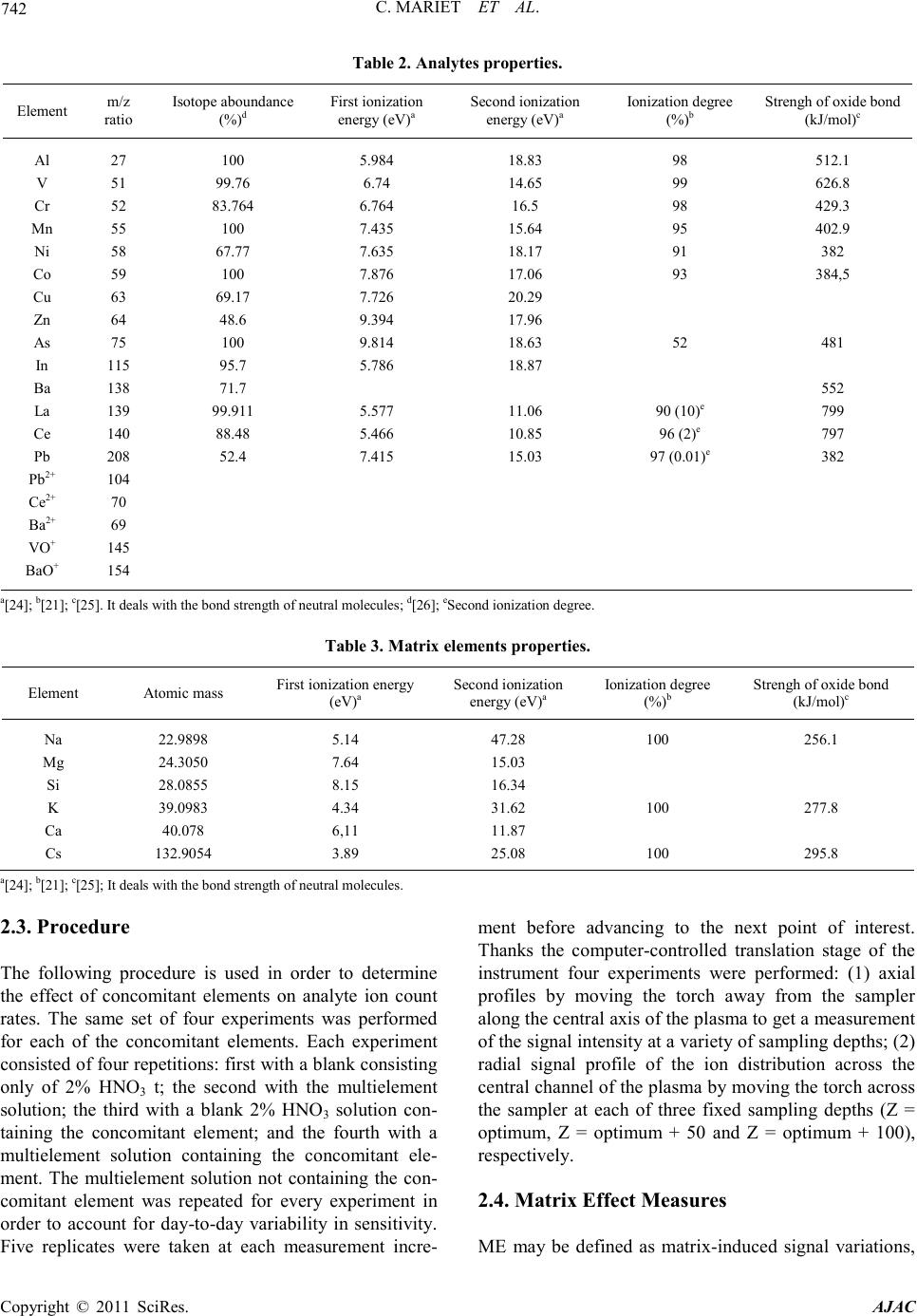
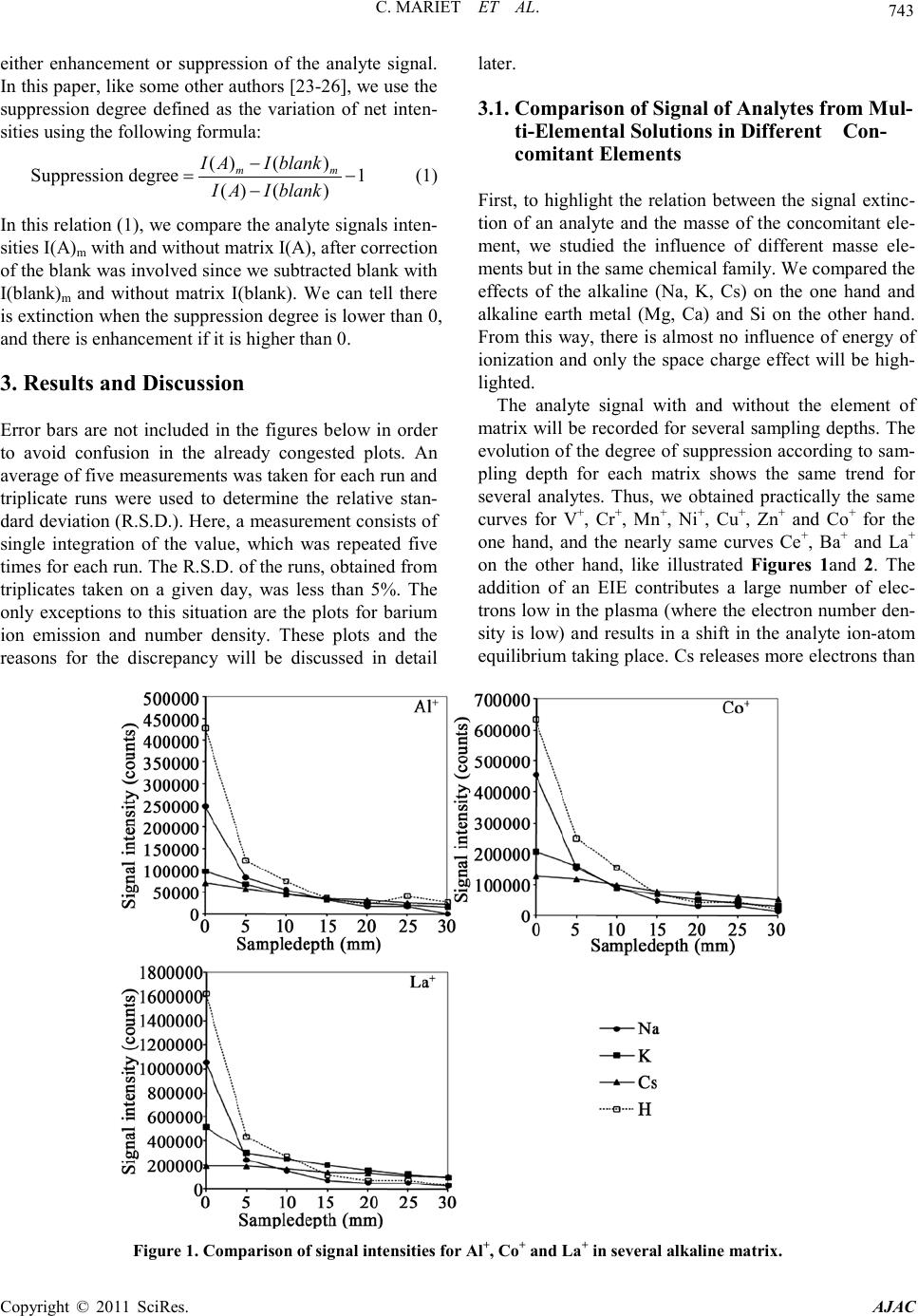
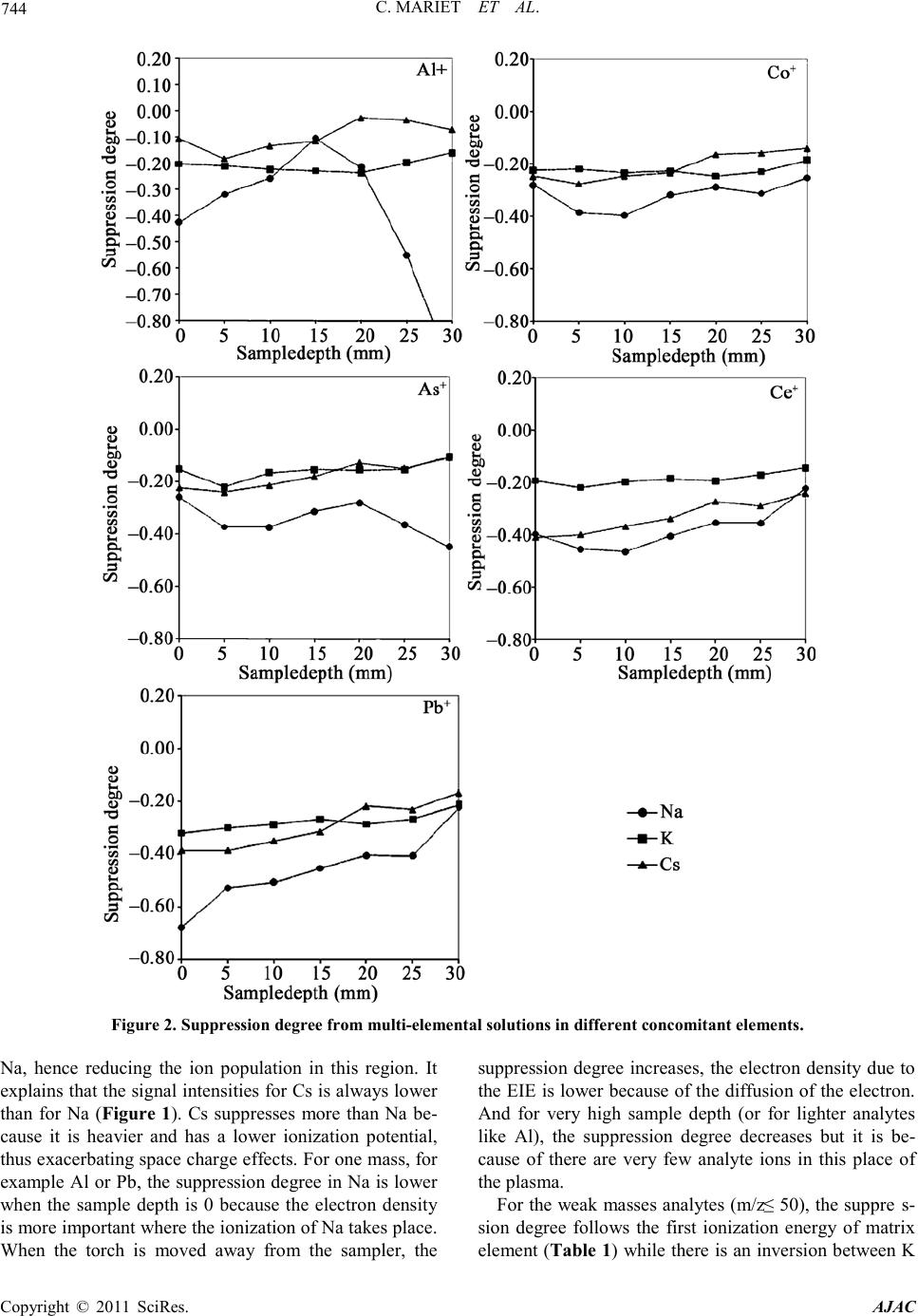
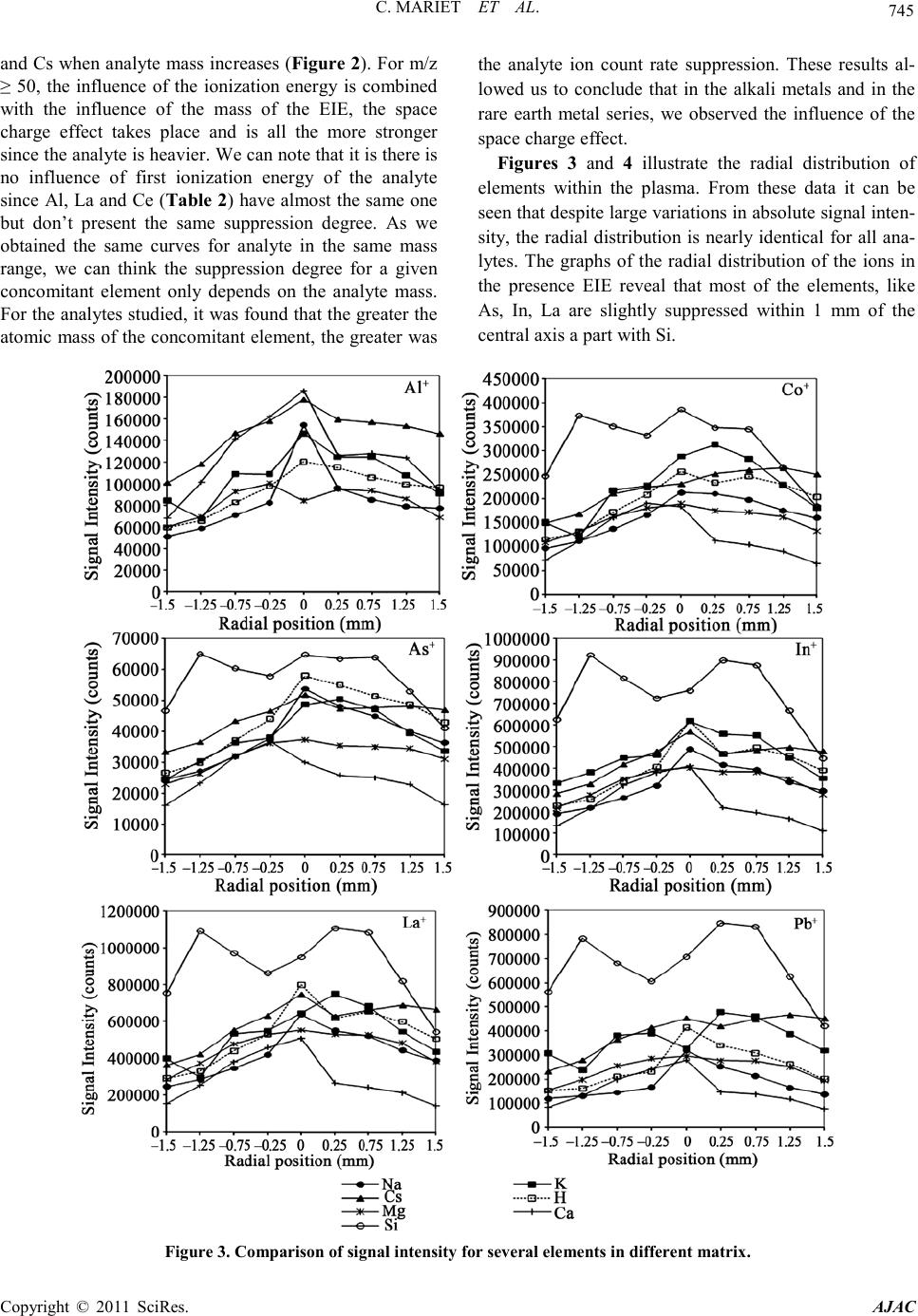
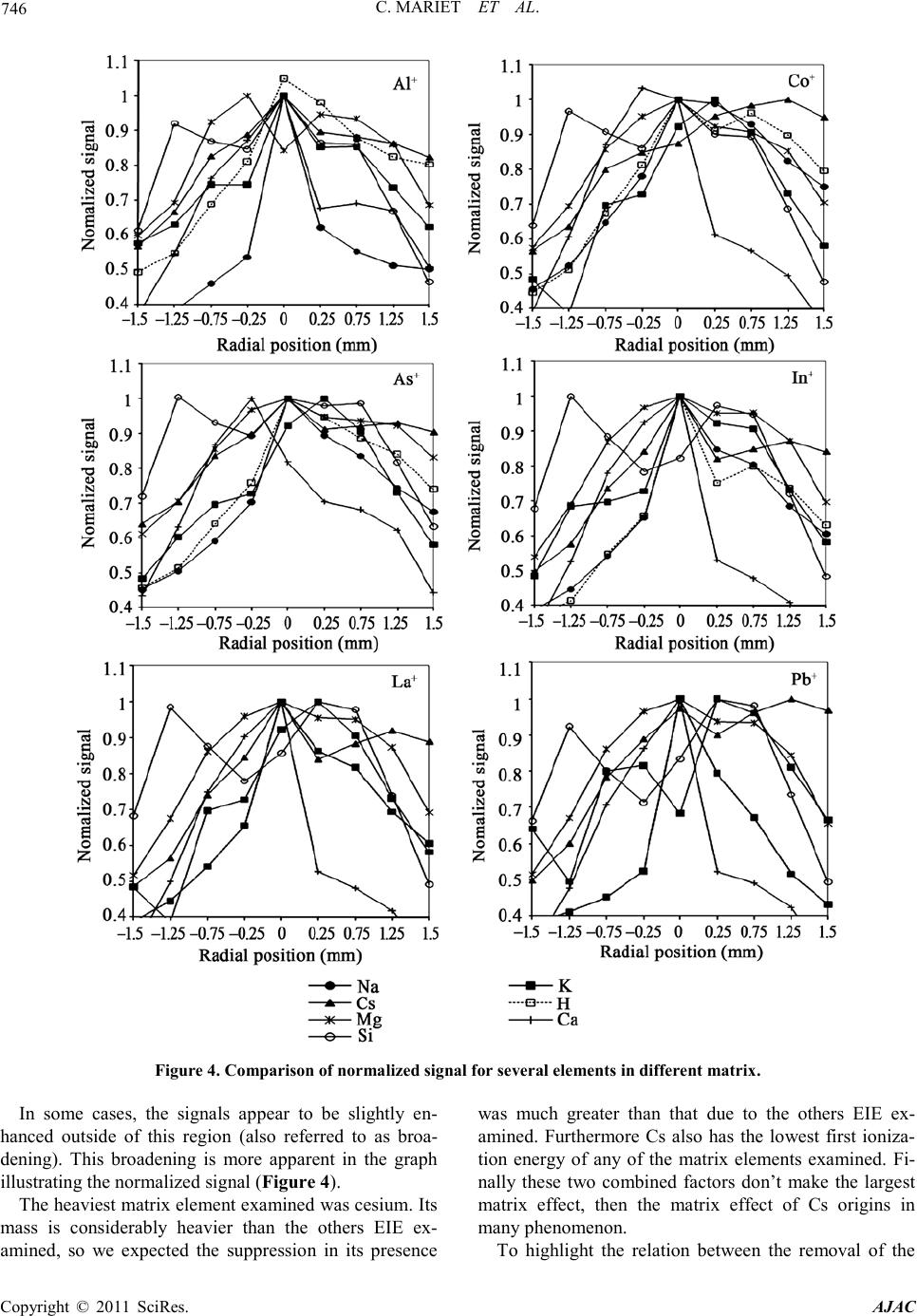
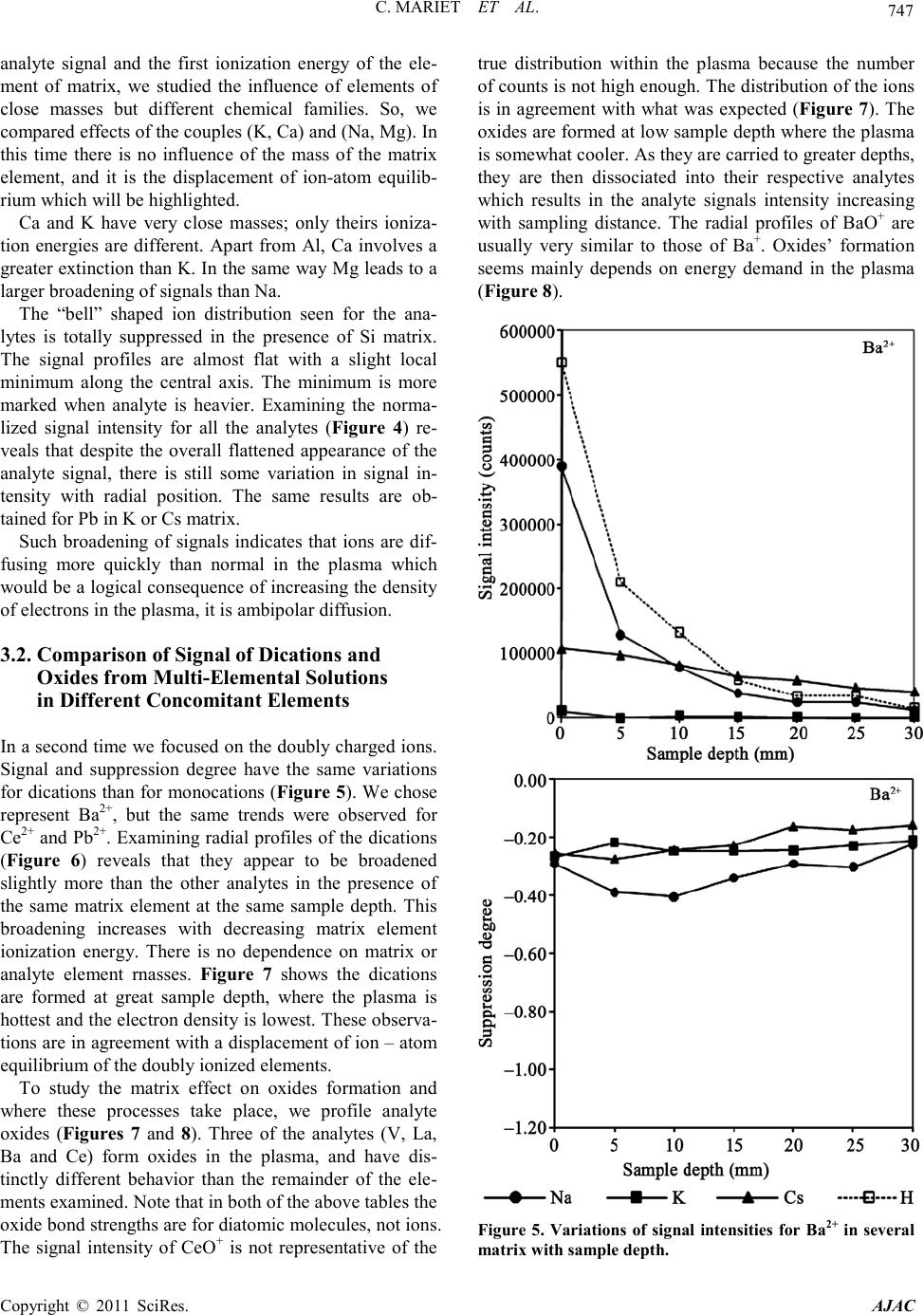
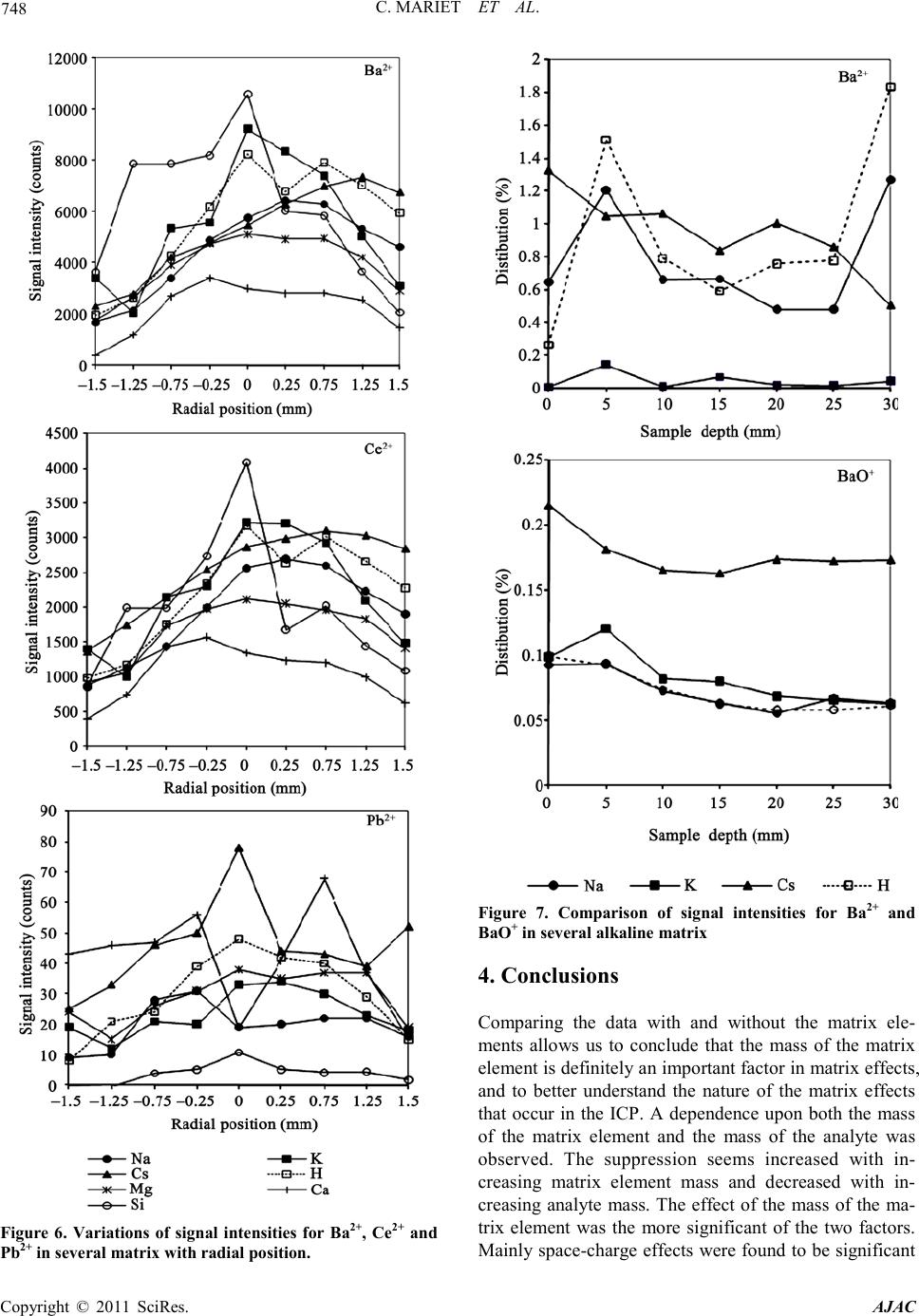
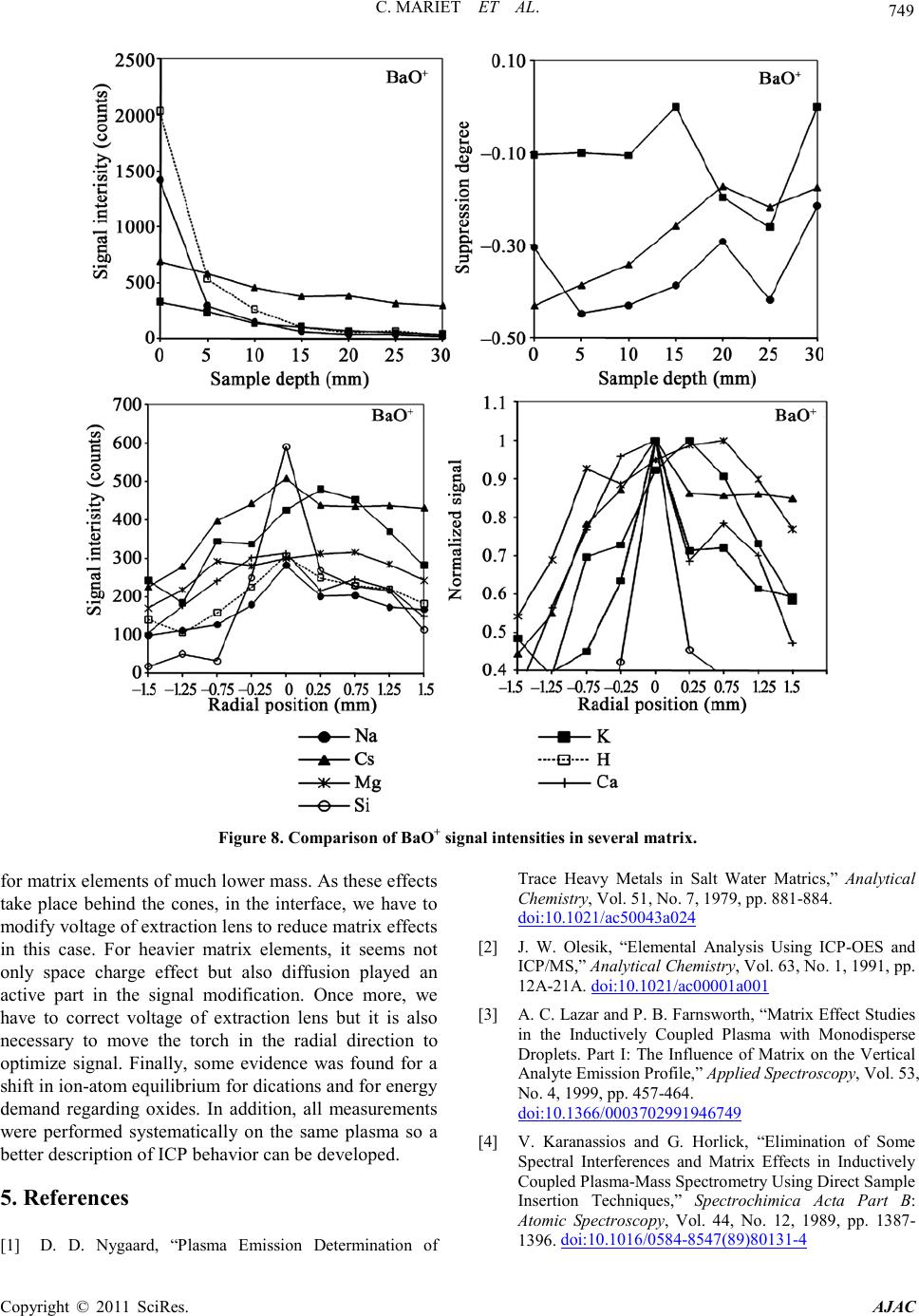
C. MARIET ET AL.
Copyright © 2011 SciRes. AJAC
[5] J. L. Venzie an d R. K. Marcus, “Effects of Easily Ionisa-
