted with a rabbit FE65 Ab (a), an S-nitrocystein rabbit antibody (b) or an anti-Tip60 antibody (c) in HEK293 cells. (d) The effects of FE65 S-nitrosylation on Tip60 HAT activity were assessed v ia an ELISA assay with the same sample. The ELISA assays revealed an inverse correlation be- tween levels of Tip60 and magnitudes of FE65 dependent. 4.3. Figure 3. The Increase of FE65 Expression Promoted its Binding with Tip60 but Inhibited the HAT Activity of Tip60. After transfection with GFP-FE65 WT DNA as indicated above (0, 2, 4 ug), the quantification of the Tip60 (a) or FE65 (b) expression level in western blot was performed with anti-Tip60 Ab or anti-FE65 Ab (c) The effects of FE65 on Tip60 HAT activity were assessed via an ELI- SA assay with the same sample in Figure 3 (a) and (b). The ELISA assays revealed an inverse correlation be- tween levels of Tip60 and magnitudes of FE65-depend- ent transfection. (d) Amount of cell protein was analyzed via Western blot with anti-actin antibody for the negative control. These results were replicated at least 3 times. 4.4. Figure 4. The Subcellular Localization of FE65 by S-Nitrosylation S-nitrosylation of FE65 by NO inhibits nuclear localiza- tion (a and b). HEK293 cells transfected with GFP- FE65 and GFP-FE65 C440A were incubated with either 100 μM NOBF4 for 6 h. GFP-FE65 WT was observed to merge with Tip60 (yellow color) in the nuclear speckles (dots) around the nuclear pore but not in the cytoplasm (a). However, the transfected GFP-FE65 C440A mutant was not merged with Tip60 (b) in the treatment of 100 μM NOBF4 for 6 h. The localization of GFP-FE65 WT or C440A was compared via direct immunofluorescence microscopy (X 400). S-nitrosylation of FE65 appears to be related to its nuclear subcellular localization. These results were replicated at least 3 times. 4.5. Figure 5. Protein Stability of FE65 or C440A Mutant. 1) EGFP-FE65 or the FE65 C440A mutant was trans- fected into HEK293 cells and the cells were treated with cyclohexamide. The FE65 proteins were chased for the indicated time periods. Western blotting with a mono- clonal FE65 antibody of SDS-PAGE subjected EGFP- FE65 proteins is shown. In order to monitor the amount of protein, equal quantities of cell lysates were subjected to Western blotting using an actin antibody. The results shown are one representative of five repeated experi- ments. 2) Quantification of the pulse-chase experiment is shown by image analysis with Fuji Image Quant soft- ware. 5. Discussion The results of this study demonstrated, for the first time, that FE65 can be S-nitrosylated on its C440 residue. The mechanism by which the S-nitrosylation of FE65 im- pairs its protective functions appears to occur through the inhibition of its Tip60 activity. This idea contrasts sharply with the mechanism underlying the S-nitrosyla- tion-mediated impairment of FE65’s Tip60 binding function. FE65’s binding function to Tip60 is increased by the S-nitrosylation of FE65 via an enhancement of the binding affinity of FE65 to Tip60. S-nitrosylation is a nitric oxide (NO)-induced post- translational modification in which a cysteinyl thiol (R- SH) is converted to a nitrosothiol; this modification is known to function as a regulatory mechanism of various classes of proteins, including ion channels such as TRPA1, TRPC5, the skeletal muscle-type ryanodine re- captor (ryanodine receptor type 1) channel, the N-me- thyl-D-aspartate receptor channel, the cardiac L-type Ca2+channel, and the cardiac Na+ channel. Thus, protein S-nitrosylation may convey a broad spectrum of cellular signals. NO is a pleiotropic cell signaling molecule that controls a variety of biological processes [16]. Accord- ing to the classical view, cyclic GMP mediates NO sig- naling [7,8]. The importance of a cGMP- independent pathway occurring via protein S-nitrosylation is being increasingly recognized by researchers in the field of NO signal transduction. Thus, protein direct S-nitrosylation appears to be an important NO-mediated regulatory me- chanism relevant to various classes of proteins. However, the direct link between protein S-nitrosylation and its functional relevance has been demonstrated only for limited examples. Our result which S-nitrosylation regu- lates FE65 function seems to be the good example for this researcher field, even the mechanism and the func- tional significance underlying the preferential S-nitrosy- lation of the target Cys remain incompletely understood. Protein S-nitrosylation, like phosphorylation, seems to be emerging as an important player in regulating protein activity and signal transduction in response to DNA damage. Tip60 appears to be quite relevant to acetylation, and activates several key DNA damage-responsive pro- teins, such as H2AX, ATM, and p53. Interestingly, the S-nitrosylation of FE65 occurs somewhat later, thus C opyright © 2011 SciRes. