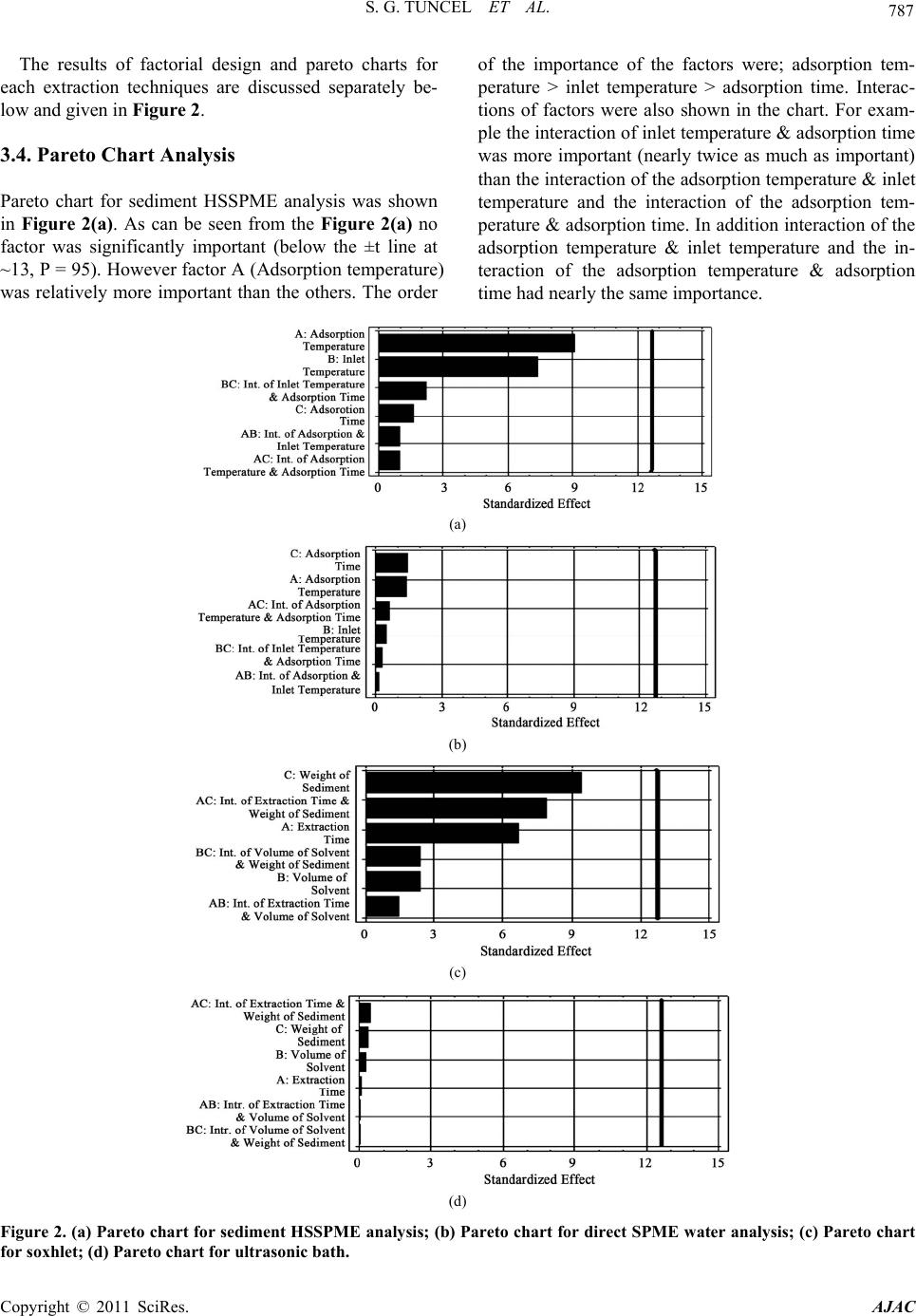
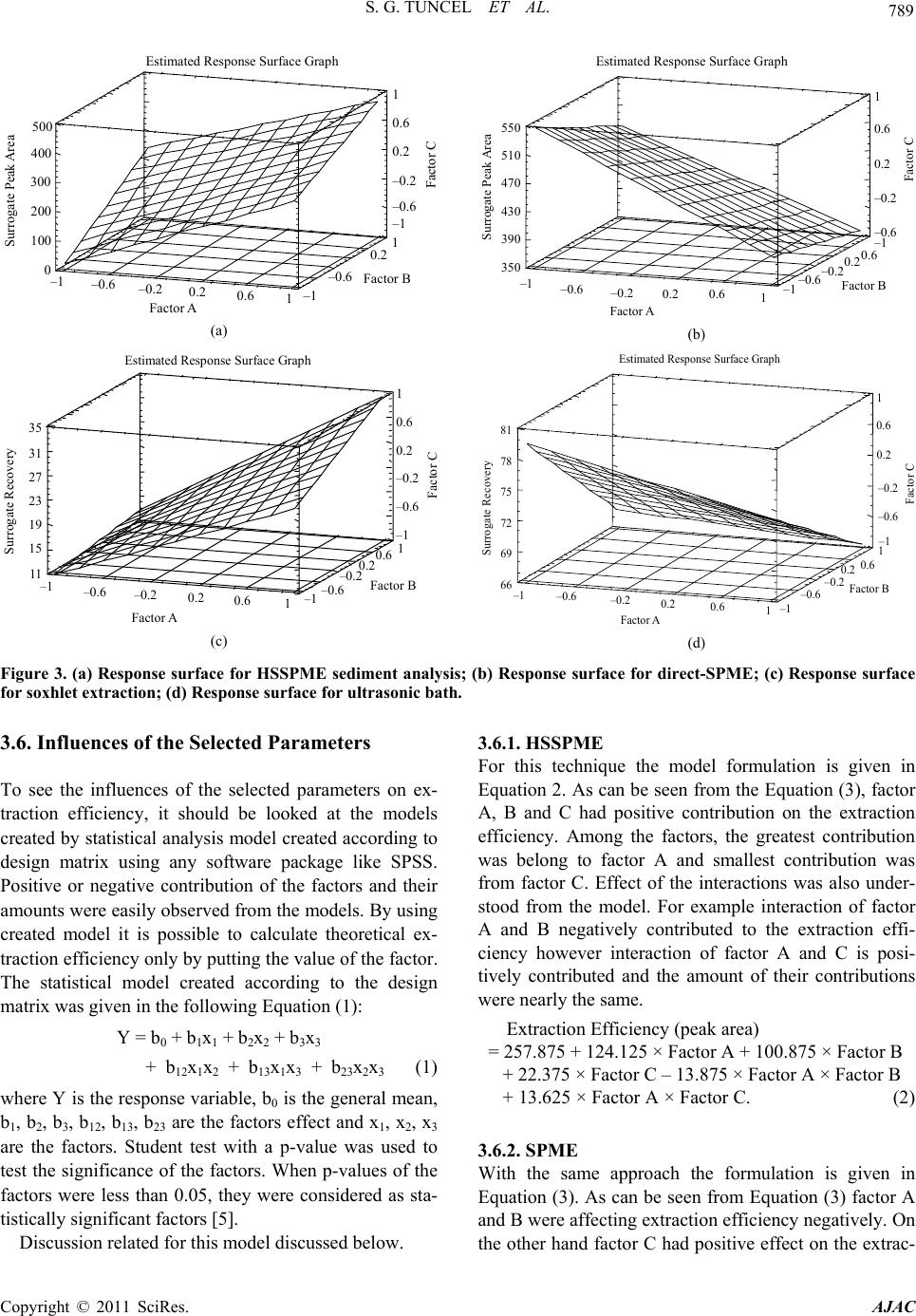
S. G. TUNCEL ET AL.
794
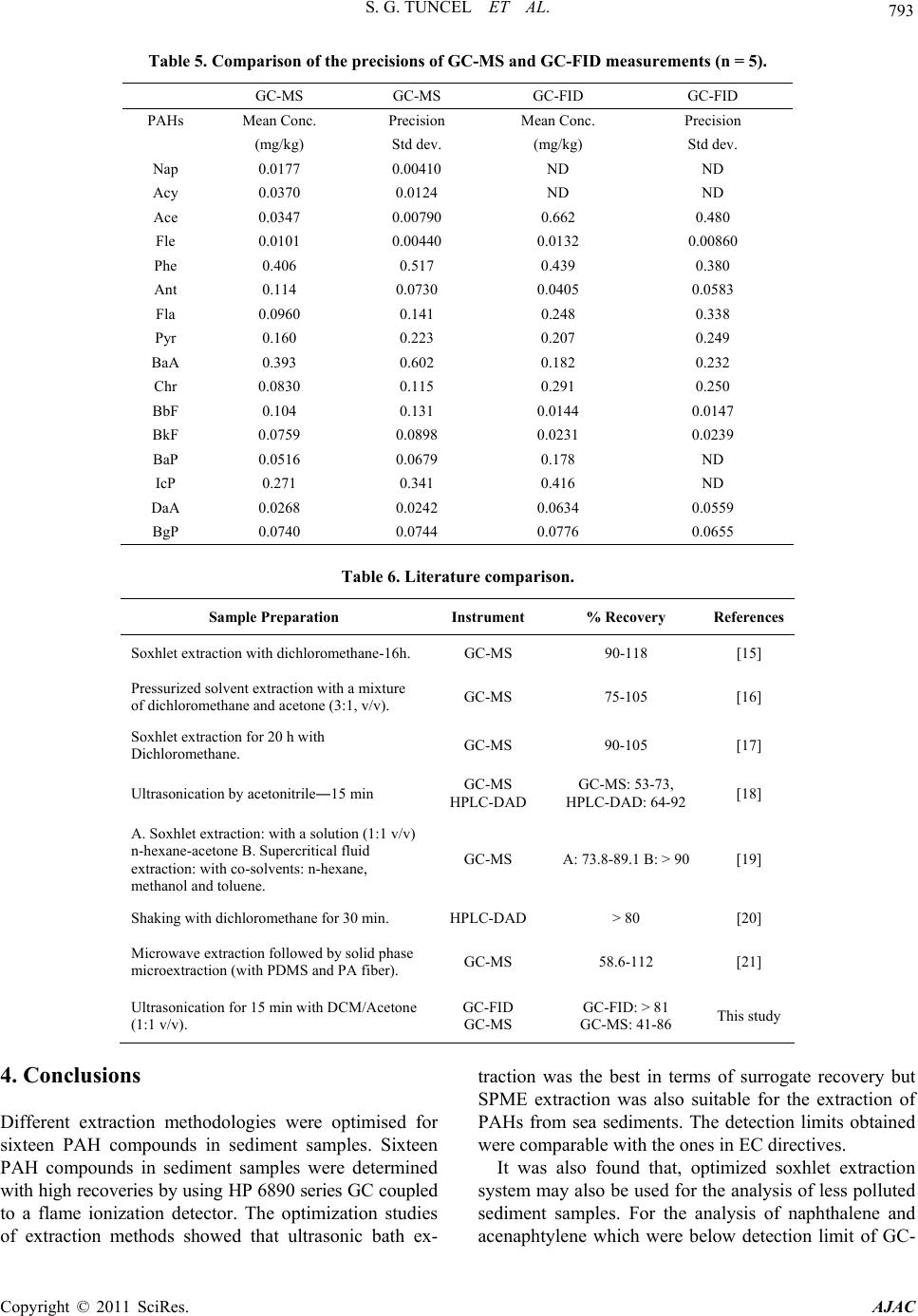
FID, GC-MS was offered as a better alternative.
Concentrations of PAHs in sediments were generally
moderate in Oludeniz sediment samples. Acenaphtene
and chrysene were dominant ones among the sixteen
PAHs analysed and their concentrations were 0.620 and
0.515 mg·kg–1 relatively.
5. References
[1] J. Xu, Y. Yu, P. Wang, W. Guo, S. Dai and H. Sun,
“Polycyclic Aromatic Hydrocarbons in the Surface Sedi-
ments from Yellow River, China,” Chemosphere, Vol. 67,
No. 7, 2007, pp. 1408-1414.
doi:10.1016/j.chemosphere.2006.10.074
[2] L. Culotta, C. D. Stefano, A. Gianguzza, M. R. Mannino
and S. Orecchio, “The PAH Composition of Surface
Sedi- ments from Stagnone Coastal Lagoon, Marsala
