Open Access Library Journal
Vol.05 No.02(2018), Article ID:82688,12 pages
10.4236/oalib.1104277
Study on the Interactions of Two Isomer Selaginellins as Novel Small Molecule Inhibitors Targeting PTP1B by Docking and Molecular Dynamics Simulations
Li Wang, Yi Wang, Dan Meng, Honglei Li*
Department of Pharmacy, Kangda College of Nanjing Medical University, Lianyungang, China
Copyright © 2018 by authors and Open Access Library Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: December 20, 2017; Accepted: February 24, 2018; Published: February 27, 2018
ABSTRACT
Protein Tyrosine Phosphatase 1B (PTP1B) plays a key role in down-regulation of insulin and leptin signaling pathways. Therefore, development of novel inhibitors of PTP1B has becoming research focus in reducing blood sugar levels for Diabetes Mellitus and Obesity. Hence, we selected two isomer Selaginellins (1 and 2) to explain and analyze the binding mechanism of the two potential inhibitors against the PTP1B by molecule docking and molecular dynamics simulations. Firstly, the two isomers (1 and 2) and the initial ligand 19 were docked to the receptor of PTP1B by using molecular docking technology. Secondly, taken the 19 as an indicator, molecular dynamic (MD) simulations and molecular mechanics/generalized born surface area (MM/GBSA) methods were requested to assess the binding affinity and complex stability of the two chemicals towards PTP1B. Finally, we explained that the interactions of compound 1 and 2 take up the active binding site PTP1B. Simultaneously, energy decomposition analysis recognized several amino acid residues situated in substrate-binding site that might provide clues for future inhibitor exploitation towards PTP1B. In conclusion, these results may explain the reasons of differences of optical isomer biological activity and provide valuable information for developing novel inhibitors targeting PTP1B-mediated glucose transport and metabolism for Diabetes Mellitus and Obesity therapeutics.
Subject Areas:
Drugs & Devices, Pharmacology
Keywords:
PTP1B, Molecular Docking, Selaginellins, Molecular Dynamics Simulations

1. Introduction
Protein Tyrosine Phosphatase 1B (PTP1B), belongs to a large family of signaling enzymes ,which modulate several fundamental cellular functions by phosphorylation and dephosphorylation reactions that can contribute to treat diabetes mellitus and obesity [1] [2] [3] . PTP1B plays a key role in down regulating of insulin and has caused as a potential target for the treatment of type II diabetes [4] [5] . Developing selective and potent depressors of PTP1B is a major challenge, for its high homogeneity to other cellular PTPs and polar nature of active site pocket. Thus, developing novel suppressants of PTP1B that concurrently occupies both binding active site A and B (the adjacent pTyr binding site) partially addressed the selectivity issue [6] . The hydrophobic property is critical for the chemical’s cell permeability and enhanced by creating compounds that target allosteric binding site (Site-C) with reduced negative charge [7] [8] .
Selaginella tamariscina, a traditional Chinese medicine, has been reported to reduce the blood glucose levels and accelerate the repair the β-cells of pancreatic islet which injured by alloxan [9] [10] . Woo’s team has isolated and identified two isomer natural products (selaginellin (1) and Selariscinin A (2)) from the methanol extract of S. tamariscina as insulin-mimetic inhibitors of PTP1B by significant different enzyme activities in vitro [11] . Some progress has been finished in experimental research by Woo’s, but the theoretical researches on the interactions of the two isomer chemicals toward PTP1B and the structural features influencing their PTP1B inhibitory activity are still undefined.
Molecular modeling techniques likely molecular docking and molecular dynamic (MD) simulations, are useful tools to demystify detailed information of the protein-ligand interactions at the molecular and atomic levels [12] [13] . Molecular docking is common simulation software to predict the experimental binding orientations and affinities of small chemicals within the receptor binding sites [14] [15] . Besides, MD simulation is a useful technology affording vivid pictures to describe the undulations and conformational transitions of protein-ligand complex, and permitting further investigation of the interaction mechanisms of the complex at the atomic levels [16] . Hence, combined studies of molecular docking and MD simulation can provide sharp insight into comprehending the characters of structure of ligand-receptor interactions.
