Open Access Library Journal
Vol.05 No.03(2018), Article ID:82832,12 pages
10.4236/oalib.1104335
Electronic Properties of Mo(1−x)W(x)S2-Ni Grown over Graphene
D. H. Galvan1, J. Antunez-Garcia1, S. Fuentes1, M. Shelyapina2
1Centro de Nanociencias y Nanotecnologia, Universidad Nacional Autonoma de Mexico, Ensenada, Mexico
2Saint-Petersburg State University, St. Petersburg, Russia

Copyright © 2018 by authors and Open Access Library Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 12, 2018; Accepted: March 3, 2018; Published: March 6, 2018
ABSTRACT
A proposal of the theoretical adsorption of 2H-Mo(1−x)W(x)S2-Ni over graphene using extended Hückel tight-binding method was investigated. It is well known that a theoretical prior investigation is well accepted by the scientific community due to that provides information regarding electronic as well as magnetic properties of the material under investigation. Energy bands for 2H-Mo(1−x)W(x)S2-Ni indicate a soft metal, while graphene with oxygen vacancies and 2H-Mo(1−x)W(x)S2-Ni yields indication of a semiconductor behavior with a measured energy gap Eg~0.98 eV between the Valence and Conduction bands respectively. In addition, an electronic charge transfer from 2H-Mo(1−x)W(x)S2-Ni toward graphene is presented. The new hybrid system presents high dispersion of 2H-Mo(1−x)W(x)S2-Ni on graphene. Total and partial Density of States yield indication that an enhancement of the contribution to the total DOS, in the vicinity of the Fermi level for the structure over graphene has occurred, when compared to 2H-MoS2 original. These results could indicate that the new proposed system could be used as a catalyst for HDS in the petrochemical industry.
Subject Areas:
Nanometer Materials
Keywords:
MoS2 Clusters, Graphene, Extended Hückel, Energy Bands, TDOS

1. Introduction
During the latest few years, many researchers have been devoted to the investigation of MoS2 due to its variety of physical [1] [2] , mechanical [3] , optical [4] and chemical properties [5] [6] .
Molybdenum and other transition-metal sulfide-based (TMS) hydro treating catalysts were developed early in the twentieth century [7] [8] and soon became extensively used in refineries worldwide, since they present activity toward hydrogenation and sulfur removal. Their tolerance for sulfur represents a big advantage over noble metal catalysts, which are easily contaminated by small amounts of sulfur, and more over are less expensive when compared to the noble metals.
Molybdenum and tungsten sulfides (separately or mixed on different proportions) have been shown a good catalytic activity in hydrotreating processes [9] ; moreover, according to recent theoretical studies Mo(1−x)W(x)S2 prefers to form an alloy, opposite to Mo(1−x)Cr(x)S2 and Mo(1−x)V(x)S2, in which a phase segregation occurs [10] , offering a homogeneous distribution of substituting atoms. Ni alloying to unsupported Mo(1−x)W(x)S2 has shown significant improvement in the catalytic activity as compared to any other hydrotreating catalyst available [11] . Among the properties determining the activity of these Mo(1−x)W(x)S2 catalysts, their morphology plays a great importance [12] in their electronic properties. The theoretical studies of unsupported Mo(1−x)W(x)S2 doped with M = Ni, Co, Fe or Cu reveal that the configuration becomes more metallic when M is intercalated randomly between two sulfur layers [13] . The fact of the enhancement in the DOS indicates that there exists a correlation in the increment in the catalytic activity of the new material.
Confinement of MoS2 nanoparticles or nanolayers within carbon materials has proven effective in improving their catalytic properties [14] [15] .
