 American Journal of Anal yt ical Chemistry, 2011, 2, 795-808 doi:10.4236/ajac.2011.27091 Published Online November 2011 (http://www.SciRP.org/journal/ajac) Copyright © 2011 SciRes. AJAC Comparison of Chromatographic Performance for L-Phenylalanine-Derived Organic Phases on Silica by “Grafting fro m ” a n d “Grafting to” Strategies Mohammed Mizanur Rahman1,2*, Makoto Takafuji2, Hirotaka Ihara2 1Department of Applied Chemistry and Chemical Engineering, Faculty of Engineering and Technology, University of Dhaka, Dhaka, Bangladesh 2Department of Applied Chemistry and Biochemistry, Faculty of Engineering, Kumamoto University, Kumamoto, Japan E-mail: *mizan607@gmail.com Received April 30, 2011; revised June 7, 2011; accepted June 24, 2011 Abstract L-phenylalanine-derived polymerizable organogel, N’-octadecyl-Nα-(4-vinyl)-benzoyl-L-phenylalanineami- de (4) has been prepared according to the procedure described elsewhere. Compound 4 was successfully po- lymerized by surface initiated atom transfer radical polymerization (ATRP) from the initiator grafted silica particles (sil-poly4). It was also telomerized with 3-mercaptopropyltrimethoxysilane (MPS) and the telomer (T4) was grafted on to silica (sil-T4). TGA and elemental analysis measurement revealed that higher amount of polymer can graft by ATRP process than that of “grafting to” strategy. The results of 13C CP/MAS NMR measurement showed that the N-alkyl chain of the grafted polymers for both sil-poly4 and sil-T4 remained as less ordered gauche conformational form on silica surface and no inversion to trans form was occurred until temperature is increased up to 50˚C. The retention of alkylbenzene samples showed that sil-poly4 prepared by “grafting from” method yielded extremely higher retention than conventional C18 phase however, sil-T4 prepared by conventional “grafting to” method showed lower retention than C18 phase. Aspects of molecular recognition were evaluated by the retention studies of a series of polycyclic aromatic hydrocarbons (PAHs) and aromatic positional isomers. We have observed sil-T4 yielded slightly higher selectivity for PAHs than sil-poly4 regardless the fact that it has low surface coverage and lower hydrophobic interactions. The en- hanced selectivity observed for sil-T4 than C18 phases and sil-poly4 can be explained by the π - π interactions between the guest PAHs and carbonyl groups present in the polymer chain. In addition the aromatic moieties of compound 4 that aggregates through π - π interactions also contribute to the separation of PAHs for both sil-poly4 and sil-T4. The minimal π - π interactions between the carbonyl groups and guest molecules for sil-poly4 probably due to the presence of long chain initiator which restrict the polymer to form order thin layer over silica surface. Keywords: Polymerizable Organogel, Atom Transfer Radical Polymerization, Grafting from, Grafting to, HPLC, Molecular Recognition 1. Introduction Polymeric thin films are of increasing importance be- cause of their applications to nonlinear optical materials [1], microfabrication [2], biocompatible medical implants [3], and capillaries for electrophoresis [4-7]. In all appli- cations, controlling the degree of the polymerization is an issue in the preparation of these films, and the inabil- ity to control growth of polymer films has diminished their applicability to porous silica gel for liquid chroma- tographic stationary phases. Stationary phases prepared by surface modification of porous silica with organic interactive layers are the foundation of modern liquid chromatography [8]. Preparation of bonded stationary phases based on silica has been carried out almost exclu- sively by the “grafting to” technique, by attachment of alkyl chains (typically octadecyl) to porous silica parti- cles through silane coupling reactions. This method is to  M. M. RAHMAN ET AL. 796 graft polymers to the surface by forming a covalent bond between the polymer and the surface. This has been ac- complished in many ways, including the use of thiol- derivatized polymer for grafting to gold [9], the grafting of peptides [10] and dendritic polymers [11] through an amide linkage to the surface, and attaching a silane cou- pling agent to the terminus of a polymer to allow grafting to a silica surface [12,13]. Steric interactions limit the density of the polymer that often leads to low grafting density and low film thickness, as the functional moieties must diffuse through the existing polymer film to reach the reactive sites on the surface [14]. The major difference between the conventional radical polymerization techniques and the CRP is the lifetime of the propagating radicals. While radicals derived from thermal decomposition of conventional radical polym- erization initiators (AIBN-type) may undergo termina- tion reactions within a few seconds the lifetime of the propagating radicals can be extended to several hours in controlled processes [15]. Owing to the low radical con- centration maintained by equilibrium between the dor- mant initiator and the propagating radicals (macroinitia- tor) in CRP the probability of the termination reactions is a negligible providing polymer with low polydispersity (Mw/Mn). Among the CRP increasing attention has been placed to atom transfer radical polymerization (ATRP) since its discovery by Matyjaszewski and Sawamoto [16,17], because this method does not require precise experimental conditions like other controlled polymeri- zation processes. The reaction between the activator complex (often CuBr chelated by N-donor ligands) and the dormant initiator results in the formation of propa- gating radical and deactivator complex via reversible halogen atom transfer reaction (Scheme 1). A low stationary concentration of propagating radical (< 10–8 M) is maintained by the dynamic equilibrium which is established after a short period of time (in a few seconds). Using surface-initiated ATRP does not only improve the grafting density of the inorganic particles but may provide polymer chains with controlled mo- lecular weight and molecular weight distribution. While this approach affords the advantages of controlled po- lymerization, it still suffers from the pH instability of conventional silica-based separation materials because the initial bond is silicon-carbon ether. Further, since most ATRP initiators used also contain an ester linkage as part of the initiator moiety, the pH stability will be Scheme 1. Basic mechanism of ATRP process. even lower. Self-assembled systems such as lipid membrane ag- gregates can provide a highly ordered microenvironment leading to unique host-guest chemistry exceeding the functions of the original lipid [18]. Dialkyl L-glutamide- derived lipids form nanotubes [19], nanohelices [20-22], and nanofibers [23] based on bilayer structures in water or in organic solvents and on the fact that intermolecular hydrogen bonding among the amide moieties contributes to self-assembly. We have reported [24,25] the applica- tion of double alkyl self-assembling L-glutamide deriva- tive for HPLC that was prepared by covalent linkage with silica. Up to date there is no report on the synthesis of polymerizable organogel from L-phenylalanine and their application in separation science especially in high performance liquid chromatographic stationary phases. In this study we will present the synthesis, self-assem- bling properties of octadecyl L-phenylalanine derived polymerizable organogel and its application for separa- tion science especially in high performance liquid chro- matography. The grafted particles were prepared by sur- face initiated ATRP (grafting from) and conventional “grafting to” strategies. The complete characterization of the grafted silica particles in both approaches were done and compared with each other. Evaluation of chroma- tographic performance has been done for the separation of poly aromatic hydrocarbons and aromatic positional isomers and will be discussed in the following sections. 