In this study, we chose the two isomer chemicals (1 and 2) acting as PTP1B inhibitors to do a molecular modeling study by utilizing docking methodology and MD simulations to appraise the stability of the PTP1B-ligand complexes. Moreover, molecular mechanics/generalized born surface area (MM/GBSA) method was used to calculate the binding free energy and energy decomposition [17] . Finally, we explained that compound 2 might have the higher affinity to occupy the binding pocket of PTP1B, compared to 1. Overall, our results might afford valuable information and enrich new ideas for developing novel molecule targeting PTP1B in treatment of diabetes mellitus and obesity.
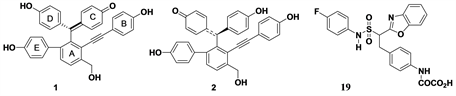
Figure 1. Shows the structures of 1 and 2 from Selaginella tamariscina and 19.
2. Materials and Methods
2.1. Data Preparation
The X-ray crystal complex of PTP1B-IN1834146C (Named: Compound 19 in this study) was downloaded from the database of RCSB Protein Data Bank (PDB) (http://www.pdb.org/pdb/home/home.do), and the PDB code was 2VEU [18] . The structure was prepared in the following procedures: 1) removing the co-crystallized ligand of 19 and structural water molecules from the crystal structure, 2) adding hydrogen atoms to the protein by the Biopolymer module implemented in the SYBYL X1.3, and 3) assigning the Kollman-All atom charges to the protein atoms. Finally, the resultant structure was converted from the PDB format to MOL2 and used for the molecular docking experiments.
The two isomer compounds (1 and 2) were collected for docking in reports by Woo and co-workers [11] , and their structures was shown in Figure 1. The three dimensional structures of the two chemicals were created and optimized in SYBYL X1.3. Structure energy minimization was executed by using the Powell gradient algorithm and the Tripos force fields with a convergence criterion of 0.001 kcal∙mol−1∙Å−1 and a maximum of 1000 iterations. MMFF94 charges were distributed to each chemical compound [19] . The minimized structure was used as the original configuration for molecular docking.
2.2. Molecular Docking
The PTP1B and the candidate compounds (1 and 2) were prepared using SYBYL X 1.3, the fabrication process included addition hydrogen atoms and charges, as well as the elimination of solvent molecules in the receptor of PTP1B. The accuracy of specified parameters docking was validated by re-docking the original co-crystallized ligand of 19 into the binding site of PTP1B, and the value of root mean square deviation (RMSD) was 0.552 Å.
To generate the starting models for PTP1B in complex with two natural chemicals (1 and 2) and one co-crystallized ligand [20] , the docking process was executed to obtain the docking-binding models by using the surflex-dock procedure applied in Tripos SYBYL X1.3. The threshold and bloat values were transferred to 0.5 and 0, severally. Twenty poses of the three ligands were considered in the docking process. The search grid extended 6 Å beyond the protein dimension. Ring flexibility and soft grid treatment were turned on. Docking is regarded successful in case of the RMSD value of the optimum form is less than a given threshold value of 0.1 Å from the crystal pose.
2.3. MD Simulations
To determine the molecular docking results, the MD simulation was executed with AMBER 9.0 software package. The three docking systems (1-2VEU, 2-2VEU, and 19-2VEU) were used as the primal structures for MD simulations. The FF03 AMBER force field and the general AMBER force field (gaff) were used to depict the complexes of protein-ligand, respectively. Each system was neutralized by adding Na+ and solvated in a truncated octahedron box of water molecules with a border distance of 12 Å [21] [22] .