ble Elements on the Liquid Sampling―Atmospheric
Pressure Glow Discharge,” Spectrochimica Acta Part B,
Vol. 61, No. 6, 2006, pp. 715-721.
doi:10.1016/j.sab.2006.02.005
[6] G. C. -Y. Chan and G. M. Hieftje, “Investigation of
Plasma-Related Matrix Effects in Inductively Coupled
Plasma-Atomic Emission Spectrometry Caused by Ma-
trices with Low Second Ionization Potentials― Identi-
fication of the Secondary Factor,” Spectrochimica Acta
Part B, V ol. 61, No. 6, 2006, pp . 642 -659.
doi:10.1016/j.sab.2005.09.007
[7] G. Gamez, S. A. Lehn, M. Huang and G. M. Hieftje,
“Effect of Mass Spectrometric Sampling Interface on the
Fundamental Parameters of an Inductively Coupled
Plasma as a Function of Its Operating Conditions Part I.
Applied r.f. Power and Vacuum,” Spectrochimica Acta
Part B, V ol. 62, N o. 4, 2007, pp . 357 -369.
doi:10.1016/j.sab.2007.03.015
[8] G. Gamez, S. A. Lehn, M. Huang and G. M. Hieftje,
“Effect of Mass Spectrometric Sampling Interface on the
Fundamental Parameters of an Inductively Coupled
Plasma as a Function of Its Operating Conditions Part II.
Central-Gas Flow Rate and Sampling Depth,” S pectro-
chimica Acta Pa r t B, Vol. 62, No. 4, 2007, pp. 37 0-377.
doi:10.1016/j.sab.2007.03.016
[9] S. A. Lehn, K. A. Warner, M. Huang and G. Hieftje,
“Effect of Sample Matrix on the Fundamental Properties