Graphene is a zero gap semiconductor [16] . A Dirac cone is formed at the interaction point of the π and π* bands on the Fermi surface in the K direction. Growth of graphene on a surface distorts its electronic structure by the interaction with the substrate. These distortions depend on whether the system is physisorbed or chemisorbed; chemisorptions cause disappearance of the Dirac cone due to hybridization of bonds, such as on surface of Co (111) [17] . Physisorption on graphene presents two various behaviors of the energy bands. In the first type, as found in graphene grown on Cu and Boron Nitride [18] , mini physisorption gaps appear in the band structure of graphene. In the second type, which can be realized in graphene supported on Au (111) or Al (111) surface [19] , the Dirac cone can still be distinguished in the energy bands but it is displaced upward in the Au (111) and downward in the Al (111). Displacement of the Dirac cone of graphene establishes a charge transfer between the metal surface and the graphene.
The upward shift of the Dirac cone is owing to an electron donation from metal to graphene, whereas the downward shift results from a vacancy donated by the metal to graphene [19] . The charge transfer from the metal to graphene is due to the π bands which is more than half filled and provides a low density of states for graphene, as compared to the high density of states from the metal. Such a change in the density of states (DOS) requires to be equilibrated at the Fermi level.
The aim of this paper is to propose a new kind of tri-metallic catalyst based on 2H-MoS2 when one of the atomic positions of Mo is substituted by W, creating bimetallic 2H-Mo(1−x)W(x)S2, and subsequently, Ni was intercalated in between S-Mo-S sub unit , originating our final 2H-Mo(1−x)W(x)S2-Ni tri metallic structure. In addition, the new tri-metallic catalyst is located above a graphene sheet.
2. Theoretical Calculations
The calculations reported in this study, had been carried out by means of tight-binding approach [20] within the framework of extended Hückel [21] method using YAeHMOP (Yet Another Extended Hückel Molecular Orbital Program) computer package with f-orbitals [22] . It is necessary to stress that the extended Hückel method is a semi empirical approach for solving Schrödinger’s equation for a system of electrons, based on the variational theorem. In this approach, explicit electron correlation is not considered except for the intrinsic contribution included in the parameter set. More details about the mathematical formulation of the method have been described elsewhere [23] and will be omitted here.
Despite its simplicity this method has proven itself as a trustworthy and reliable one to study electronic properties of molecules adsorbed on a surface [24] metal complexes [25] including MoS2 nanostructures both free [26] and supported on graphene surface [13] [27] .
Theoretical calculations were performed on a system selected as a repeated cluster originated from a super cell. The super cell was generated from a crystalline structure which arouse from crystalline 2H-MoS2 with the following vectors a = 3.1604 Å, c = 12.294 Å and space group P63/mmc (194) [28] . Table 1 provides the atomic parameters used in the Extended Hückel tight-binding calculations. Also, the Hii(eV) (Valence orbital Ionization potentials) as well as ζ (exponents for the Slater type orbitals) for Mo, W, S, O, C and Ni atoms are provided from S. Alvarez [29] .
To create a single graphene sheet, we call it further “graphene sheet”, a super cell consisted of 24 carbon atoms with a honeycomb arrangement obtained from the infinite 2D graphene hexagonal lattice (P6mm plane group) with two carbons in the primitive unit cell was used. This primitive unit cell will be referred as “graphene original”. In order to simulate a real scenario comparative to the experimental, a defect was introduced in the graphene sheet: carbons C14 and C19 were substituted by oxygen atoms, respectively adding vacancies to the calculations, see Figure 1.
For the Mo(1−x)W(x)S2-Ni, first one of the atomic positions of Mo was substituted by W atom. Then a Ni atom was intercalated in between the two half structures of 2H-MoS2 original structure. This structure was located 2 Å above the graphene sheet with a vacancy defect considered.

Figure 1. Graphene sheet with oxygen vacancies with 2H-Mo(1−x)W(x)S2-Ni cluster grown over it. Turquoise balls are carbon atoms, Red balls are oxygen, gray balls are Ni, Yellow are Suphur, Tungten are black balls while Mo are violet balls.

Table 1. Atomic parameters used in the extended Hückel tight-binding calculations, Hii (eV) and ζ (Valence Orbital ionization potential and exponent of Slater type orbitals). The d-orbitals for Mo, W and Ni are given as linear combination of two Slater type orbitals. Each exponent is followed by a weighting coefficient in parentheses. A modified Wolfsberg-Helmholtz formula was used to calculate Hij [42] .