2. Experimental Section 2.1. Materials and Methods Triethylamine (Wako, 99+ %) was distilled over potas- sium hydroxide before use. L (-) phenylalanine (Wako, 99+ %), stearylamine (Wako 99%), N-carbobenzoxy- chloride (Z-Cl) (Wako, assay min 95%), diethyl phos- phorocyanidate (DEPC) (Wako, assay min 90%) palla- dium carbon (Pd 5%, Wako) were used as received. Tri- chlorosilane (TCA, 97%), 2-bromoisobutyryl bromide (Aldrich, 98%), ω-undecene-1-ol (Aldrich, 98%), plati- num(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane (Karstedt catalyst) (Aldrich, 0.1 M in xylene), 1,1,4,7,7-pentame- thyldiethylenetriamine (PMDETA) (Wako, 98.0%) and copper(I) bromide (Aldrich, 99.999%) were used as re- ceived. Toluene (Wako, 99+ %) and diethyl ether (Wako, 99.5+ %) were distilled from sodium/benzophenone and stored under nitrogen when not used. Porous silica parti- cles (YMC-GEL) were purchased from YMC (Kyoto, Japan) whose average diameter, pore size and surface area are 5 µm, 12 nm, and 300 m2·g–1 respectively. HPLC grade methanol as well as PAHs samples was obtained from Nacalai Tesque (Japan). Analytical thin-layer chro- Copyright © 2011 SciRes. AJAC  M. M. RAHMAN ET AL. Copyright © 2011 SciRes. AJAC 797 2.2. TGA Measurements matography (TLC) was performed on 0.25 mm silica gel plates, and silica gel column chromatography was carried out with silica gel 60 (Wakogel C-300, Silica Gel). The other chemicals used in this work were obtained from the commercial sources and used without further purification. An ordinary commercial monomeric C18 column (Inertsil, ODS 3, column size 250 mm × 4.6 i.d. with particle size 5.5 μm, pore size 10 nm, and surface area of silica parti- cles 450 m2·g–1 with 13.8% C in the bonded octadecyl phase from G. L. Science, Tokyo, Japan) and a poly- meric C18 column (Shodex, C18 P, particle size 5 μm, pore size 10 nm, surface area 300 m2·g–1 with end cap of the unreacted silanol group, containing 17.5% C pur- chased from Shodex, Tokyo, Japan) were used as refer- ences for chromatographic analysis. Thermogravimetic analyses were performed on a Seiko EXSTAR 6000 TG/DTA 6300 thermobalance in static air from 30˚C to 800˚C at a heating rate 10˚C/min. 2.3. 13C-CP/MAS NMR, 29Si-CP/MAS NMR and Suspended-State 1H NMR Meas- urement NMR spectra was measured by Varian UnityInova AS400 at a static magnetic field of 9.4 T using nanoprobe GHX for suspension-state NMR and solid probe for CP/MAS NMR as spin rate of 2000 - 3500 Hz for suspension-state NMR and 4000 - 4500 Hz for solid-state NMR. The sam- ples for suspension-state 1H NMR were made by using 10 mg of sil-poly4 and sil-T4 in 100 μL of CD3OD in- cluding 0.03% tetramethylsilane. 1H NMR spectra were measured at 20˚C - 50˚C at every 5˚C interval using a GHX Varian AS400 nanoprobe. The parameters used for measurement were delay time 1.5 s, pulse width 2.2, transient numbers 32, and spectral width 6000 Hz. Water was suppressed using a presaturation pulse sequence with saturation delay of 1.5 s and saturation power of 2 db. For assigning peaks, after determination of pulse width of 90˚ simple RELAY COSY (correlation spec- troscopy test) was done and the chemical shifts of the terminal methyl and methylene proton of alkyl chain were determined. For solid-state 13C CP/MAS, the NMR measuring parameters are spectral width 50000 Hz, pro- ton pulse width PW 90 = 11.6 μs, contact time for cross polarization 5 ms, and delay before acquisition was 2 s. High-power proton decoupling of 63 db with fine at- tenuation of dipole r = 2500 was used only during detec- tion periods. 29Si Cross-polarization magic angle spin- ning (CP-MAS) NMR spectra were collected with the Synthesis of L-phenylalanine derived polymerizable organogel N’-octadecyl-Nα-(4-vinyl)-benzoyl-L-phenyla- lanineamide (4) has been done according to the process described in Rahman et al. [26] and is given in synthesis Scheme 2. IR measurements were conducted on a JASCO (Japan) FT/IR-4100 Plus instrument in KBr. For DRIFT measurement accessory DR PRO410-M (Jasco, Japan) was used. Thermogravimetic analyses were per- formed on a Seiko EXSTAR 6000 TG/DTA 6300 ther- mobalance in static air from 30˚C to 800˚C at a heating rate 10˚C/min. For characterization of synthesis 1H and 13C NMR spectra were recorded on a JEOL JNM-LA400 (Japan) instrument was used. Chemical shifts (δ) of 1H, 13C expressed in parts per million (ppm) with use of the internal standards Me4Si (δ = 0.00 ppm) respectively. Coupling constants (J) are reported in Hertz (Hz). Mass spectra were recorded on a PerSeptive Biosystems Voy- ager-DE STR spectrometer. Elemental analyses were carried out on a Perkin–Elmer CHNS/O 2400 apparatus. UV/Vis spectra were measured on a JASCO V-560 spec- trophotometer using quartz cell of 1 cm width. Scheme 2. Synthesis of L-pheny lala nine-derived self-a ssembled monomer.  M. M. RAHMAN ET AL. 798 same instrument. Representative samples of 200 - 250 mg were spun at 3500 Hz using 7 mm double bearing ZrO2 rotors. The spectra were obtained with a cross-po- larization contact time of 5 ms. The pulse interval time was 1.5 s. The transmitter frequencies of 29Si and 1H were 59.59 MHz and 300.13, respectively. Typically, 1.5 k FIDs with an acquisition time of 30 ms were accumu- lated in 1 kilobytes (kb) data points and zero-filling to 8 kb prior to Fourier transformation. The line broadening used was 30 Hz and the spectral width for all spectra was about 25 kHz. 2.4. HPLC Measurement The chromatographic system consists of a Gulliver PU- 980 intelligent HPLC pump with a Rheodyne sample injector having 20 μL loops. A Jasco multi-wavelength UV detector MD 2010 plus was used. The column tem- perature was maintained by using a column jacket with a circulator having a heating and a cooling system. A per- sonal computer connected to the detector with Jasco- Borwin (Ver 1.5) software was used for system control and data analysis. As the sensitivity of UV detector is high, 5 μL of sample solution was used for each injection. To avoid overloading effects, special attention was given in this study to the selection of optimum experimental conditions. Chromatographic measurements were per- formed using HPLC grade methanol and water mixture (90:10) as mobile phase at a flow rate 1.00 mL·min–1. The retention factor (k) measurement was done under isocratic elution conditions. The separation factor (α) is the ratio of the retention factor of two solutes that are being analyzed. The retention time of D2O was used as the void volume (t0) marker (the absorption for D2O was measured at 400 nm, which actually considered as injec- tion shock). All data points were derived from at least triplicate measurements; with retention time (tR) value varying 1%. Water/1-octanol partition coefficient (P) was measured by the retention studies with octadecylated silica, ODS (monomeric) (Inertsil ODS, i.d. 250 mm × 4.6 mm, GL Science, Tokyo, Japan): log P = 3.579 + 4.207 log k (r) 0.999 997) [25]. 