Thereafter, two-phase energy minimizations were performed to avoid feasible steric stress. Firstly, each complex was fixed with a restraint constant of 2.0 kcal∙mol−1∙Å−1 and the hydrone and Na+ were minimized with the steepest descent (SD) method for 2000 steps followed by conjugated gradient (CG) method for 3000 steps. Second, the whole relaxed complexes (1-2VEU, 2-2VEU, and 19-2VEU) were optimized by 5000 steps via SD and CG, respectively. Then each of the protein-ligand system was slowly heated from 0 to 300 K in 200 ps with a weak constraint of 1.0 kcal∙mol−1∙Å−1 at a constant volume and equilibrated for 500 ps at 300 K and 1 atm. Finally, a 5 ns production MD simulation was performed in a NPT (constant composition, T = 300 K and P = 1 atm) ensemble. During these steps, the Particle Mesh Ewald (PME) method was applied to treat the long-range electrostatic interactions with a non-bonded cutoff of 8.0 Å, and the SHAKE algorithm was turned on to constrain all covalent bonds involving hydrogen atoms with 2 fs time step [23] . Coordinated trajectories were recorded per 1 ps and the stability of each complex was checked from the RMSD.
2.4. Binding Free Energy Calculations
In this study, the MM-PBSA approach implemented in the AMBER 9.0 software was applied to estimate the binding free energies ( , kcal/mol) of the protein-ligand systems [24] [25] . For every complex, a total of 200 snapshots of the simulated structures drawed from the last 2 ns stable MD trajectory were used for calculations. The PTP1B-ligand binding free energy was evaluated by Equation (1)
(1)
Gcomplex, GPTP1B, and Gligand are the free energies of the complex, the PTP1B protein, and the ligand, respectively. Each of them can be calculated by Equation (2)
(2)
Where ΔGMM is the molecular mechanics free energy, ΔGsol is the solvation free energy, and −TΔS represents the entropy term. The molecular mechanics free energy is estimated as follows by Equation (3):
(3)
ΔGele and ΔGvdw express the electrostatic and Van der Waals coactions, respectively. The solvation free energy is consisted of two modules by Equation (4).
(4)
ΔGele,sol is the polar contribution to solvation and can be acquired by solving the Poisson-Boltzmann equation for molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method or generalized Born models for MM-GBSA. ΔGnonpol,sol is the nonpolar solvation term and is decided by using Equation (5).
(5)
where γ and β respectively representing the surface tension and constant, were set to 0.0072 kcal/(mol∙Å2) and 0, separately. This was exploited by Still et al. The solvent accessible surface area (SASA) is the solvent accessible surface area (Å2) that is evaluated by using the MSMS algorithm with a probe radius of 1.4 Å [26] . As the calculation of the entropy term was time-consuming and its value seldom converged, the entropy contribution has been omitted in this study.
For distinguishing the contrast of the binding systems of these complexes, the binding free energies were disintegrated to each of residue by using the MMGMSA method. Each inhibitor-residue pair include four energy terms: ΔGvdw, ΔGele, ΔGele,sol and ΔGnonpol,sol, which can be summarized in the following Equation (6):
(6)
where ΔGvdw and ΔGele were computed with the sander program in AMBER 9.0. The polar contribution was decided by the generalized Born (GB) model (GBOBC, igb = 2). The nonpolar contribution was calculated using the SASA [26] [27] .
3. Result and Discussion
3.1. Molecular Docking
To confirm the docking accuracy, the original ligand compound 19 in the crystal structure was docked back into the binding active site of PTP1B. The detailed structures of the compound 19 in the binding site predicted by the above mentioned studies are shown in Figures 2(A)-(B). Figure 2(A) indicates that compound 19 interacts extensively with PTP1B in a defined manner such as the phenyl carbamoyl group occupies the active site and interacts with the catalytic (Site-A) residues,while benzoxazole moiety is positioned at second aryl phosphate binding (Site-B) residues. Figure 2(B) shows the phenyl carbamoyl group of the compound 19 immerges into a pocket (Site-A)constructed and the oxygen of the carbamoyl group interacts with residues of Ala-217, Ser-216, Gly-218, Asp-181, Cys-215, Arg-221 and Phe-182 thought eight hydrogen-bonds (3.0 Å, 3.3 Å, 3.3 Å, 3.2 Å, 2.9 Å, 3.2 Å, 3.2 Å and 3.5 Å, respectively). Similarly extensive interaction with Asp-48 with the amine group in Site-B by one hydrogenbond (3.0 Å). And the docking results are consistent with the experimental results [18] . This result showed that the docking technique and parameters used in the present study were similar in the PTP1B system.