3. Energy Bands
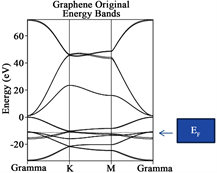
Figures 2(a)-(e) provide information regarding Energy Bands (eV) vs k points in the reciprocal space spanning the First Brillouin zone from Γ(0 0 0) to Κ (π/3a π/3a 0) to Μ(π/2a 0 0) to Γ(0 0 0) for each one of the structures enunciated formerly. The Fermi level (EF) is indicated by a horizontal dotted line separating the Valence (VB) to the Conduction bands (CB) respectively. Note, for Figure 2(a), that the Fermi level (at K point) yields the typical Dirac cone separating two bands (multiple degenerate) which are π and π* in behavior, which yield indication of the 0-gap semiconductor which originally was reported by Geim and Novoselov [30] [31] . The summary is provided in Table 2. Table 2 provides the analysis for the forbidden Energy gap (eV), Fermi level location (eV) and behavior for each configuration like graphene original, 2H-Mo(1−x)W(x)S2-Ni, graphene sheet, graphene sheet with Oxygen vacancies (defect), and graphene sheet with oxygen defect and with 2H-Mo(1−x)W(x)S2-Ni over it.
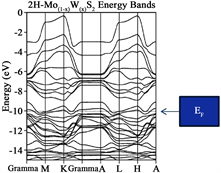
In addition, it has been reported that crystalline 2H-MoS2 is a semiconductor reported by several groups of investigators. Moreover, the reported forbidden energy gap was in the order between 1 - 1.9 eV from different authors [32] [33] . Although, when the 2H-Mo(1−x)W(x)S2-Ni our result yield a soft metal behavior as provided in Figure 2(b). Notice also, that only two bands (multiple degenerate crosses the Fermi level. Due that we have experience with similar systems like 2H-MoS2 nanoparticles grown over Reduced graphene oxide [34] [35] we propose that the most likely place to locate the 2H-Mo(1−x)W(x)S2-Ni above O14-O19 defect and 2 Å above the graphene sheet.
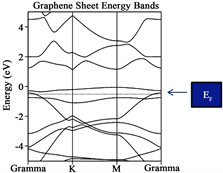
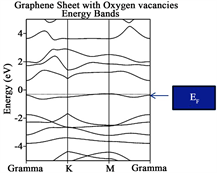
When the graphene sheet was constructed, see Figure 2(c), the system behaves as a semiconductor with a measured Eg~0.90 eV. Also notice that three bands are located in the vicinity of the Fermi level, the top band is almost flat indicating small interaction between the atoms and small velocity. When the Oxygen vacancies are introduced into the graphene sheet C14-C19 substituted by oxygen atoms, a similar scenario appears see Figure 2(d), the forbidden Energy gap is wide open with a reported Eg~1.29 eV. Notice that only two bands are located in the vicinity of the Fermi level and one of the bands barely touches it being almost a flat band. Last, Figure 2(e) corresponds to graphene with oxygen defect (Vacancies) and with 2H-Mo(1−x)W(x)S2-Ni over it. A measured Energy gap of the
Table 2. Analysis for the forbidden energy gap Eg (eV), Fermi level location (eV) and behavior for each configuration under investigation.
 (a)
(a)
 (b)
(b)
 (c)
(c)
 (d)
(d)
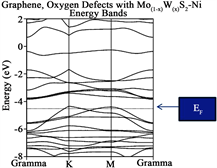 (e)
(e)
Figure 2. (a) corresponds to Band structure calculations for Graphene original, the Fermi level is indicated by an arrow; (b) corresponds to 2H-Mo(1−x)W(x)S2, the Fermi level is indicated by an arrow; (c) corresponds to Graphene sheet, the Fermi level is indicated by a horizontal arrow; (d) yields Graphene sheet with oxygen vacancies, the Fermi level is again indicated by an arrow; (e) yields indication of Graphene sheet with vacancies (defect) with 2H-Mo(1−x)W(x)S2-Ni cluster grown over it, as the former cases, Fermi level is indicated by a horizontal arrow.
order of 0.98 eV is reported providing a semiconductor behavior. Two bands touch the Fermi level and two or more bands (multiple degenerated) form the forbidden gap. The top bands are almost flat indicating the slower velocity provided by the electrons that form the band.