of the Inductively Coupled Plasma,” S pect rochi mica Acta
Part B, V ol. 58, No. 10, 2003, pp. 1786-1806.
doi:10.1016/S0584-8547(03)00159-9
[10] D. Lariviere, V. F. Taylor, R. D. Evans and R. J. Cornett,
“Radionuclide Determination in Environmental Samples
by Inductively Coupled Plasma Mass Spectrometry,”
Spectrochimica Acta Part B, Vol. 61, No. 8, 2006, pp.
877-904. doi:10.1016/j.sab.2006.07.004
[11] A. M. Desaulty, C. Mariet, P. Dillmann, J. L. Joron and P.
Fluzin, “A Provenance Study of IroN Archaeological Ar-
tefacts by ICP-MS Multi-Elemental Ana lysi s,” Spectro-
chimica Acta Part B, Vol. 63, No. 11, 2008, pp. 1253-
1262. doi:10.1016/j.sab.2008.08.017
[12] M. He, B. Hu, Y. Zeng and Z. Jiang, “ICP-MS Direct
Determination of Trace Amounts of Rare Earth Impuri-
ties in Various Rare Earth Oxides with Only One Stan-
dard Series,” Alloys and Compounds, Vol. 390, No. 1-2,
2005, pp . 168-174. doi:10.1016/j.jallcom.2004.06.107
[13] S. Kozono and H. Haraguchi, “Determination of Ultra-
trace Impurity Elements in High Purity Niobium Mate-
rials by on-Line Matrix Separation and Direct Injec-
tion/Inductively Coupled Plasma Mass Spectrometry,”
Talanta, Vol. 72, No. 5, 2007, pp. 1791-1799.
doi:10.1016/j.talanta.2007.02.021
[14] T. Duan, X. Song, P. Guo, H. Li, L. Pan, H. Chena and J.