2.5. Immobilization of ATRP Initiator on Silica Surface 5 g silica was suspended in 30 mL toluene in a round bottomed flask and 2.150 g (4.72 mmol) (11-(2-bromo-2- methyl)propionyloxy)undecyltrichlorosilane (5) was added and the suspension was rotated for five minutes. Then 1.44 g Et3N (14.2 mmol) was added and the rotation was continued under inert atmosphere for 24 hours. Silica particles were separated washed with toluene, methanol, water, methanol and diethyl ether (each three times) and were stored at room temperature before polymerization. 2.6. General Procedure for “Grafting from” Method 4.1 g initiator-grafted silica (sil-5), 3.94 g (7.2 mmol) of 4 and 0.536 g (3.1 mmol) PMDETA was suspended in 17 mL dry toluene and purged with nitrogen. 0.085 g (2.02 mmol) CuBr was added and the mixture was de- gassed by three-freeze-pump thaw cycles. The flask was then placed in an oil bath with a preset temperature of 90˚C and rotate with slow velocity was maintained for 24 hours. The reaction mixture was let cool to room tem- perature, filtered and washed with hot toluene, hot chlo- roform and methanol repeatedly. For separation of the remaining catalyst particles were placed to a round bot- tomed flask, suspended in the mixture of methanol and aqueous solution of K2EDTA (0.25 M) and the flask was rotated at 40˚C for 6 hours. After filtration silica particles were washed with water, methanol, and diethyl ether and dried under vacuum and finally packed in 25 cm and 4.6 mm i.d. stainless steel column for HPLC measurement. The synthesis scheme of surface-initiated ATRP process of compound 4 is given in reaction Scheme 3. Scheme 3. Surface-initiated ATRP process of compound 4 from silica. Copyright © 2011 SciRes. AJAC  799 M. M. RAHMAN ET AL. 2.7. Procedure for “Grafting to” Method 4.0 g (7.32 mmol) of compound 4 was dissolved in 50 ml dry toluene by heating and 0.15 g (0.73 mmol) of 3- mercaptopropyltrimethoxysilane (MPS) and 40 mg of initiator AIBN was added into the solution and purged with N2 gas. The mixture was degassed by three-freeze- pump thaw cycles. It was then placed in oil bath and heated at 60˚C for 24 hrs to complete the polymerization reaction. The reaction mixture was concentrated and dissolved in minimum amount of chloroform and re- precipitated from methanol. The white crystal appeared were filtered washed with methanol several times and dried under vacuum to obtain T4. For grafting on to sil- ica surface 4.0 g of T4 and 4.0 g silica were taken in a 100 mL three-necked flask and 40 mL dry toluene was added. The suspension was refluxed for 72 hours to complete the grafting process. After filtration the silica particles were washed with hot toluene, methanol, and chloroform and again with methanol, and diethyl ether and dried under vacuum to get sil-T4. The synthesis scheme of telomerization and grafting processes are shown in Scheme 4. 3. Results and Discussion 3.1. Application of Self-Assembling Organogel to Use in HPLC Stationary Phases We have developed poly (octadecyl acrylate) derivatives as lipid membrane analogues for HPLC stationary phase in which the ordered structure induced the orientation of the carbonyl groups that work as π - π interaction sources Scheme 4. “Grafting to” process for the preparation sta- tionary phase from compound 4. with solute molecules. The aligned carbonyl groups were found effective for molecular recognition through multi- ple π - π interactions [27]. Dialkyl L-glutamide-derived lipids form nano-tubes, -helices, and -fibers based on bilayer structures in water and in organic solvents, fur- thermore on the fact that intermolecular hydrogen bond- ing among the amide moieties contribute to self-assembly [27,28]. L-glutamide-derived self-assembling organogel (without a polymerizable head group) has been directly immobilized onto silica surface though covalent linkage for reversed phase high performance liquid chromatog- raphy. We have observed higher molecular shape selec- tivity which was further enhanced by the multiple π - π interaction with ordered carbonyl groups present in the glutamide-derived self-assembling organogel. The dial- kyl glutamide derived self-assembling molecule was rigidly immobilized and form a condensed layer over silica surface which make the carbonyl group in a or- dered state favorable for multiple π - π interactions. The inter- and/or intramolecular hydrogen bonding promote this assembly, that was considered as a driving force for multiple carbonyl π-benzene π interactions [24,25]. In the L-phenylalanine-derivative (4) similar self-assem- bling system was observed in which the cohesion was mainly attributed to the hydrogen bonding among the amide moieties, and the π - π interactions among the phenyl groups would be advantageous for further non- covalent interactions between the stationary phase and the guest molecules. We anticipate that these possible interactions would be recipient for HPLC separation our attention shifted towards the polymerization of compound 4 on silica surface by “grafting from” method with sur- face-initiated ATRP and “grafting to” technique. 3.2. Grafting Density and Surface Coverage The molar amount of octadecyl moieties per 1 g silica (M) can be calculated as according to following equation (from carbon percentages of the polymer grafted silica particles) as mentioned in our previous paper [28] 16 mol g10P10012 C n (1) where PC is the percentage of carbon element according to elemental analysis, and n is the number of carbon at- oms present per molecule of monomer (in this case n = 36, since the monomer contain 36 carbons). The amount of polymer grafted on to silica (Pw) can be calculated as: 4 W10Pm M (2) m is the molecular mass of each molecule of grafted monomer. In addition surface coverage (N) can be calculated as: Copyright © 2011 SciRes. AJAC 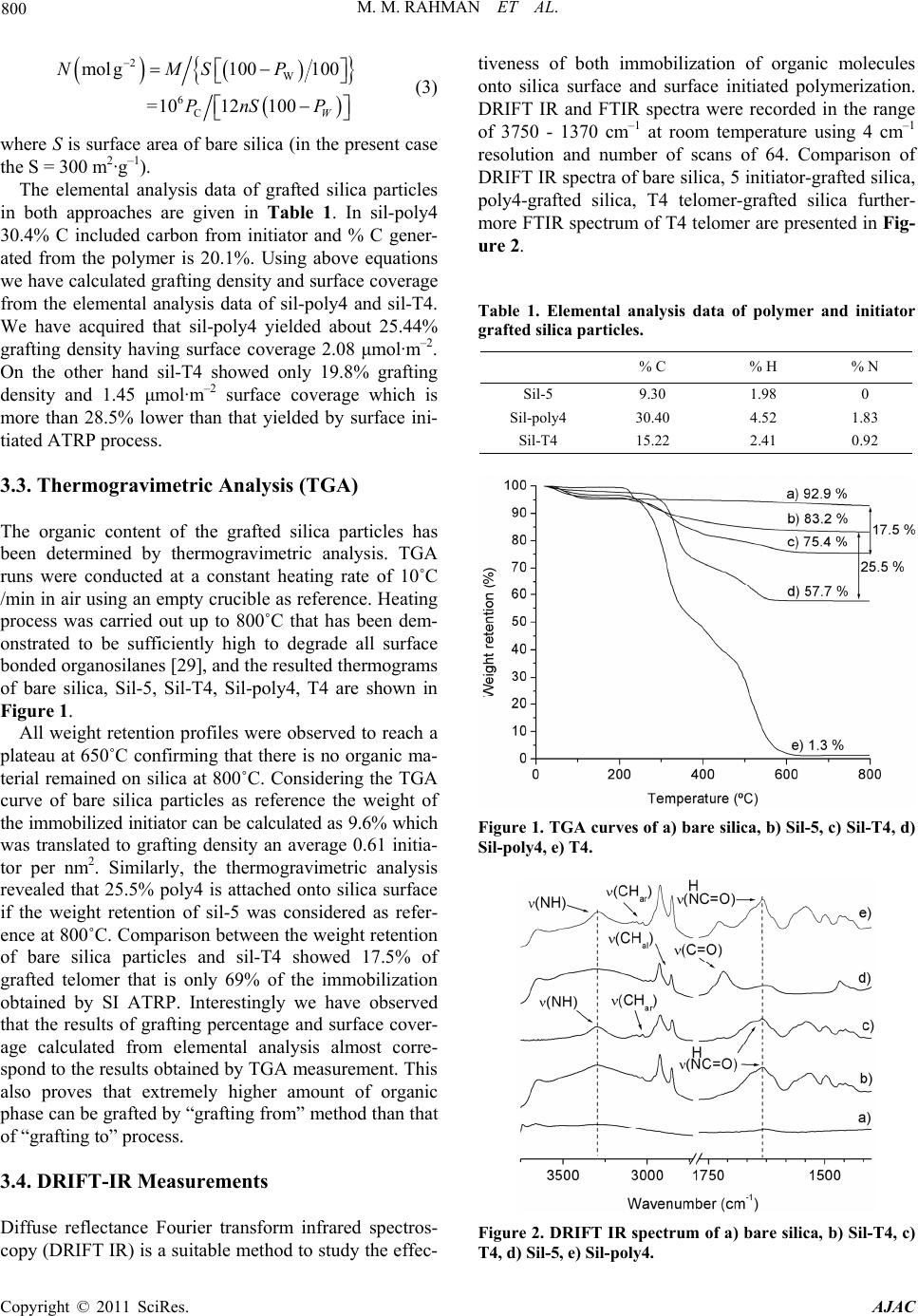 M. M. RAHMAN ET AL. 800 2 W 6 C mol g100100 =1012100W NMSP PnS P (3) where S is surface area of bare silica (in the present case the S = 300 m2·g–1). The elemental analysis data of grafted silica particles in both approaches are given in Table 1. In sil-poly4 30.4% C included carbon from initiator and % C gener- ated from the polymer is 20.1%. Using above equations we have calculated grafting density and surface coverage from the elemental analysis data of sil-poly4 and sil-T4. We have acquired that sil-poly4 yielded about 25.44% grafting density having surface coverage 2.08 μmol· m–2. On the other hand sil-T4 showed only 19.8% grafting density and 1.45 μmol· m–2 surface coverage which is more than 28.5% lower than that yielded by surface ini- tiated ATRP process. 