A very important fact considered when the Dirac cone is careful monitored throughout the research when it travels up or down with respect of the Fermi level establish a charge transfer between the metal surface and the graphene. When the cone travels up electrons are provided to the system under consideration, while the cone travels down, vacancies are added instead. Let us concentrate in the location of the Fermi level for graphene (original). Notice that it is located at −11.2450 eV, and take this value as reference. When the graphene sheet was constructed, the Fermi level was located at 0.90 eV. The Fermi level traveled up a clear indication that electrons was provided to the new system. In addition, for graphene sheet with oxygen defect, the Fermi level is shifted up to 1.29 eV an indication similar to the former case. When the 2H-Mo(1−x)W(x)S2-Ni cluster was located over the graphene sheet with oxygen vacancies, the location of the Fermi level is shifted down to 0.98 eV. This fact indicates that vacancies were added to the graphene sheet. This important fact allows us to predict in advance, new materials suited for special purposes.
4. Total and Partial Density of States
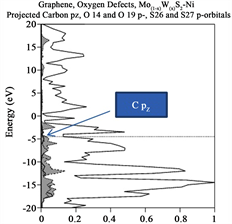
Due that we also are interested in identifying which atoms contributes most to the Total DOS, a Partial DOS analysis in order to see which atoms contribute to those orbitals close to the Fermi level was performed. This information is provided in Table 2 and arises from Figures 3(a)-(e), respectively, for each case enunciated formerly in the manuscript. Each figure provides Energy (eV) vs percentage of contribution, considering that the highest peak is 100% and the other peaks are rescaled with respect to it. The Fermi level is indicated by a horizontal dotted line, while the selected projected contribution from each atom is indicated by a hatched line and the Total DOS is indicated by a solid line in the respectively figure. Due that we are interesting in monitor the carbon pz orbital contribution, and indicated by an arrow, in Figure 3(a) provides information regarding graphene original structure. Notice that carbon pz orbitals contribute with 36% to the total DOS. It is necessary to underline, that we have projected only 6 out of 24 Carbon atoms, just to save time. This contribution appears in the band bellow the Fermi level, due to its semiconductor behavior. In order to obtain the information provided in Table 3, it is necessary to underline that for each structure under investigation, a projected DOS was calculated for the atoms that form such structure like the structures provided on the next paragraph. Henceforth, Mo d-, p- and s-orbitals, W d-, p- and s-orbitals and Ni d-, p- and s-orbitals were calculated separately, and from each graph the contribution from each orbital to the Total DOS was obtained. Table 3 provides information regarding the contribution (in %) to the total DOS from each atom in the selected structure, concentrating only on 2H-MoS2 (original) and 2H-Mo(1−x)W(x)S2-Ni. Notice that for the first case, 2H-MoS2 mo d- and S p- contribution is of the order
Table 3. Contribution (in %) to the total DOS from each atom in the selected structure. The analysis was performed in the vicinity of the Fermi level.
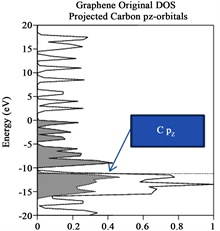 (a)
(a)
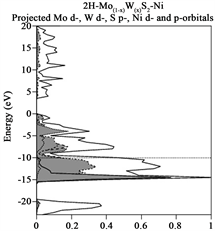 (b)
(b)
 (c)
(c)
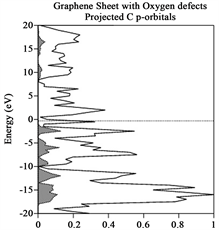 (d)
(d)
 (e)
(e)
Figure 3. (a)-(e) Total (solid line) and projected (hatched lines) DOS for the cases enunciated in the former paragraph. (a) yields indication of Total and projected DOS Graphene original, notice that for some cases Carbon pz is indicated by an arrow; (b) yield information regarding Total and Projected DOS for 2H-Mo(1−x)W(x)S2, Graphene sheet; (c) Total and Projected DOS for Graphene sheet with oxygen vacancies, notice that Carbon pz DOS is highlighted. (d) and (e) yields Total and Projected Carbon pz DOS for Graphene sheet with vacancies (defect) and with 2H-Mo(1−x)W(x)S2-Ni cluster grown over it, respectively.