To clarify the interaction mechanism of the two natural compounds (1 and 2), the average structures were produced by averaging 100 snapshots from the last
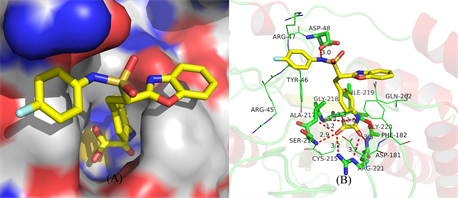
Figure 2. (A)-(B) show docking structure of compound 19 and the corresponding surface of PTP1B(A). Interaction between the PTP1B active binding site and compound 19(B). Hydrogen bonds are depicted by red dotted lines.
1ns MD trajectories. The binding modes of the two isomers (1 and 2) in the active pocket of PTP1B are given in Figures 3(A)-(D).
From Figures 3(A)-(B), it is can be seen that the detailed structure of the compound 1 in the binding site A and B predicted by the above mentioned studies, compare to compound 19, the compound 1 interacts extensively with PTP1B in a defined manner such that the phenol group (A-ring) occupies the active site and interacts with the catalytic (Site-A) residues, while the phenol group (D-ring) is positioned binding with residues Arg-47 (Site-B) by one hydrogen-bond (3.1 Å). We find that the B-ring of compound 1 immerges into a pocket (Site-A) constructed by amino acid residues and interacting with Ala-217, Asp-181, Arg-221 and Phe-182 thought by two hydrogen-bonds (3.5 Å, 3.1 Å), the hydroxyl of D-ring and A-ring group in compound 1 are hydrogen bonded to Arg-47 (3.1 Å) in Site-B, Gln-262 (3.0 Å), respectively. Although the compound 2 docking results at the same binding active site of PTP1B from Figures 3(C)-(D), we can find the difference between compound 1 and 2, the hydroxyl of E-ring group is hydrogen bonded to Asp-181 in Site-A by two hydrogen-bonds (3.0 Å, 3.1 Å), and the hydroxyl of A-ring group interacts with Arg-47 by two hydrogen-bonds (3.0 Å, 3.2 Å), while the hydroxyl of D-ring in compound 2 binding to Gln-262 by one hydrogen-bond (3.4 Å). Above all, the hydrogen bonds acquired from the molecular docking proved important information on the binding patterns of ligand-receptor (1-2VEU, 2-2VEU and 19-2VEU), further insights are to be revealed through the following MD simulations.
3.2. MD Simulations
To further study the detailed ligand-receptor interactions in the binding procedure, the three docking complexes (1-2VEU, 2-2VEU and 19-2VEU) were executed for 5 ns MD simulations. The RMSD plots show that the system is stable throughout the MD simulations in Figure 4.
In Figure 4, the complex of 19-2VEU reach equilibrium around 1 ns, whereas

Figure 3. (A)-(B) show docking structures of compound1 and the corresponding surface of PTP1B(A). Interaction between the PTP1B binding active site and compound 1(B). (V)-(D) Docking structures of compound 2 and the corresponding surface of PTP1B(C). Interaction between the PTP1B binding active site and compound 2(D). Hydrogen bonds are described by red dotted lines.

Figure 4. shows the RMSD of the backbone atoms of the complexes (1-2VEU, 2-2VEU and 19-2VEU) during MD simulations, in which the black line symbolize the system of compound 19, while blue lines and red line indicate the system of compound 1 and 2, respectively.
the others achieve equilibrium around after 0.5 ns. The mean RMSD values of the binding modes were 1.2 Å, 1.3 Å and 1.1 Å, severally. And the relative RMSD fluctuations were very small, suggesting that the systems were stable during the MD simulations.