3.3. Thermogravimetric Analysis (TGA) The organic content of the grafted silica particles has been determined by thermogravimetric analysis. TGA runs were conducted at a constant heating rate of 10˚C /min in air using an empty crucible as reference. Heating process was carried out up to 800˚C that has been dem- onstrated to be sufficiently high to degrade all surface bonded organosilanes [29], and the resulted thermograms of bare silica, Sil-5, Sil-T4, Sil-poly4, T4 are shown in Figure 1. All weight retention profiles were observed to reach a plateau at 650˚C confirming that there is no organic ma- terial remained on silica at 800˚C. Considering the TGA curve of bare silica particles as reference the weight of the immobilized initiator can be calculated as 9.6% which was translated to grafting density an average 0.61 initia- tor per nm2. Similarly, the thermogravimetric analysis revealed that 25.5% poly4 is attached onto silica surface if the weight retention of sil-5 was considered as refer- ence at 800˚C. Comparison between the weight retention of bare silica particles and sil-T4 showed 17.5% of grafted telomer that is only 69% of the immobilization obtained by SI ATRP. Interestingly we have observed that the results of grafting percentage and surface cover- age calculated from elemental analysis almost corre- spond to the results obtained by TGA measurement. This also proves that extremely higher amount of organic phase can be grafted by “grafting from” method than that of “grafting to” process. 3.4. DRIFT-IR Measurements Diffuse reflectance Fourier transform infrared spectros- copy (DRIFT IR) is a suitable method to study the effec- tiveness of both immobilization of organic molecules onto silica surface and surface initiated polymerization. DRIFT IR and FTIR spectra were recorded in the range of 3750 - 1370 cm–1 at room temperature using 4 cm–1 resolution and number of scans of 64. Comparison of DRIFT IR spectra of bare silica, 5 initiator-grafted silica, poly4-grafted silica, T4 telomer-grafted silica further- more FTIR spectrum of T4 telomer are presented in Fig- ure 2. Table 1. Elemental analysis data of polymer and initiator grafted silica particles. % C % H % N Sil-5 9.30 1.98 0 Sil-poly4 30.40 4.52 1.83 Sil-T4 15.22 2.41 0.92 Figure 1. TGA curves of a) bare silica, b) Sil-5, c) Sil-T4, d) Sil-poly4, e) T4. Figure 2. DRIFT IR spectrum of a) bare silica, b) Sil-T4, c) T4, d) Sil-5, e) Sil-poly4. Copyright © 2011 SciRes. AJAC  801 M. M. RAHMAN ET AL. The spectra of sil-T4 and T4 are nearly identical pro- viding an evidence that fairly high amount of telomere has been grafted onto silica particles. Successful immo- bilization of 5 initiator is confirmed by the two strong bands at 2924 cm–1 and 2853 cm–1, respectively attrib- uted to the asymmetric and symmetric CH2 stretching furthermore the band at 1717 cm–1 arising from carbonyl group of the ester moiety. Analysis of spectrum 2e) shows a broad band at 1633 cm–1 with a shoulder at 1656 cm–1 derived from the overlapped amide carbonyl stretching vibrations and a band at 3290 cm–1 due to the N-H stretching vibration indicating the presence of poly4 on silica surface. These findings clearly proved that silica particles could be coated using two approaches, namely grafting to technique and grafting from technique (SI ATRP). 3.5. Solid-state NMR Spectroscopic Measurements 29Si CP/MAS NMR spectroscopy is well suited for as- sessing the surface reaction of silanes with silica. Reso- nances for the silica species appears from –90 to –110 ppm. The 29Si CP/MAS NMR spectra of the polymer grafted silica for sil-poly4 and sil-T4 are shown in Fig- ure 3. The 29Si CP/ MAS NMR spectrum for bare silica and ATRP initiator grafted silica (sil-initiator) are also included for comparison. The two polymer grafted silica showed that large extent of silanol groups remain un- functionalized by both grafting methods. Figure 3 shows the differentiation between free si- lanol groups (Q3) and geminal silanol groups (Q2) be- sides the siloxane groups (Q4) that are indicated by sig- nals at –92, –102 and –111 ppm, respectively. In ATRP polymer-grafted silica the signal corresponding to resid- ual geminal silanols (Q2) is not seen while its intensity is very less in ATRP initiator grafted silica (sil-initiator), however this signal is quite intense for sil-T4 which was prepared by “grafting to” method. In the spectra after immobilization of ATRP initiator (b) and after polym- erization (c) emphasize signals for Tn species that are related to the number of siloxane bonds. When the ini- tiator was reacted with silica surface a large amount of cross-linked T2 type silicon species (–57 ppm) was ob- served while polymer grafting increased cross-linked surface indicating by the appearance of T3 signals (–65 ppm). The disappearance of signal for T1 in the spectrum (b) and (c) is due to the reaction of the silanol with self-assembling monolayer formed by ATRP initiator [30]. For sil-T4 the signal corresponds to T2 and T3 are completely absent however, T1 signal is slightly ap- peared which indicates that relatively higher amount of silanol groups remain unfunctionalized in sil-T4 than sil- poly4. In liquid- or suspended-state NMR, only those mole- cules or parts of molecules are detectable that has very fast rotational motions. The suspension-state 1H NMR of sil-poly4 and sil-T4 were measured from 25˚C to 50˚C. Neither half-height width (line width) of methylene groups nor spin-spin relaxation time (T2) showed any significant change with temperature (20˚C - 50˚C) for both sil-poly4 and sil-T4. We observed that intensity of the NMR peaks representing terminal methyl and me- thylene groups of octadecyl moieties increased slightly and detectable when a very high vertical scale was used for graphical presentation. These results indicate that the organic phase on the silica surface is in a solid state at room temperature. Under the condition of magic angle spinning and di- polar coupling of protons, the chemical shift of methyl- ene groups in 13C CP/MAS NMR spectroscopy depends largely on the conformation of octadecyl chains [31]. For each central carbon atom in an octadecyl chain with the trans conformation, a chemical shift of around 33 ppm is expected. On the other hand, for conformations with rapid changes between gauche and trans, a chemical shift of about 30 ppm is expected [32]. Solid-state 13C Figure 3. 29Si CP/MAS NMR spectra (a) bare silica, (b) sil-ATRP initiator, (c) sil-poly4 and (d) sil-T4. Copyright © 2011 SciRes. AJAC  M. M. RAHMAN ET AL. 802 th sil-poly4 .6. Evaluation of Retention Mode is known that conventional reversed phase HPLC stance, Engelhardt [33] proposed using the se- le t of hydropho- bi CP/MAS NMR spectra were measured for bo and sil-T4 at room temperature. We have observed an intense signal at 30.6 ppm which indicated that N-alkyl moieties appeared at less ordered gauche conformational form and its shoulder which is almost negligible at around 32.8 ppm results that there are some parts of the alkyl moiety representing trans or ordered form. How- ever, in sil-T4 the N-alkyl moiety also appear by domi- nating gauche form at somewhat up field than sil-poly4 and at 30.0 ppm. In this case a shoulder at around 32.4 ppm which is more intense than sil-poly4 indicates it contains more ordered N-alkyl chain than sil-poly4. 