of <1% due that these values were considered to those orbitals in the vicinity of the Fermi level. On the other hand for the second case, 2H-Mo(1−x)W(x)S2-Ni Mo d-, S p- and W d-contributions are 12%, 18% and 14%. A considerable enhancement obtained in the new structure.
Using the reported results provided by Topsøe et al. [36] [37] [38] [39] [40] from the University of Aarhus for the existence of metallic one dimensional states on MoS2 clusters which established a new view of catalytic active site which yielded new concepts used in catalysis. This new concept indicates that whenever you will have ordinary 2H-MoS2 with different crystalline 1T-MoS2 the catalysis is increased considerably. Afterwards, recently has been reported a catalyst of MoS(2+x) (x~0.5) [41] which yielded selectivity toward hydrogenation when compared with the activity of a reference industrial and other prepared catalysts which contained one-dimensional structures. This indication provides support that the new structure presented could be considered a likely candidate for HDS catalysis.
5. Conclusions
The conclusions obtained in this investigation are as follow: from the Energy band analyses, the original graphene yields indication for a “zero: gap semiconductor, as expected. In addition for graphene sheet, graphene sheet with oxygen vacancies (defect) and graphene with vacancies and with 2H-Mo(1−x)W(x)S2-Ni located over the defect of the Eg ranging between 0.90, 1.29 and 0.98 eV respectively. On the other hand, for the new proposal material 2H-Mo(1−x)W(x)S2 yielded indication for a soft metal.
Total and projected DOS analysis on each case enunciated formerly, provided information on the contributions from each orbital from each atom to the total DOS. A considerable enhancement on the contributions from Mo-, S p- and W d-orbitals was obtained for 2H-Mo(1−x)W(x)S2-Ni when compared to 2H-MoS2 original. This fact provides indication that the new hybridized set of orbitals could indicate that the new material could be exploited toward a catalytic material in the HDS process.
Furthermore, sulphur atoms adjacent to the graphene sheet form an electronic cloud sheared between these two structures generating that the electronic conduction increased. The active sites from 2H-Mo(1−x)W(x)S2-Ni will perform as attractive centers for the charge carriers and take them from it to the graphene sheet with a greater facility.
Acknowledgements
D. H. Galvan acknowledges Departamento de Supercomputo, Universidad Nacional Autonoma de Mexico, for providing CPU time in order to perform this research. Proyecto SC16-1-IG-4.