Xu, “Elimination of Matrix Effect and Spectroscopic In-
terference by Two Compactly Combined Separations in
the Determination of Cd in Geological Samples with
High Mo, Zr or Sn Contents by ICP-MS,” Journal of
Analytical and Atomic Spectrometry, Vol. 22, No. 4, 2007,
pp. 403-406. doi:10.1039/b610685d
[15] B. U. Peschel, W. Herdering and J. A. C. Broekaert, “A
Radiotracer Study on the Volatilization and Transport
Effects of Thermochemical Reagents Used in the Analy-
sis of Alumina Powders by Slurry Electroth ermal Vapo-
rization Inductively Coupled Plasma Mass Spectrome-
try,” Spectrochimica Acta Part B, Vol. 62, No. 2, 2007,
pp. 109-115. doi:10.1016/j.sab.2007.01.006
[16] J. Mora, L. Gras, E. H. van Veen and M. T. C. de
Loos-Vollebregt, “Electrothermal Vaporization of Miner-
al Acid Solutions in Inductively Coupled Plasma Mass
Spectrometry: Comparison with Sample Nebulization,”
Spectrochimica Acta Part B, Vol. 54, No. 6, 1999, pp.
959-974. doi:10.1016/S0584-8547(99)00029-4
[17] T. Ka´ntor, S. Maestre and M. T. C. D. Loos-Vollebregt,
“Studies on Transport Phenomena in Electrothermal Va-
porization Sample Introduction Applied to Inductively
Coupled Plasma for Optical Emission and Mass Spec-
tro me try,” Spectrochimica Acta Part B, Vol. 60, No. 9 -10,
2005, pp . 1323-1333. doi:10.1016/j.sab.2005.06.011
[18] D. C. Gregoire, “The Effect of Easily Ionisable Conco-
mitant Elements on Non-Spectroscopic Interferences in
Inductively Coupled Plasma Mass Sp ectrometry,” Spec-
trochimica Acta Part B, Vol. 42, No. 6, 1987, pp. 895-
907. doi:10.1016/0584-8547(87)80100-3
[19] M. M. Fraser and D. Beauchemin, “Effect of Concomi-
tant Elements on the Distribution of Ions in Inductively
Coupled Plasma Mass Spectrometry. Part 1 Elemental
Ions,” Spectrochimi ca Acta Part B, V o l. 55 , No . 11 , 2000,
pp. 1705 -1731. doi:10.1016/S0584-8547(00)00273-1
[20] J. A. Olivares and R. S. Houk, “Ion Sampling for Induc-
tively Coupled Plasma Mass Spectrometry,” Analytical
Chemistry, Vol. 57, No. 13, 1985, pp. 2674-2679.
doi:10.1021/ac00290a054
[21] X. Chen and R. S. Houk, “Spatially Resolved Measure-
ments of Ion Density behind the Skimmer of an Induc-
tively Coupled Plasma Mass Spectrometer,” Spectrochi-
mica Acta Part B, Vol. 51, No. 1, 1996, pp. 41-54.
doi:10.1016/0584-8547(95)01387-3
[22] A. E. Holliday and D. Beauchemin, “Spatial Profiling of
Analyte Signal Intensities in Inductively Coupled Plasma
Mass Spectrometry,” Spectrochimica Acta Part B, Vol.
59, No. 3, 2004, pp. 291-311.
doi:10.1016/j.sab.2003.12.018
[23] M. M. Fraser and D. Beauchemin, “Effect of Concomi-
tant Elements on the Distribution of Ions in Inductively
Coupled Plasma Mass Spectrometry. Part 2 Polyatomic
Ions,” Spect rochi mica Acta Part B, Vol. 56, No . 12 , 2001,
pp. 2479 -2495. doi:10.1016/S0584-8547(01)00346-9
[24] R. S. Houk, “Mass Spectrometry of Inductively Coupled
Plasmas,” Analytical Chemistry, Vol. 58, No . 1 , 19 86, pp.
97A-105A. doi:10.1021/ac00292a003
[25] H. Ying, M. Antler, J. W. Tromp and E. D. Salin, “Sam-
ple Diagnosis Using Non-Anal yte Signals for Inductively
Coupled Mass Spectrometry,” Spectrochimica Acta Part
B, Vol. 57, No. 2, 2002, pp. 277-290.
doi:10.1016/S0584-8547(01)00382-2