3 It packing materials or C18 or alkyl phases can recognize the hydrophobicity of elutes and this hydrophobicity is measured by the methylene activity of the stationary phases. For in ctivity factor (α) between ethyl benzene and toluene in a mobile phase of methanol/water mixture as an indicator of hydrophobicity. We observed sil-poly4 yielded slightly higher selectivity (αethylbenzene/toluene = 1.23) than sil-T4 (αethylbenzene/toluene = 1.20) for this sample set. Ki- mata et al.51 suggested that hydrophobicity value be de- termined from the selectivity (α) between amylbenzene and butylbenzene in a mobile phase of methanol/water. We have also observed that sil-poly4 (αamylbenzene/butylbenzene = 1.30) showed higher selectivity than sil-T4 (αamylbenzene/ butylbenzene = 1.25) for this test mixture too. The retention mode as well as the exten c interaction between the elutes and the packing mate- rials in HPLC can be determined by retention studies of alkylbenzenes as elutes [35-38]. Figure 4 shows the re- lationship between log k and log P for sil-poly4, sil-T4 and C18-M (monomeric C18) phase. The figure clears us that both sil-poly4 and sil-T4 phases trailed the retention mode similar to that of conventional reversed-phase ma- terials. As observed, sil-T4 showed extremely low reten- tion for alkylbenzenes comparing to sil-poly4 and C18. It was also observed that log k and log P plots of alkylben- zenes and PAHs in C18 are parallel and almost coincided with each other. This happened because monomeric C18 can recognize only the hydrophobicity of elutes. Never- theless, both sil-poly4 and sil-T4 gives higher retention for PAHs compared to its values for alkylbenzenes. For instance, the log P of naphthacene (5.71) is much smaller than dodecylbenzene (8.43), but the log k value of naph- thacene (0.68) is much higher than dodecylbenzene (0.53) in sil-T4. In contrast log k for dodecylbenzene (3.25) is higher than naphthacene (2.95) in sil-poly4 which di- vulged that sil-T4 can recognize molecular hydrophobic- ity inferior than sil-poly4. These results indicate that the sil-T4 phase provides specific interactive sites for PAHs that can recognize aromaticity relatively high regardless it has low surface coverage and less hydrophobic interac- tions than sil-poly4 and C18 phase. The chromatogram for four PAHs on sil-T4 is given in Figure 5. Figure 4. Relationships between log k and log P of diffent er stationary phases. Mobile phase: methanol-water (90/10 v/v), column temperature: 30˚C. Elutes: a, benzene; b, tolu- ene; c-h, ethyl-, butyl-, hexyl-, octyl-, decyl-, and dodecyl- benzene; i, naphthalene; j, anthrac e ne ; and k, naphthac e ne . Figure 5. Chromatogram of four PAHs of sil-T4. Mob flow rate: 1.0 mL/min. ile phase: methanol/water (90/10), column temperature: 30 C, Copyright © 2011 SciRes. AJAC  M. M. RAHMAN ET AL. 803 ation was performed 3.7. Separation Performance The first chromatographic evalu using the Tanaka test mixture containing hydrophobic probes which give information about shape and methyl- ene selectivity, ion-exchange capacities from acidic and neutral media and hydrogen bonding capacities [33]. Figure 6 shows the chromatogram obtained for the above mentioned test mixture on sil-poly4 and sil-T4, we observed that all of the compounds were well separated by both phase. However, sil-T4 requires less than half time than that of sil-poly4 to get well resolved separation. It also observed that peak shape is rather better in sil-T4 than sil-poly4. This characterization protocol is a well- developed approach that is recommended to obtain in- formation about functionality of the silylant reagent, and the methylene selectivity as well as to establish the re- peatability and reproducibility of the separation behavior of commercially available stationary phases. The chro- matogram (in Figure 6) shows the separation of two homologous alkyl benzenes and non-planar polyaromatic hydrocarbons (PAHs), where it was observed that all compounds are well resolved. Figure 6. Separation of Tanaka test mixture by sil-poly4 and sil-T4. Mobile phase: methanol/ water (90/10), flow rate phase can be ism the aqueous binary mo- 1.0 mL/min, column temperature 30˚C. 3.8. Selectivity for PAHs The shape selectivity of a given stationary measured by the separation factor (α) value for pairs of test solutes having the same carbon number but different shape, such as perylene/1, 1’-binaphthyl, triphenylene/ o-terphenyl, chrysene/pyrene and trans-stilbene/cis- stil- bene. On a typical monomeric C18 phase (C18-M) the separation factor αtriphenylene/o-terphenyl ranges from 1.0 to 1.7, while the value on a polymeric C18 (C18-P) phase lies between 2.0 to 2.7. Jinno et al. [39,40] suggested that a separation factor αtriphenylene/o-terphenyl ≥ 2.0 is an indication of significant solute planarity recognition. Table 2 lists some of the α values measured on col- umns packed with sil-T4, sil-poly4 and C18 phases. It is clear that the sil-T4 and sil-poly4 phase with a selectivity factor αtriphenylene/ o-terphenyl of 3.30-3.20 possesses enhanced shape selectivity for PAHs. The selectivity factor αperylene/ 1,1’-binaphthyl 16.5 and 13.5 on the sil-T4 and sil-poly4 fur- ther exemplifies this extremely high planarity recogni- tion. Interestingly, for the isomeric aromatic pair, chry- sene and pyrene, the sil-T4 phase yielded a selectivity factor of αchrysene/pyrene = 1.55 while sil-poly4 yielded slightly lower value 1.52 demonstrated that on both phases shape recognition includes not only planarity but also the geometry of solutes. As suggested from the data in Ta b le 2, compared with C18 phases, sil-T4 phases pro- vide a greater degree of shape selectivity. The retention data for PAHs and aromatic positional isomers for sil-poly4 and sil-T4 in two different mobile phase sys- tems are given in Table 3. 3.9. Separation Mechan Under isoeluotropic conditions, bile phases of methanol, acetonitrile and tetrahydrofuran possess equal solvent strength and similar retention be- havior on alkyl-based stationary phases [41]. If π - π in- teraction sites are present, the differences between methanol and acetonitrile become significant, as acetoni- trile [42-44] shows π - π solvent interactions. Acetonitrile as an electron-rich organic modifier suppresses the π - π interaction between the solute and the aromatic π - π ac- tive moiety of the stationary phase through formation of electron donor-acceptor complexes with either the aro- matic ligand or the solute, depending on whether the one or the other is the more electron-deficient counterpart. Non-functional planar and non-planar PAHs are com- monly employed test solutes to demonstrate the presence of π - π interaction, since they only allow hydrophobic and π – π type interactions, without further interaction possibilities [45-48]. Retention factors of planar and non-planar PAHs for methanol and acetonitrile mobile Copyright © 2011 SciRes. AJAC 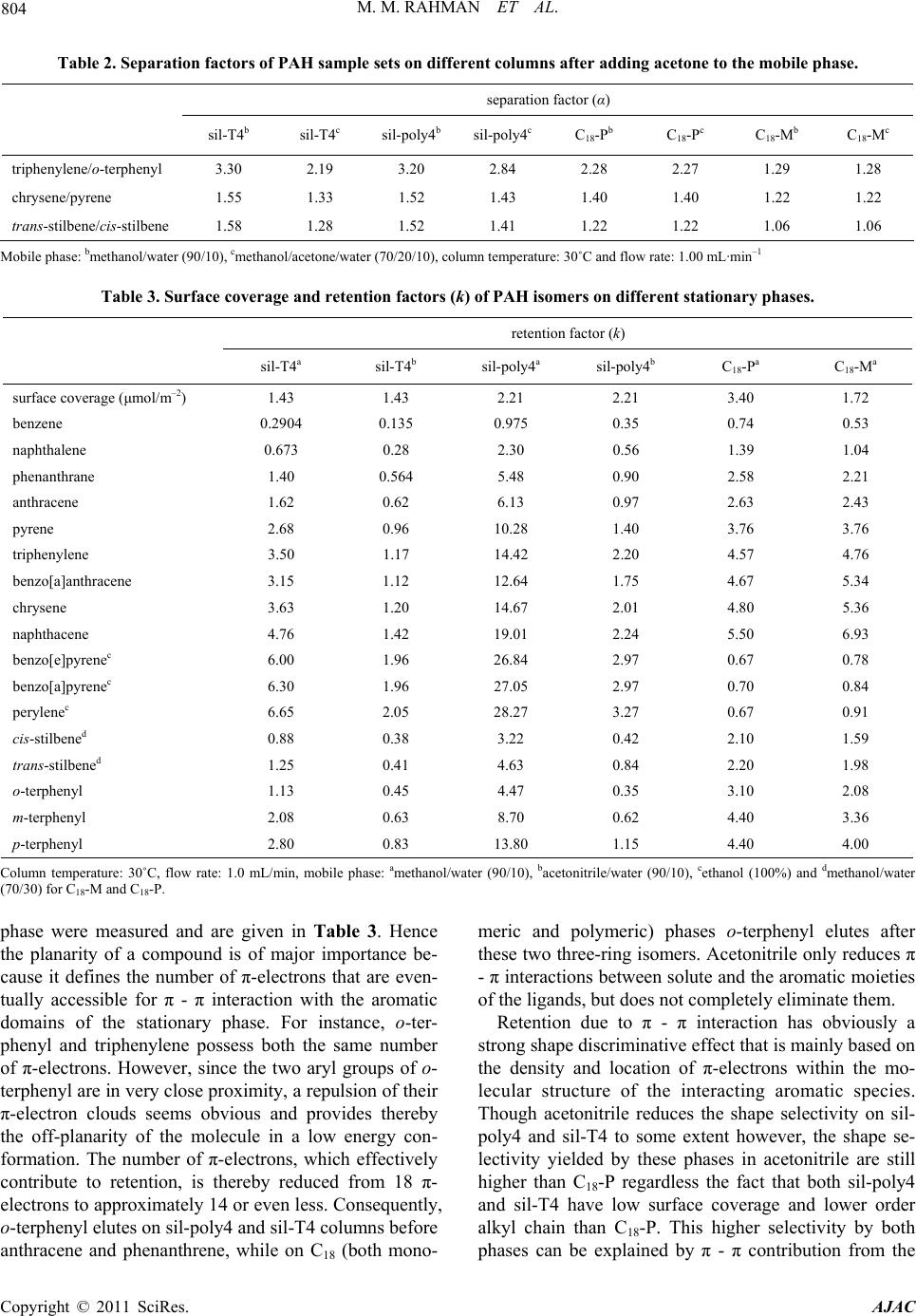 M. M. RAHMAN ET AL. Copyright © 2011 SciRes. AJAC 804 erent columns after adding acetone to the mobile phase. Table 2. Separation factors of PAH sample sets on diff separation factor (α) sil-T4b sil-T4c sil-poly4b s18-Pc C 18-Mb C 18-Mc il-poly4c C 18-Pb C triphenylene/ terphenyl o-3.30 2.19 3.20 2.84 2.28 2.27 1.29 1.28 chrysene/pyr1.55 1.33 1.52 1.43 1.40 1.40 1.22 1.22 ene trans-stilbene/cis-stilbene 1.58 1.28 1.52 1.41 1.22 1.22 1.06 1.06 Mo anol/water (90/10hanol/aceter (70/), column tature: 30˚ow rateL·min–1 hases. bile phethase: bm), cmetone/wat 20/10emperC and fl: 1.00 m Table 3. Surface coverage and retention factors (k) of PAH isomers on different stationary p retention factor (k) sil-T4a sil-T4b sil-poly4a b C 18-Pa C 18-Ma sil-poly4 surface coverage (μmol/m–2) 40 1.1.43 1.43 2.21 2.21 3.72 benzene ane ]anthracene yrenec c ened 0.2904 0.135 0.975 0.35 0.74 0.53 naphthalene 0.673 0.28 2.30 0.56 1.39 1.04 phenanthr1.40 0.564 5.48 0.90 2.58 2.21 anthracene 1.62 0.62 6.13 0.97 2.63 2.43 pyrene 2.68 0.96 10.28 1.40 3.76 3.76 triphenylene3.50 1.17 14.42 2.20 4.57 4.76 benzo[a3.15 1.12 12.64 1.75 4.67 5.34 chrysene 3.63 1.20 14.67 2.01 4.80 5.36 naphthacene 4.76 1.42 19.01 2.24 5.50 6.93 benzo[e]p 6.00 1.96 26.84 2.97 0.67 0.78 benzo[a]pyrene 6.30 1.96 27.05 2.97 0.70 0.84 perylenec 6.65 2.05 28.27 3.27 0.67 0.91 cis-stilbened 0.88 0.38 3.22 0.42 2.10 1.59 trans-stilb 1.25 0.41 4.63 0.84 2.20 1.98 o-terphenyl 1.13 0.45 4.47 0.35 3.10 2.08 m-terphenyl 2.08 0.63 8.70 0.62 4.40 3.36 p-terphenyl 2.80 0.83 13.80 1.15 4.40 4.00 Cure: 30˚C, flow rate: 1.0 mLobile phasenol/water ( bacetonitrile0/10), cetha00%) and dm/water (7 and C18-P. nd are given in Table 3. Hence e planarity of a compound is of major importance be- meric and polymeric) phases o-terphenyl elutes after these two three-ring isomers. Acetonitrile only reduces π s. Th olumn temperat/min, m: ametha 90/10),/water (9nol (1ethanol 0/30) for C18-M phase were measured a th cause it defines the number of π-electrons that are even- tually accessible for π - π interaction with the aromatic domains of the stationary phase. For instance, o-ter- phenyl and triphenylene possess both the same number of π-electrons. However, since the two aryl groups of o- terphenyl are in very close proximity, a repulsion of their π-electron clouds seems obvious and provides thereby the off-planarity of the molecule in a low energy con- formation. The number of π-electrons, which effectively contribute to retention, is thereby reduced from 18 π- electrons to approximately 14 or even less. Consequently, o-terphenyl elutes on sil-poly4 and sil-T4 columns before anthracene and phenanthrene, while on C18 (both mono- - π interactions between solute and the aromatic moieties of the ligands, but does not completely eliminate them. Retention due to π - π interaction has obviously a strong shape discriminative effect that is mainly based on the density and location of π-electrons within the mo- lecular structure of the interacting aromatic specie ough acetonitrile reduces the shape selectivity on sil- poly4 and sil-T4 to some extent however, the shape se- lectivity yielded by these phases in acetonitrile are still higher than C18-P regardless the fact that both sil-poly4 and sil-T4 have low surface coverage and lower order alkyl chain than C18-P. This higher selectivity by both phases can be explained by π - π contribution from the 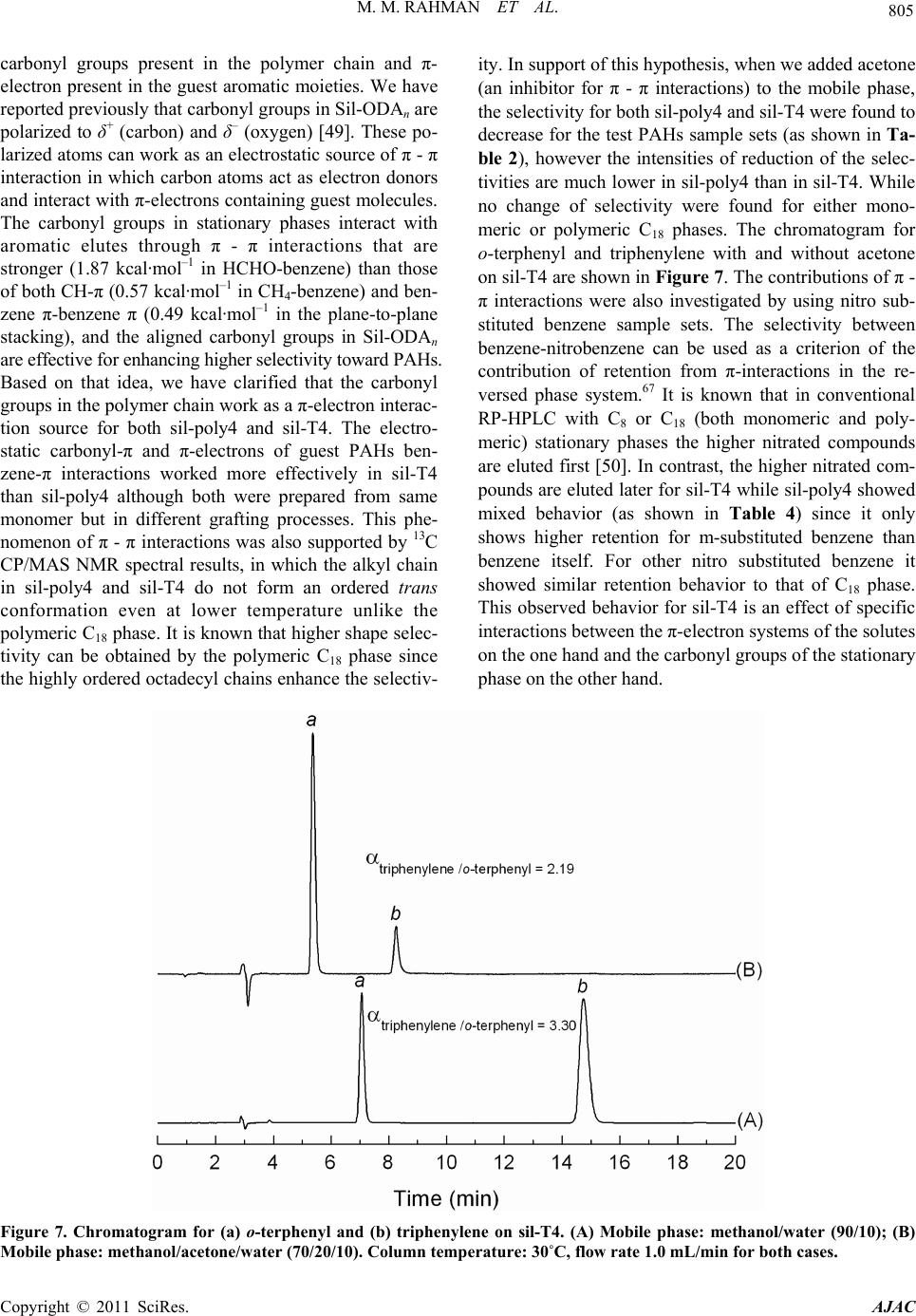 M. M. RAHMAN ET AL. 805 carbonyl groups present in the polymer chain and π- electron present in the guest aromatic moieties. We have reported previously that carbonyl groups in Sil-ODAn are polarized to δ+ (carbon) and δ– (oxygen) [49]. These po- larized atoms can work as an electrostatic source of π - π interaction in which carbon atoms act as electron donors and interact with π-electrons containing guest molecules. The carbonyl groups in stationary phases interact with aromatic elutes through π - π interactions that are stronger (1.87 kcal·mol–1 in HCHO-benzene) than those of both CH-π (0.57 kcal·mol–1 in CH4-benzene) and ben- zene π-benzene π (0.49 kcal·mol–1 in the plane-to-plane stacking), and the aligned carbonyl groups in Sil-ODAn are effective for enhancing higher selectivity toward PAHs. Based on that idea, we have clarified that the carbonyl groups in the polymer chain work as a π-electron interac- tion source for both sil-poly4 and sil-T4. The electro- static carbonyl-π and π-electrons of guest PAHs ben- zene-π interactions worked more effectively in sil-T4 than sil-poly4 although both were prepared from same monomer but in different grafting processes. This phe- nomenon of π - π interactions was also supported by 13C CP/MAS NMR spectral results, in which the alkyl chain in sil-poly4 and sil-T4 do not form an ordered trans conformation even at lower temperature unlike the polymeric C18 phase. It is known that higher shape selec- tivity can be obtained by the polymeric C18 phase since the highly ordered octadecyl chains enhance the selectiv- ity. In support of this hypothesis, when we added acetone (an inhibitor for π - π interactions) to the mobile phase, the selectivity for both sil-poly4 and sil-T4 were found to decrease for the test PAHs sample sets (as shown in Ta- ble 2), however the intensities of reduction of the selec- tivities are much lower in sil-poly4 than in sil-T4. While no change of selectivity were found for either mono- meric or polymeric C18 phases. The chromatogram for o-terphenyl and triphenylene with and without acetone on sil-T4 are shown in Figure 7. The contributions of π - π interactions were also investigated by using nitro sub- stituted benzene sample sets. The selectivity between benzene-nitrobenzene can be used as a criterion of the contribution of retention from π-interactions in the re- versed phase system.67 It is known that in conventional RP-HPLC with C8 or C18 (both monomeric and poly- meric) stationary phases the higher nitrated compounds are eluted first [50]. In contrast, the higher nitrated com- pounds are eluted later for sil-T4 while sil-poly4 showed mixed behavior (as shown in Table 4) since it only shows higher retention for m-substituted benzene than benzene itself. For other nitro substituted benzene it showed similar retention behavior to that of C18 phase. This observed behavior for sil-T4 is an effect of specific interactions between the π-electron systems of the solutes on the one hand and the carbonyl groups of the stationary phase on the other hand. Figure 7. Chromatogram for (a) o-terphenyl and (b) triphenylene on sil-T4. (A) Mobile phase: methanol/water (90/10); (B) Mobile phase: methanol/acetone/water (70/20/10). Column temperature: 30˚C, flow rate 1.0 mL/min for both cases. Copyright © 2011 SciRes. AJAC  M. M. RAHMAN ET AL. 806 Table 4. Retention factors (k) and separation factors ( ) for nitrobenzenes/benzene on sil-T4 and sil-poly4 revealing -electron donating-accepting properties under RP conditions. sil-T4 Sil-poly4 Retention factor (k) Separation factor (α) Retention factor (k) Separation factor (α) benzene 0.290 0.9750 1.09 0.98 nitrobenzene 0.315 0.9584 1.14 0.80 o-dinitrobenzene 0.33 0.7850 1.24 0.98 p-dinitrobenzene 0.358 0.9530 1.35 1.13 m-dinitrobenzene 0.392 1.100 Mobile phase: methanol/water (90/20), column temperature: 30˚C, flow rate: 1.00 mL/min, UV detection 254 nm. The solutes are substituted with electron-drawing ni- tro-groups therefore; their aromatic ringsystems possess a low π-electron density. In contrast, both sil-T4 and sil- ceptors. teractions that can lead to donor-acceptor lexes. The stability of these complexes he electron density of both the solutes a phase Since thn density of thecreases with increasing nmbers of nitro groupsnsity of π - π interaces in the sas discussed above thyl group in sil-T4 poy formed a thin condensing layer over silica surface by forming hy- drogeng the amide able for muπ - π interactions. Conversely due to the prese ain initiator ithe polymer might be unable to form such a cd layer over silica surface less auspicious foiple interac- tionsower π - π interactions with guest 4. to” technique although it has much lower grafting den- sity than that prepared by “grafting from” technique. The new stationary phase prepared from amino acid derived [1] S. B. Roscoe, A. K. Kakkaarks, A. Malik, M. K. Durbin, W. Lin, G. K. Wong and P. Dutta, “Self-Assem- ed Chrctive Me- flectivity and Second-Harmonic Generation as Comple- mentary Prob Block-Film Microstructure Relationships,” Vol. 12, No1996, pp. 4218-4223. 