In order to check the amino acid residue contribution of the receptor in the binding procedure, results of the analyses of the root-mean-square fluctuation (RMSF) versus the residue number for the three systems (1-2VEU, 2-2VEU and 19-2VEU) were expounded in Figure 5. We can find that the protein structures of three complexes in each graph display the similar RMSF allocations and similar trends of dynamic characteristics from Figure 5. For instance, the active site, especially the regions around residues Tyr-47, Asp-48, Val-49, Asp-181, Phe-182, IIe-219 and Gln-262, has a larger conformational drift for 2-2VEU system than that for the 1-2VEU, suggesting that 2 should have a more stabilized interaction with the receptor than 1. Over all, these analyses results of binding stabilization shows consistent with the experimental activities.
3.3. Binding Free Energy Analysis
To appraise the binding stability of the complexes, the binding free energy calculations of each binding system were executed by the MM-PBSA program in the software of AMBER 9.0. From Table 1, the calculated ΔGbind value for the 2-2VEU complex (−23.15 kcal/mol) was lower than the value of the 1-2VEU (−10.11 kcal/mol), nearly the value of the 19-2VEU (−28.74 kcal/mol), advising that 2 can form stronger binding to the receptor of PTP1B than 1, which was in

Figure 5. Shows the RMSF of every residue of the protein for the three complexes (1-2VEU, 2-2VEU and 19-2VEU).

Table 1. Shows binding free energy (kcal/mol) of compounds 1, 2 and 19 complexes.
accordance with the order of experimental activities. As shown in Table 1, the Van der Waals interactions (ΔGvdw) and the electrostatic interactions (ΔGele) make a significant contribution to the binding interaction, and there are a great difference in Van der Waals and electrostatic energies between 1 and 2. The nonpolar solvation free energy (ΔGnp) mildly favor the affinity, while the polar solvation free energy (ΔGp) opposes the binding strongly. From the above results, it can be further validated that the hydrophobic interaction and hydrogen bond play a conclusive role for establishing the ligand to the receptor.
To obtain further insight into the detailed ligand-receptor interactions, the binding free energy in the three complexes was decomposed to individual residue located within 6 Å of the ligand by using the MM-GMSA approach. From Figure 6, it can be seen that almost all residues energetically contribute more for the binding of compound 2 than that of compound 1, especially residues (large than 0.5 kcal/mol) Tyr-47, Asp-48, Val-49, Asp-181, Phe-182, IIe-219, Gly-220, Arg-221 and Gln-266, and the two isomer compounds have the similarly free energy decomposition plots with compound 19. As most of the previous residues are nonpolar, it is evident that ligands can form strong Van der Waals coactions with these amino acid residues and these Van der Waals interactions make more contributions to the binding free energy. The amino acid residues of ASP-181, Arg-47 and Gln-262 make strong contacts with the two isomers not only via Van der Waals interactions but also by forming H-bonds with 2VEU. Simultaneously, it is awared that almost all residues energetically contribute more for the binding of compound 2 than 1, suggesting that the interactions of compound 2 with 2VEU is stronger than compound 1. From these results, we can infer that hydrophobic interactions and hydrogen bond play a critical role in the binding affinity of the inhibitors to the receptor of PTP1B.
4. Conclusions
In this study, molecular docking, MD simulations and binding free energy calculations were performed to explain the interactions mechanism of the two isomer compounds (1 and 2) to PTP1B. After forming five hydrogen bonds with Arg-47, Asp-181 and Gln-262, compound 2 is in close to hydrophobic residues

Figure 6. shows free energy distribution plots for the three complexes (1-2VEU, 2-2VEU and 19-2VEU).
like Asp-48, Val-49, Phe-182, Ala-217, Gly-220, Ile-219 and Arg-221, result in stronger inhibitory affinity than 1. It was notarized by the subsequent binding free energy analysis for the two complexes. Furthermore, enhancing stacking coactions with site A may be helpful to raise the inhibitory affinities of newly designed inhibitors.