Cite this paper
Galvan, D.H., Antunez-Garcia, J., Fuentes, S. and Shelyapina, M. (2018) Electronic Properties of Mo(1−x)W(x)S2-Ni Grown over Graphene. Open Access Library Journal, 5: e4335. https://doi.org/10.4236/oalib.1104335
References
- 1. Eknapakul, T., King, P.D.C., Asakawa, M., Buaphet, P., He, R.-H., Mo, S.-K., Takagi, H., Shen, K.M., Baumberger, F., Sasagawa, T.T., Jungthawan, S. and Meevasana, W. (2014) Electronic Structure of Quasi-Freestanding MoS2 Monolayer. Nano Letters, 14, 1312-1316. https://doi.org/10.1021/nl4042824
- 2. Gong, Y., Liu, Z., Lupini, A.R., Shi, G., Lin, J., Najmaei, S., Lin, Z., Elías, A.L., Berkdemír, A., You, G., Terrones, H., Terrones, M., Vajtai, R., Pantelides, S.P., Pennycook, S.J., Lou, J., Zhou, W. and Ajayan, P.M. (2014) Band Gap Engineering and Layer-by-Layer Mapping, of Selenium-Doped Molybdenum Disulfide. Nano Letters, 14, 442-449. https://doi.org/10.1021/nl4032296
- 3. Castellanos-Gomez, A., Poot, M., Steel, G.A., Van der Zant, H.S.J., Agrait, N. and Rubio-Bollinger, G. (2012) Elastic Properties of Freely Expanded MoS2 Nanosheet. Advanced Materials, 24, 772-775. https://doi.org/10.1002/adma.201103965
- 4. Yadgarov, L., Choi, C.L., Sedova, A., Cohen, A., Rosentsveig, R., Bar-Elli, O., Oron, D., Dai, H. and Tenne, R. (2014) Dependence of the Absorption and Optical Surface Plasmon Scattering of MoS2 Nanoparticles on Aspect Ratio, Size and Media. ACS Nano, 8, 3575-3583. https://doi.org/10.1021/nn5000354
- 5. Yan, Y., Xia, B., Xu, Z. and Wang, X. (2014) Recent Development of Molybdenum Sulfides as Advanced Electrocatalysts for Hydrogen Evolution Reaction. ACS Catalysis, 4, 1693-1705.
- 6. Chianelli, R.R., Siadati, M.H., Perez de la Rosa, M., Berhault, G., Wilcoxon, J.P., Bearden Jr., R. and Abrams, B.L. (2006) Catalytic Properties of Single Layers of Transition Metal Sulfides Catalytic Materials. Catalysis Reviews, 48, 1-41. https://doi.org/10.1080/01614940500439776
- 7. Donath, E.E. (1982) History of Catalysis in Coal Liquafactions. In: Anderson, J.R. and Boudart, M., Eds., Catalysis, Vol. 3, Springer, Berlin, p. 1-37.
- 8. Song, C. (2003) An Overview of New Approach to Deep Desulfurization for Ultra-Clean Gasoline, Disel Fuel and Jet Fuel. Catalysis Today, 86, 211-263. https://doi.org/10.1016/S0920-5861(03)00412-7
- 9. Nishimura, S. (2001) Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis. John Wiley & Sons, New York.
- 10. Wei, X.-L., Zhang, H., Guo, G.-C., Li, X.-B., Lau, W.-M. and Lieu, L.-M. (2014) Modulating the Atomic and Electronic Structures through Alloying and Heterostructure of Single-Layer MoS2. Journal of Materials Chemistry, A2, 2101-2109. https://doi.org/10.1039/C3TA13659K
- 11. Soled, S.L., Miseo, S., Krikak, R., Vroman, H., Ho, T.H. and Riley, K.L. (2001) US Patent 6 299760 [1B], to Exxon Research and Engineering Company.
- 12. Hein, J., Gutierrez, O.Y., Schachtl, E., Xu, P., Browning, N.D., Jentys, A. and Lercher, J.A. (2015) Distribution of Metal Cations in Ni-Mo-W Sulfide Catalysts. ChemCatChem Catalysis, 7, 3692-3704. https://doi.org/10.1002/cctc.201500788
- 13. Olivas, A., Antúnez-García, J., Fuentes, S. and Galvan, D.H. (2014) Electronic Properties of Unsupported Trimetallic Catalysts. Catalysis Today, 220-222, 106-112. https://doi.org/10.1016/j.cattod.2013.09.055
- 14. Zheng, X., Xu, J., Yan, K., Wang, H., Wang, Z. and Yang, S. (2014) Space-Confined Growth of MoS2 Nanosheets within Graphite: The Layered Hybrid of MoS2 and Graphene as an Active Catalyst for Hydrogen Evolution Reaction. Chemistry of Materials, 26, 2344-2353. https://doi.org/10.1021/cm500347r
- 15. Zhao, X., Zhu, H. and Yang, X. (2014) Amorphous Carbon Supported MoS2 Nanosheets as Effective Catalysts for Electrocatalytic Hydrogen Evolution. Nanoscale, 6, 10680-10685. https://doi.org/10.1039/C4NR01885K
- 16. Castro-Neto, A.H., Guinea, F., Peres, N.M.R., Novoselov, K.S. and Geim, A.K. (2009) The Properties of Graphene. Reviews of Modern Physics, 81, 109-162. https://doi.org/10.1103/RevModPhys.81.109
- 17. Pletikosic, I., Kralj, M., Pervan, P., Brako, R., Coraux, J., N’Diaye, A.T., Busse, C. and Michely, T. (2009) Dirac Cones and Minigaps for Graphene on Ir (111). Physical Review Letters, 102, Article ID: 056808. https://doi.org/10.1103/PhysRevLett.102.056808
- 18. Giovannetti, G., Khomyakov, P.A., Brocks, G., Kelly, P.J. and van den Brink, J. (2007) Substrate-Induced Band Gap in Graphene on Boron Nitride: An ab Initio Density Functional Calculations. Physical Review B, 76, Article ID: 073103.