2k poly4 possess carbonyl groups and π-electron rich ben- zene ring in each molecule, which increases the π-elec- - polymeric oganogel and by “grafting to” method exhibits good separation properties for application in RP-HPLC. tron density in the stationary phases, can act as an elec tron donor, while the nitrated solutes act as electron ac- These functionalities result in specific π - π in- 5. References comp depends on t nd the stationary [52]. bl e electro solutes de , the inte der. A ssibl oly4 ndense r mult u tion also increas e carbon me or n bonding amo moieties which favor- ltiple nce of long chn sil-p o and is and showed l PAHs samples than sil-T4. Conclusions L-phenylalanine based polymerizable organogel has been grafted on silica surface by “grafting from” and “grafting to” strategies. Elemental analysis and TGA measure- ments showed that extremely higher amount of organic phase can be grafted by surface-initiated ATRP process than “grafting to” method. Chromatographic results re- vealed that slightly higher selectivity for PAHs was ob- tained by the stationary phase prepared from “grafting r, T. J. M omophoric NLO-Aonolayers. X-Ray R es of Building Langmuir, doi:10.1021/la95014 . 17, [2] Y. N. Xia, M. MrksicE. Kim and G. M. sides, “Microcontact Prtadecylsiloxn the Sur- face of Silicon Its Application in Microfab- rication,” Journafrican Chemiciety, Vol. 117, No. 37, 1995, pp7. doi:10.1021/ja h, ing of Oct e and the Ame . 9576-957 031 White ane oin Dioxid l ocal So 00142a . J. Wirt[3] X. Y. Huang and Mh, “Surface- Radical Polymerizationrous Silica,” Analytical Chemistry, Vol. 69, No. 22, . 4577-4580. 23 Initiated on Po 1997, pp doi:10.1021/ac97045 [4] C. A. Chang, H. Abdel-Aziz, N. Melchor, Q. H. Wu, K. H. Pannell and D. W. Armstrong, “Liquid Chroma- tographic Retention Behavior of Organometallic Com- pounds and Ligands with Amine-, Octadecylsilica- and Beta-Cyclodextrin-Bonded Phase Columns,” Journal of Chromatography A, Vol. 347, 1985, pp. 51-60. doi:10.1016/S0021-9673(01)95468-8 [5] A. Zlatkis, R. P. J. Ranatunga and B. S. Middleditch, “Concentration of Organics from Aqueous Solutions Us- ing Uncoated Capillary Columns,” Analytical Chemistry, Vol. 62, No. 22, 1990, pp. 2471-2478. doi:10.1021/ac00221a012 Copyright © 2011 SciRes. AJAC  M. M. RAHMAN ET AL. 807 Journal of Chromatography -159. 1-9673(93)80655-R [6] D. Schmalzing, C. A. Piggee, F. Foret, E. Carrilho and B. L. Karger, “Characterization and Performance of a Neu- tral Hydrophilic Coating for the Capillary Electrophoretic Separation of Biopolymers,” A, Vol. 652, No. 1, 1993, pp. 149 doi:10.1016/002 ] M. Chiari, N. Dell’Orto and A. GelainChiari, “Synth[7 esis and Characterization of Capillary Columns Coated with Glycoside-Bearing Polymer,” Analytical Chemistry, Vol. 68, No. 17, 1996, pp. 2731-2736. doi:10.1021/ac960158v [8] Unger and K. K. P. Silica, “Its Properties and Use as Support in Column Liquid Chromatography,” Elsevier, Amsterdam, 1979. [9] B. Zhao and W. J. Brittain, “Synthesis of Polystyrene Brushes on Silicate Substrates via Carbocationic Polym- erization from Self-Assembled Monolayers,” Macro- molecules, Vol. 33, No. 2, 2000, pp. 342-348. doi:10.1021/ma9910181 [10] M. Hessemann, D. Mecerreyes, C. J. Hawker, J. L. Hed- rich, R. Shah and N. L. Abbott, “Polymerization for Am- plification of Self-Assembled Monolayers Patterned by Microcontact Printing,” try, Vol. 38, N General & Introductory Chemis- o. 5, 1999, pp. 647-649. doi:10.1002/(SICI)1521-3773(19990301)38:5<647::AID- ANIE647>3.0.CO;2-0 [11] J. M. Song, F. M. Winnik and J. L. Brash, “Synthesis and Solution Properties of Fluorescently Labeled Amphiphilic (N-alkylacrylamide) Oligomers,” Macromolecules, Vol. 31, No. 1, 1998, pp. 109-115. doi:10.1021/ma971111u [12] G. J. Kluth, M. M. Sung and R. Maboudian, “Interaction of H(D) Atoms with Octadecylsiloxane Self-Assembled Monolayers on the Si(100) Surface,” Langmuir, Vol. 13, No. 24, 1997, pp. 6491-6496. doi:10.1021/la970641g [13] R. Zajac and A. Chakrabarti, “Irreversible Polymer Ad- sorption from Semidilute and Moderately Dense Solu- tions,” Physical Review E, Vol. 5 6536-6549. 2, No. 6, 1995, pp. ysRevE.52.6536doi:10.1103/Ph C., 2000. [14] K. Matyjaszewski, “Controlled-Living Radical Polym- erization: Progress in ATRP, NMP and RAFT,” Ameri- can Chemical Society, Washington, D. [15] J.-S. Wang and K. Matyjaszewski, “Controlled/“Living” Radical Polymerization. Atom Transfer Radical Polym- erization in the Presence of Transition-Metal Com- plexes,” Journal of the American Chemical Society, Vol. 117, No. 20, 1995, pp. 5614-5615. doi:10.1021/ja00125a035 [16] M. Kato, M. Kamigaito, M. Sawamoto and T. Higashi- , No. 5, 1995, mura, “Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(triphenylphosphine) ruthenium(II)/Methylaluminum Bis (2,6-di-tert-butylphe- noxide) Initiating System: Possibility of Living Radical Polymerization,” Maromolecules, Vol. 28 pp. 1721-1723. doi:10.1021/ma00109a056 [17] A. M. Balachandra, G. L. Baker and M. L. Bruening, “Preparation of Composite Membranes by Atom Transfer Radical Polymerization Initiated from a Porous Support,” pp. 1-14. Journal of Membrane Science, Vol. 227, No. 1-2, 2003, 009doi:10.1016/j.memsci.2003.07. Fukumoto and C. nged Monomeric Dyes on [18] K. Yamada, H. Ihara, T. Ide, T. Hirayama, “Formation of Helical Super Structure from Single-Walled Bilayers by Amphiphiles with Oligo-L-Glutamic Acid-Head Group,” Chemistry Letters, 1984, pp. 4821-4822. [19] H. Hachisako, H. Ihara, C. Hirayama and K. Ya- mada, “Chirally Arra Helical Bilayer Membranes,” Liquid Crystals, Vol. 13, No. 2, 1993, pp. 307-311. doi:10.1080/02678299308026304 [20] H. Hachisako, T. Yamazaki, H. Ihara, C. Hiraya Yamada, “Formation of Specif ma and K. ic Hydrophobic Sites for Incorporation of Methylene Blue by Laterally Arranged L-Glutamate Residues in Anionic, Crystalline Bilayer Aggregates,” Journal of the Chemical Society, Perkin Transactions 2, No. 7, 1994, pp. 1671-1680. doi:10.1039/p29940001671 [21] H. Ihara, M. Takafuji, C. Hirayama and D. F. O’Brien, “Effect of Photopolymerization on the Morphology of Helical Supramolecular Assemblies,” Langmuir, Vol. 8, No. 6, 1992, pp. 1548-1553. doi:10.1021/la00042a010 [22] H. Ihara, M. Takafuji and T. Sakurai, “Encyclopedia of Nanoscience and Nanotechnology,” American Scientific se Publishers, Stevenson Ranch, 2004, p. 473. [23] M. Takafuji, M. M. Rahman, M. Derakshan, H. R. An- sarian and H. Ihara, “Dioctadecyl L-Glutamide-Derived Lipid- Grafted Silica as a