The predicted ΔGbind values are −10.11, −23.15 and −28.74 kcal/mol for the 1-2VEU, 2-2VEU and 19-2VEU complexes, respectively, which is agreed with the inhibitory abilities as expected. Between the predicted ΔGbind and the experimental ΔGbind,exp approximately estimated from the IC50 values, [11] advises that the predicted ΔGbind for PTP1B/inhibitor systems can be used to assess the experimental inhibitory abilities for novel PTP1B inhibitors of similar structural features.
The results obtained from these computational approaches revealed the binding modes of the two ligand-receptor complexes and the main interaction mechanisms, and it can be useful for the optimization of new-style PTP1B inhibitors based on selaginellins and other similar natural products.
Acknowledgements
This work is supported by the Construction Foundation of National Innovative City of Lianyungang in 2015 (No. SH 1514) and the Research Foundation of Kangda College of Nanjing Medical University (No. KD2015KYJJZD001 & KD2017KYJJZD004).
Cite this paper
Wang, L., Wang, Y., Meng, D. and Li, H.L. (2018) Study on the Interactions of Two Isomer Selaginellins as Novel Small Molecule Inhibitors Targeting PTP1B by Docking and Molecular Dynamics Simulations. Open Access Library Journal, 5: e4277. https://doi.org/10.4236/oalib.1104277
References
- 1. Asante-Appiah, E. and Kennedy, B.P. (2003) Protein Tyrosine Phosphatases: The Quest for Negative Regulators of Insulin Action. American Journal of Physiology Endocrinology and Metabolism, 284, E663-E670.
https://doi.org/10.1152/ajpendo.00462.2002 - 2. Lund, I.K., Hansen, J.A., Andersen, H.S., Moller, N.P.H. and Billestrup, N. (2005) Mechanism of Protein Tyrosine Phosphatase 1B-Mediated Inhibition of Leptin Signalling. Journal of Molecular Endocrinology, 34, 339-351.
https://doi.org/10.1677/jme.1.01694 - 3. Tonks, N.K. and Neel, B.G. (2001) Combinatorial Control of the Specificity of Protein Tyrosine Phosphatases. Current Opinion in Cell Biology, 13, 182-195.
https://doi.org/10.1016/S0955-0674(00)00196-4 - 4. Blume-Jensen, P. and Hunter, T. (2001) Oncogenic Kinase Signaling. Nature, 411, 355-365.
https://doi.org/10.1038/35077225 - 5. Thareja, S., Aggarwal, S., Bhardwaj, T.R. and Kumar, M. (2012) Protein Tyrosine Phosphatase 1B Inhibitors: A Molecular Level Legitimate Approach for the Management of Diabetes Mellitus. Med Res Rev., 32, 459-517.
https://doi.org/10.1002/med.20219 - 6. Zhang, Z.Y. (2002) Protein Tyrosine Phosphatases: Structure and Function, Substrate Specificity, and Inhibitor Development. Annual Review of Pharmacology and Toxicology, 42, 209-234.
https://doi.org/10.1146/annurev.pharmtox.42.083001.144616 - 7. Lau, C.K., Bayly, C.I., Gauthier, J.Y., Li, C.S., Therien, M., Asante-Appiah, E., Cromlish, W., Boie, Y., Forghani, F., Desmarais, S., Wang, Q., Skorey, K., Waddleton, D., Payette, P., Rama-chandran, C., Kennedy, B.P. and Scapin, G. (2004) Structure Based Design of a Series of Potent and Selective Non Peptidic PTP-1B Inhibitors. Bioorganic & Medicinal Chemistry Letters, 14, 1043-1048.