- 19. Khomyakov, P.A., Giovannetti, G., Rusu, P.C., Brocks, G., van der Brink, J. and Kelly, P.J. (2009) First-Principles Study of the Interaction and Charge Transfer between Graphene and Metals. Physical Review B, 79, Article ID: 195425. https://doi.org/10.1103/PhysRevB.79.195425
- 20. Wangbo, M.-H. and Hoffmann, R. (1978) The Band Structure of the Tetracyanoplatinate Chain. Journal of the American Chemical Society, 100, 6093-6098. https://doi.org/10.1021/ja00487a020
- 21. Hoffmann, R. (1963) An Extended Hückel Theory. I. Hydrocarbons. The Journal of Chemical Physics, 39, 1397-1412. https://doi.org/10.1063/1.1734456
- 22. Glassey, W.V., Popoian, G.A. and Hoffmann, R. (1999) Total Energy Partitioning with One-Electron Formalism: A Hamilton Population Study of Surface-CO Interaction in the c(2 × 2)-CO/Ni(100) Chemisorption System. The Journal of Chemical Physics, 111, 893-910. https://doi.org/10.1063/1.479200
- 23. Galvan, D.H. (1998) Extended Hückel Calculations on Cubic Boron Nitride and Diamond. Journal of Materials Science Letters, 17, 805-810. https://doi.org/10.1023/A:1006630320896
- 24. Hallmark, V.M., Chiang, S. and Predicting, S. (1995) STM Images of Molecular Adsorbates. Surface Science, 329, 255-268. https://doi.org/10.1016/0039-6028(95)00047-X
- 25. Eisenberg, R. and Gray, H.B. (2011) Noninnocence in Metal Complexes: A Dithiolene Dawn. Inorganic Chemistry, 50, 9741-9751. https://doi.org/10.1021/ic2011748
- 26. Ghosh, R.K. and Mahapatra, S. (2013) Direct Band-to-Band Tunneling in Reversed Biased MoS2 Nanoribbon p-n Junctions. IEEE Transactions on Electron Devices, 60, 274-279. https://doi.org/10.1109/TED.2012.2226729
- 27. Galvan, D.H., Fuentes-Moyado, S., Estrada-Cruz, J.F.D.R. and Shelyapina, M.G. (2016) Electronic Properties of 1H-MoS2 Clusters Grown on Graphene Oxide. International Journal of Nanotechnology, 13, 60-72. https://doi.org/10.1504/IJNT.2016.074523
- 28. Fleischauer, P.D., Lince, R., Bertrand, P.A. and Bauer, R. (1989) Electronic Structure and Lubrication Properties of Molybdenum Disulfide: A Quantitative Molecular Orbital Approach. Langmuir, 5, 1009-1015. https://doi.org/10.1021/la00088a022
- 29. Alvarez, S. (1993) Tables of Parameters for Extended Hückel Calculations. Universitat de Barcelona, Barcelona.