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 117 suggesting that FE65 S-nitrosylation may be directly regulated in some fashion. We are pursuing now the pos- sibility that both FE65 is also modified by NO as the target proteins to respond DNA damage. Further, be- cause we also notice S-nitrosylation site in Tip60 and observed this modification, we do not rule out the possi- bility that our result in Figure 4 is due to the direct S-nitrosylation on Tip 60 by NOBF4 treatment. Because we determined that FE65 S-nitrosylation was increased in both an MPTP animal model of AD and in AD patients, these results indicate that neurons may be more susceptible to cell death in the face of unfavorable conditions such as proteasomal dysfunction and protein aggregation-induced toxicity. Our findings also indicate that a more thorough understanding of how nitrosative stress can contribute to AD will help us to develop new therapeutic approaches for this disease or for senescence. In addition to its ability to impair neuroprotection pro- teins, S-nitrosylation has also been shown to be involved in mediating cell death via FE65 (our unpublished data). Our studies also demonstrated that the S-nitrosylation of FE65 can mediate the translocation of the FE65-Tip60 protein complex to the nucleus, and initiate apoptosis (Figure 5). Our finding that the S-nitrosylation of FE65 can affect its Tip60 binding function further suggests that nitrosative stress can affect neuronal survival via the targeting of a number of pathways. FE65 is also known to form dimers in the cell, and this dimerization is important for its physiological func- tions and scaffolding activity with its WW domain. The results of our study did not reveal whether the S-nitro- sylation of FE65 induces its monomerization for a less active form. In addition to FE65, several other proteins, including actin, creatine kinase, glycogen phosphorylase, and the homodimeric HIV-1 protease are targets for S- nitrosylation in intact cells after exposure to oxidative stress. Several recent studies have also shown that other neuroprotective proteins, including peroxiredoxin and protein-disulphide isomerase, are modified by S-nitrosy- lation, and this modification compromises their normal protective functions. Thus, the repertoire of activities via which S-nitrosylation can compromise cellular survival is rather diverse. Along this line, it has been recently demonstrated that the treatment of endothelial cells with the combination of NOBF4 and cysteine, a system that generates GSSG and nitric oxide, resulted in the loss of intracellular glutathione, probably as the result of the formation of protein-mixed disulfides. NO enhances the effects of FE65’s Tip60 binding ability against a variety of cell death stimuli. In summary, this study describes the interaction be- tween FE65 and Tip60, which is enhanced by a post- translational modification of FE65 (through S-nitrosy- lation) by NO, promoting the association of the FE65- Tip60 protein complex and inhibiting both the HAT ac- tivity of Tip60 and cell death. Greater insight into the FE65 and Tip60 signal cascade is expected to contribute to our molecular understanding of a role of both FE65 and Tip60 in neurodegenerative disorders, and is also expected to help in the development of novel strategies for the treatment of such disorders. 