https://doi.org/10.1016/j.bmcl.2003.11.076 - 8. Dufresne, C., Roy, P., Wang, Z., Asante-Appiah, E., Cromlish, W., Boie, Y., Forghani, F., Desmarais, S., Wang, Q., Skorey, K., Waddleton, D., Ramachandran, C., Kennedy, B.P., Xu, L., Gordon, R., Chan, C.C. and Leblanc, Y. (2004) The Development of Potent Non-Peptidic PTP-1B Inhibitors. Bioorganic & Medicinal Chemistry Letters, 14, 1039-1042.
https://doi.org/10.1016/j.bmcl.2003.11.048 - 9. Zheng, X.K., Zhang, L., Wang, W.W., Wu, Y.Y., Zhang, Q.B. and Feng, W.S. (2011) Anti-Diabetic Activity and Potential Mechanism of Total Flavonoids of Selaginella tamariscina (Beauv.) Spring in Rats Induced by High Fat Diet and Low Dose STZ. Journal of Ethnopharmacology, 137, 662-668.
https://doi.org/10.1016/j.jep.2011.06.018 - 10. Zheng, X.K., Wang, W.W., Zhang, L., Su, C.F., Wu, Y.Y., Ke, Y.Y., Hou, Q.W., Liu, Z.Y., Gao, A.S. and Feng, W.S. (2013) Antihyperlipidaemic and Antioxidant Effect of the Total Flavonoids in Selaginella tamariscina (Beauv.) Spring in Diabetic Mice. J Pharm Pharmacol., 65, 757-766.
https://doi.org/10.1111/jphp.12035 - 11. Nguyen, P.H., Zhao, B.T., Ali, M.Y., Choi, J.S., Rhyu, D.Y., Min, B.S. and Woo, M.H. (2015) Insulin-Mimetic Selaginellins from Selaginella tamariscina with Protein Tyrosine Phosphatase 1B (PTP1B) Inhibitory Activity. Journal of Natural Products, 78, 34-42.
https://doi.org/10.1021/np5005856 - 12. Lu, Q., Cai, Z., Fu, J., Luo, S., Liu, C., Li, X. and Zhao, D. (2014) Molecular Docking and Molecular Dynamics Studies on the Interactions of Hydroxylated Polybro-minated Diphenyl Ethers to Estrogen Receptor Alpha. Ecotoxicology and Environmental Safety, 101, 83-89.
https://doi.org/10.1016/j.ecoenv.2013.12.018 - 13. Podder, A., Pandey, D. and Latha, N. (2016) Investigating the Structural Impact of S311C Mutation in DRD2 Receptor by Molecular Dynamics & Docking Studies. Biochimie, 123, 52-64.
https://doi.org/10.1016/j.biochi.2016.01.011 - 14. Selvam, C., Jachak, S.M., Thilagavathi, R. and Chakraborti ,A.K. (2005) Design, Synthesis, Biological Evaluation and Molecular Docking of Curcumin Analogues as Antioxidant, Cyclooxygenase Inhibitory and Anti-Inflammatory Agents. Bioorganic & Medicinal Chemistry Letters, 15, 1793-1797.
https://doi.org/10.1016/j.bmcl.2005.02.039 - 15. Qin, H.-L., Shang, Z.-P., Jantan, I., Tan, O.U., Hussain, M.A., Sher, M. and Bukhari, S.N.A. (2015) Molecular Docking Studies and Biological Evaluation of Chalcone Based Pyrazolines as Tyrosinase Inhibitors and Potential Anticancer Agents. RSC Advances, 5, 46330-46338.
https://doi.org/10.1039/C5RA02995C - 16. Zhou, R., Berne, B.J. and Germain, R. (2001) The Free Energy Landscape for β Hairpin Folding in Explicit Water. Proceedings of the National Academy of Sciences of the United States of America, 98, 14931-14936.
https://doi.org/10.1073/pnas.201543998 - 17. Wan, Z.N., Li, X., Sun, R., Li, Y.Y., Wang, X.Y., Li, X.R., Rong, L., Shi, Z. and Bao, J.L. (2015) Computer-Assisted Identification of Novel Small Molecule Inhibitors Targeting GLUT1. Journal of Molecular Structure, 1101, 57-65.