Novel Organic Stationary Pha for RP-HPLC,” Journal of Chromatography A, Vol. 1074, No. 1-2, 2005, pp. 223-228. doi:10.1016/j.chroma.2005.03.090 [24] M. M. Rahman, M. Takafuji, H. R. Ansarian and H. Ihara, “Molecular Shape Selectivity through Multiple Car- bonyl-π Interactions with Noncrystalline Solid Phase for RP-HPLC,” Analytical Chemistry, Vol. 77, No. 20, 2005, pp. 6671-6681. doi:10.1021/ac050851v [25] H. Ihara, T. Sagawa, Y. Goto and S. Nagaoka, “Crystal- line Polymer on Silica. Geometrical Selectivity for Azobenzenes through Highly-Oriented Structure,” Poly- mer, Vol. 40, No. 10, 1999, pp. 2555-5562. doi:10.1016/S0032-3861(98)004856- [26] M. M. Rahman, M. Czaun, M. Takafuji and H. Ihara, “Synthesis, Self-Assembling Properties, and Atom Trans- fer Radical Polymerization of an Alkylated L-Phenyla- lanine-Derived Monomeric Organogel from Silica: A New Approach To Prepare Packing Materials for High- Performance Liquid Chromatography,” Chemistry―A European Journal, Vol. 14, No. 4, 2008, pp. 1312-1321. doi:10.1002/chem.200701302 [27] H. Ihara, M. Takafuji, C. Hirayama and D. F. O’Brien, “Effect of Photopolymerization on the Morphology of Helical Supramolecular Assemblies,” Langmuir, Vol. 8, No. 6, 1992, pp. 1548-1553. doi:10.1021/la00042a010 [28] H. R. Ansarian, M. Derakshan, M. M. Rahman, T. Sa- kurai, M. Takafuji, I. Taniguchi and H. Ihara, “Evalu- Copyright © 2011 SciRes. AJAC  M. M. RAHMAN ET AL. Copyright © 2011 SciRes. AJAC 808 lidone onto Silica,” Journal of 7, No. 10, 1989, pp ation of Micro-Structural Features of a New Polymeric Organic Stationary Phase Grafted on Silica Surface: A Paradigm of Characterization of HPLC-Stationary Phases by a Combination of Suspension State 1H-NMR and Solid-State 13C-CP/MAS-NMR,” Analytica Chimica Act a, Vol. 547, 2005, pp 179-187. [29] M. Chamberg, R. Parnas and Y. Cohen, “Graft Polym- erization of Polyvinylpyrro Applied Polymer Science, Vol. 3 . 2921-2931. doi:10.1002/app.1989.070371011 [30] T. I. Korányi and J. B. Nagy, “Distribution of Aluminum in Different Periodical Building Units of MOR and BEA Zeolites,” Journal of Physical Chemistry B, Vol. 109, No. 33, 2005, pp. 15791-15797. doi:10.1021/jp051793k [31] D. M. Grant and B. V. Cheney, “Carbon-13 Resonance. VII. Steric Per Magnetic turbation of the Carbon-13 Chemical Shift,” Journal of the American Chemical So- ciety, Vol. 89, No. 21, 1967, pp. 5315-5316. doi:10.1021/ja00997a001 [32] A. E. Tonelli, F. C. Schiling and F. A. Bovey, “Confor- 7-1158. mational Origin of the Nonequivalent Carbon-13 NMR Chemical Shifts Observed for the Isopropyl Methyl Car- bons in Branched Alkanes,” Journal of the American Chemical Society, Vol. 106, No. 4, 1984, pp. 115 doi:10.1021/ja00316a079 [33] H. Englhardt and M. Jungheim, “Determination of Inor- ganic Anions, Carboxylic Acids and Amino Acids in Plant Matrices by Capillary Zone Electrophoresis,” Chromatographia, Vol. 45, No. 1, 1997, pp. 59-62. doi:10.1007/BF02505538 [34] K. Kimata, K. Iwaguchi, K. Jinno, R. Eksteen, K. Hosoya, M. Araki and N. Tanaka, The Journal of Chroma- tographic Science, Vol. 27, 1989, p. 721. [35] A. Shundo, T. Sakurai, M. Takafuji, S. Nagaoka and H. Ihara, “Molecular-Length and Chiral Discriminations by Beta-Structural Poly(L-Alanine) on Silica,” Journal of Chromatography A, Vol. 1073, No. 1-2, 2005, pp. 169- 174. doi:10.1016/j.chroma.2004.08.062 [36] H. Ihara, W. Dong, T. Mimaki, M. Nishihar M. Takafuji and S. Nagaoka, “Poly(4 a, T. Sakurai, -Vinylpyridine) as Novel Organic Phase for RP-HPLC. Unique Selectivity for Polycyclic Aromatic Hydrocarbons,” Journal of Liq- uid Chromatography & Related Technologies, Vol. 26, No. 15, 2003, pp. 2473-2485. doi:10.1081/JLC-120023796 [37] H. A. Claessens, M. A. Van Straten, C. A. Cramers, M. Jezierska and B. Buszewski, Journal of Chromatography A, Vol. 826, 1998, p. 135. [38] P. C. Sadek and P. W. Carr, The Journal of Chroma- tographic Science, Vol. 21, 1983, p. 314. [39] K. Jinno, K. Yamamoto, H. Nagashima, T. Ueda and K. Itoh, “Silicas Chemically Phenyl Groups as Station Bonded with Multidentate ary Phases in Reversed-Phase Liquid Chromatography Used for Non-Planarity Recog- nition of Polycyclic Aromatic Hydrocarbons,” Journal of Chromatography A, Vol. 517, 1990, pp. 193-207. doi:10.1016/S0021-9673(01)95721-8 [40] K. Jinno, T. Ibuki, N. Tanaka, M. Okamoto, J. C. Fretzer, W. R. Biggs, P. R. Griffiths and M. J. Olinger, The Jour- nal of Chromatographic Science, Vol. 461, 1989, p. 209. [41] H. A. Claessens, E. A. Vermeer and C. A. Cramers, se Liquid LC-GC International, Vol. 6, 1993, p. 692. [42] J. Layne, “Characterization and Comparison of the Chro- matographic Performance of Conventional, Polar- Em- bedded, and Polar-Endcapped Reversed-Pha Chromatography Stationary Phases,” Journal of Chro- matography A, Vol. 957, No. 2, 2002, pp. 149-164. doi:10.1016/S0021-9673(02)00193-0 [43] J. E. O’Gara, B. A. Alden, T. H. Walter, J. S. Petersen, C. 32 L. Niederlaender and U. D. Neue, “Simple Preparation of a C8 HPLC Stationary Phase with an Internal Polar Func- tional Group,” Analytical Chemistry, Vol. 67, No. 20, 1995, pp. 3809-3813. doi:10.1021/ac00116a0 ky, B. Buszewski, R. K. Gilpin [44] T. Czajkowska, I. Hrabovs and M. Jaroniec, “Comparison of the Retention of Or- ganic Acids on Alkyl and Alkylamide Chemically Bond- ed Phases,” Journal of Chromatography A, Vol. 691, No. 1-2, 1995, pp. 217-224. doi:10.1016/0021-9673(94)01034-C [45] W. Wielandt, A. Ellwanger, K. Albert and E. Lindner, “n-Alkyl Fluorenyl Phases in Chromatography: I. Syn- thesis and Characterization,” Journal of Chromatography A, Vol. 805, No. 1-2, 1998, pp. 71-83. doi:10.1016/S0021-9673(98)00014-4 [46] A. Ellwanger, R. Brindle, M. Kaiser, W. Wielandt, E. ions,” Journal Lindner and K. Albert, “n-Alkyl Fluorenyl Phases in Chromatography: II. Dynamic Behavior and High-Per- formance Liquid Chromatography Applicat of Chromatography A, Vol. 858, No. 2, 1999, pp. 133- 153. doi:10.1016/S0021-9673(99)00828-6 [47] R. Brindle and K. Albert, “Stationary Phases with Chemi- cally Bonded Fluorene Ligands: A New Approach for Environmental Analysis of π-Electron Containing Sol- utes,” Journal of Chromatography A, Vol. 757, No. 1-2, 1997, pp. 3-20. doi:10.1016/S0021-9673(96)00682-6 [48] K. Jinno and K. Kawasaki, “Correlation of the Retention Data of Polyaromatic Hydrocarbons Obtained on Various Stationary Phases Used in Normal- and Reversed-Phase Liquid Chromatography,” Chr 1, 1984, pp. 44-46. omatographia, Vol. 18, No. 7/BF02279465doi:10.100 [49] S. Sakaki, K. Kato, T. Miyazaki, K. Ohkubo, H. Ihara and C. Hirayama, The Journal of Chromatographic Science, Faraday Transaction 2, Vol. 9, 1993, p. 659. [50] C. S. Sychov, M. M. Ilyin, V. A. Davankov and K. O. Sochilina, “Elucidation of Retention Mechanisms on Hypercrosslinked Polystyrene Used as Column Packing Material for High-Performance Liquid Chromatography,” Journal of Chromatography A, Vol. 1030, No. 1-2, 2004, pp. 17-24. doi:10.1016/j.chroma.2003.10.098 [51] E. S. P. Bouvier and S. A. Oehrle, LC-GC International, Vol. 8, 1995, p. 338. [52] R. Brindle and K. Albert, “Stationary Phases with Chemi- cally Bonded Fluorene Ligands: A New Approach for Environmental Analysis of π-Electron Containing Sol- utes,” Journal of Chromatography A, Vol. 757, 1997, pp 3-20. doi:10.1016/S0021-9673(96)00682-6
|