6. ACKNOWLEDGEMENTS This work was supported by National Research Foundation of Korea (NRF) grants (2009-0076024 and 2009- 0069007) funded by the Korea government (MEST) to S S Kang. We also appreciated The Core Facil- ity of Chungbuk National University. REFERENCES [1] McLoughlin, D.M. and Miller, C.C. (2008) The FE65 proteins and Alzheimer’s disease. Journal of Neurosci- ence Research, 86, 744-754. doi:10.1002/jnr.21532 [2] Lee, E.J., Hyun, S.H., Chun, J. and Kang, S.S. (2007) Human NIMA-related kinase 6 is one of the Fe65 WW domain binding proteins. Biochemical and Biophysical Research Communications, 358, 783-788. doi:10.1016/j.bbrc.2007.04.203 [3] Lee, E.J., Hyun, S., Chun, J., Shin, S.H., Lee, K.E., Yeon, K.H., Park, T.Y. and Kang, S.S. (2008) The PPLA motif of glycogen synthase kinase 3beta is required for interac- tion with Fe65. Molecules and Cells, 26, 100-105. [4] Ermekova, K.S., Zambrano, N., Linn, H., Minopoli, G., Gertler, F., Russo, T. and Sudol, M. (1997) The WW do- main of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. Journal of Biological Chemistry, 272, 32869- 32877. doi:10.1074/jbc.272.52.32869 [5] Sudol, M., Sliwa, K. and Russo, T. (2001) Functions of WW domains in the nucleus. FEBS Letters, 490, 190- 195. doi:10.1016/S0014-5793(01)02122-6 [6] Gordge, M.P. and Xiao, F. (2010) S-nitrosothiols as se- lective antithrombotic agents-possible mechanisms. Brit- ish Journal of Pharmacology, 159, 1572-1580. doi:10.1111/j.1476-5381.2010.00670.x [7] Lima, B., Forrester, M.T., Hess, D.T., and Stamler, J.S. (2010) S-nitrosylation in cardiovascular signaling. Cir- culation Research, 106, 633-646. doi:10.1161/CIRCRESAHA.109.207381 [8] Sun, J. and Murphy, E. (2010) Protein S-nitrosylation and cardioprotection. Circulation Research, 106, 285- 296. doi:10.1161/CIRCRESAHA.109.209452 [9] Arnelle, D.R. and Stamler, J.S. (1995) NO+, NO, and NO-donation by S-nitrosothiols: implications for regula- tion of physiological functions by S-nitrosylation and ac- celeration of disulfide formation. Archives of Biochemis- try and Biophysics, 318, 279-285. doi:10.1006/abbi.1995.1231 [10] Stamler, J.S., Simon, D.I., Jaraki, O., Osborne, J.A., Francis, S., Mullins, M., Singel, D. and Loscalzo, J. (1992) S-nitrosylation of tissue-type plasminogen acti- C opyright © 2011 SciRes. ABC  E. J. Lee et al. / Advances in Biological Chemistry 1 (2011) 109-118 Copyright © 2011 SciRes. 118 ABC vator confers vasodilatory and antiplatelet properties on the enzyme. Proceedings of the National Academy of Sciences of the USA, 89, 8087-8091. doi:10.1073/pnas.89.17.8087 [11] Fukuda, H., Fukuda, A., Zhu, C., Korhonen, L., Swan- palmer, J., Hertzman, S., Leist, M., Lannering, B., Lind- holm, D., Bjork-Eriksson, T., Marky, I., and Blomgren, K. (2004) Irradiation-induced progenitor cell death in the developing brain is resistant to erythropoietin treatment and caspase inhibition. Cell Death and Differentiation, 11, 1166-1178. doi:10.1038/sj.cdd.4401472 [12] Lipton, S.A., Choi, Y.B., Pan, Z.H., Lei, S.Z., Chen, H.S., Sucher, N.J., Loscalzo, J., Singel, D.J. and Stamler, J.S. (1993) A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature, 364, 626-632. doi:10.1038/364626a0 [13] Lander, H.M., Ogiste, J.S., Pearce, S.F., Levi, R. and Novogrodsky, A. (1995) Nitric oxide-stimulated guanine nucleotide exchange on p21ras. Journal of Biological Chemistry, 270, 7017-7020. doi:10.1074/jbc.270.13.7017 [14] Yoshida, T., Inoue, R., Morii, T., Takahashi, N., Yama- moto, S., Hara, Y., Tominaga, M., Shimizu, S., Sato, Y. and Mori, Y. (2006) Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nature Chemical Biology, 2, 596-607. doi:10.1038/nchembio821 [15] Chander, M., and Demple, B. (2004) Functional analysis of SoxR residues affecting transduction of oxidative stress signals into gene expression. Journal of Biological Che- mistry, 279, 41603-41610. doi:10.1074/jbc.M405512200 [16] Gu, Z., Nakamura, T. and Lipton, S.A. (2010) Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurode- generative diseases. Molecular Neurobiology, 41, 55-72. doi:10.1007/s12035-010-8113-9 [17] Russo, T., Faraonio, R., Minopoli, G., De Candia, P., De Renzis, S. and Zambrano, N. (1998) Fe65 and the protein network centered around the cytosolic domain of the Alzheimer’s beta-amyloid precursor protein, FEBS Let- ters,434, 1-7. doi:10.1016/S0014-5793(98)00941-7 [18] Chen, H.I. and Sudol, M. (1995) The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proceedings of the National Acad- emy of Sciences of the USA, 92, 7819- 7823. doi:10.1073/pnas.92.17.7819 [19] Fiore, F., Zambrano, N., Minopoli, G., Donini, V., Duilio, A. and Russo, T. (1995) The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phos- phortyrosine binding domain of Shc bind the intracellular domain of the Alzheimer’s amyloid precursor protein, Journal of Biological Chemistry, 270, 30853-30856. [20] Zambrano, N., Minopoli, G., de Candia, P. and Russo, T. (1998) The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/LSF/ LBP1. Journal of Biological Chemistry, 273, 20128-20133. doi:10.1074/jbc.273.32.20128
|