https://doi.org/10.1016/j.molstruc.2015.07.074 - 18. Reddy, M.V.V.V.S., Ghadiyaram, C., Panigrahi, S.K., Krishnamurthy, N.R., Hosahalli, S., Chandrasekharappa, A.P., Manna, D., Badiger, S.E., Dubey, P.K. and Mangamoori, L.N. (2014) X-Ray Structure of PTP1B in Complex with a New PTP1B Inhibitor. Protein & Peptide Letters, 21, 90-93.
https://doi.org/10.2174/09298665113209990089 - 19. Ma, S., Zeng, G., Fang, D., Wang, J., Wu, W., Xie, W., Tan, S. and Zheng, K. (2015) Studies of N9-Arenthenyl Purines as Novel DFG-In and DFG-Out Dual Src/Abl Inhibitors Using 3D-QSAR, Docking and Molecular Dynamics Simulations. Molecular BioSystems, 11, 394-406.
https://doi.org/10.1039/C4MB00350K - 20. Jain, A.N. (2007) Surflex-Dock 2.1: Robust Performance from Ligand Energetic Modeling, Ring Flexibility, and Knowledge-Based Search. Journal of Computer-Aided Molecular Design, 21, 281-306.
https://doi.org/10.1007/s10822-007-9114-2 - 21. Bertran, O., Zhang, B., Schlueter, A.D., Halperin, A., Kroeger, M. and Aleman, C. (2013) Computer Simulation of Dendronized Polymers: Organization and Characterization at the Atomistic Level. RSC Advances, 3, 126-140.
https://doi.org/10.1039/C2RA22034B - 22. Salomon-Ferrer, R., Case, D.A. and Walker, R.C. (2013) Previous Article in Issue: State-Specific Multireference Coupled-Cluster Theory. Wiley Interdisciplinary Reviews: Computational Molecular Science, 3, 198-210.
https://doi.org/10.1002/wcms.1121 - 23. Batcho, P.F., Case, D.A. and Schlick, T. (2001) Optimized Particle-Mesh Ewald/ Multiple-Time Step Integration for Molecular Dynamics Simulations. The Journal of Chemical Physics, 115, 4003-4018.
https://doi.org/10.1063/1.1389854 - 24. Kollman, P.A., Massova, I., Reyes, C., Kuhn, B., Huo, S.H., Chong, L., Lee, M., Lee, T., Duan, Y., Wang, W., Donini, O., Cieplak, P., Srinivasan, J., Case, D.A. and Cheatham, T.E. (2000) Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Accounts of Chemical Research, 33, 889-897.
https://doi.org/10.1021/ar000033j - 25. Mhlongo, N.N. and Soliman, M.E.S. (2015) Single H5N1 Influenza A Neuraminidase Mutation Develops Resistance to Oseltamivir Due to Distorted Conformational and Drug Binding Landscape: Multiple Molecular Dynamics Analyses. RSC Advances, 5, 10849-10861.
https://doi.org/10.1039/C4RA13494J - 26. Hayes, J.M., Skamnaki, V.T., Archontis, G., Lamprakis, C., Sarrou, J., Bischler, N., Skaltsounis, A.-L., Zographos, S.E. and Oikonomakos, N.G. (2011) Kinetics, in Silico Docking, Molecular Dynamics, and MM-GBSA Binding Studies on Prototype Indirubins, KT5720, and Staurosporine as Phosphorylase Kinase ATP-Binding Site Inhibitors: The Role of Water Molecules Examined. Proteins, 79, 703-719.
https://doi.org/10.1002/prot.22890 - 27. Roe, D.R., Okur, A., Wickstrom, L., Hornak, V. and Simmerling, C. (2007) Secondary Structure Bias in Generalized Born Solvent Models: Comparison of Conformational Ensembles and Free Energy of Solvent Polarization from Explicit and Implicit Solvation. The Journal of Physical Chemistry B, 111, 1846-1857.
https://doi.org/10.1021/jp066831u