- 30. Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos, S.V., Gregorieva, I.V. and Firzov, A.A. (2004) Electrical Field Effect in Atomically Thin Carbon Films. Science, 306, 666-669. https://doi.org/10.1126/science.1102896
- 31. Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Katsnelson, M.I., Gregorieva, I.V., Dubonos, S.V. and Firsov, A.A. (2005) Two-Dimensional Gas of Massless Dirac Fermions in Graphene. Nature, 438, 197-200. https://doi.org/10.1038/nature04233
- 32. Mattheiss, L.F. (1973) Band Structure of Transition-Metal-Dichalcogenides Layered Compounds. Physical Review B, 8, 3719-3740. https://doi.org/10.1103/PhysRevB.8.3719
- 33. Grand, A.J., Griffiths, T.M., Pitt, G.D. and Yoffe, A.D. (1975) The Electrical Propertries and the Magnitude of the Indirect Gap in the Semiconducting Transition Metal Dichalcogenide Layer Crystals. Journal of Physics C: Solid State Physics, 8, L17-L23. https://doi.org/10.1088/0022-3719/8/1/004
- 34. Estrada-Cruz, J., Fuentes-Moyado, S. and Galvan, D.H. (2015) Energy Bands of the 1H-MoS2 over Reduced Graphene Oxide. Materials Today: Proceedings, 2, 108-112. https://doi.org/10.1016/j.matpr.2015.04.017
- 35. del Rosario Estrada-Cruz, J.F. (2014) Master in Science Disertation, Propiedades electronicas de nano-cumulos de 1H-MoS2 crecido sobre oxido de grafeno, Centro de Investigacion Cientifica y de Educacion Superior de Ensenada, febrero 6.
- 36. Bollinger, M.V., Lauritsen, J.V., Jacobsen, J.W., Norskov, J.K., Helveg, S. and Besenbacher, F. (2001) One-Dimensional Metallic Edge States in MoS2. Physical Review Letters, 87, Article ID: 196803. https://doi.org/10.1103/PhysRevLett.87.196803
- 37. Byskov, L.S., Norskov, J.K., Clausen, B.S. and Topsoe, H. (2000) Edge Termination of MoS2 and CoMoS Catalyst Particles. Catalysis Letters, 64, 95-99. https://doi.org/10.1023/A:1019063709813
- 38. Helveg, S., Lauritsen, J.V., Laegsgaard, E., Stensgaard, I., Norskov, J.K., Clausen, B.S., Topsoe, H. and Besenbacher, F. (2000) Atomic-Scale Structure of Single-Layer MoS2 Nanoclusters. Physical Review Letters, 84, 951-954. https://doi.org/10.1103/PhysRevLett.84.951
- 39. Carlsson, A., Brorson, M. and Topsoe, H. (2004) Morphology of WS2 Nanoclusters in WS2/C Hydrodesulfurization Catalysts Revealed by High-Angle Annular Dark-Field Transmission Electron Microscopy (HAADF-STEM) Imaging. Journal of Catalysis, 227, 530-536. https://doi.org/10.1016/j.jcat.2004.08.031
- 40. Lauritsen, J.V., Nyberg, M., Norskov, J.K., Clausen, B.S., Topsoe, H., Laegsgaard, E. and Besenbacher, F. (2004) Hydrodesulfurization Reaction Pathways on MoS, Nanoclusters Revealed by Scanning Tunneling Microscopy. Journal of Catalysis, 224, 94-106. https://doi.org/10.1016/j.jcat.2004.02.009
- 41. Camacho-Bragado, G.A., Olivas, A., fuentes, S., Galvan, D.H. and José-Yacaman, M. (2005) Structure and Catalytic Properties of Nanostructured Molybdenum Sulfides. Journal of Catalysis, 234, 182-190.
- 42. Wolfsberg, M.W. and Helmholtz, L. (1952) The Spectra and Atomic Structure of the Tetrahedral Ions MnO4, CrO4, and ClO4. The Journal of Chemical Physics, 20, 837-848. https://doi.org/10.1063/1.1700580