(Italy),” Marine Chemistry, Vol. 99, No. 1-4, 2006, pp.
117-127. doi:10.1016/j.marchem.2005.05.010
[3] Y. F. Song, X. Jing, S. Fleischmann and B. M. Wilke,
“Comparative Study of Extraction Methods for the Deter-
mination of PAHs from Contaminated Soils and Sedi-
ments,” Chemosphere, Vol. 48, No. 9, 2002, pp. 993-
1001. doi:10.1016/S0045-6535(02)00180-7
[4] N. F. Leite, P. P. Zamora and M. T. Grassi,
“Multifactorial Optimization Approach for the Determi-
nation of Polycyclic Aromatic Hydrocarbons in River
Sediments by Gas Chromatography-Quadrupole İon Trap
Selected İon Storage Mass Spectrometry,” Journal of
Chromatography A, Vol. 1192, No. 2, 2008, pp. 273-281.
doi:10.1016/j.chroma.2008.03.067
[5] M. B Deschamps, J. J. Daudin and E. Barriuso, “An
Experimental Design Approach to Optimise the Determi-
nation of Polycyclic Aromatic Hydrocarbons from
Rainfall Water Using Stir Bar Sorptive Extraction and
High Performance Liquid Chromatography-Fluorescence
Detection,” Journal of Chromatography A, Vol. 1167, No.
2, 2007, pp. 143-153. doi:10.1016/j.chroma.2007.08.025
[6] C. R. T. E. Tarley, G. Silveira, W. N. L. Santos, G. D.
Matos, E. G. P. Silva, M. A. Bezerra, M. Miró and S. L.
C. Ferreira, “Chemometric Tools in Electro Analytical
Chemistry: Methods for Optimization Based on Factorial
Design and Response Surface Methodology,” Micro
chemical Journal, Vol. 92, No. 1, 2009, pp. 58-67.
doi:10.1016/j.microc.2009.02.002
[7] J. N. Miller and J. C. Miller, “Statistics and Chemomet-
rics for Analytical Chemistry,” 4th Edition, Pearson
Education, England, 2000, Chapter 7.
[8] B. E. Berg, H. S. Lund, A. Kringstad and A. L. Kvern-
heim, “Routine Analysis of Hydrocarbons, PCB and PAH
in Marine Sediments Using Supercritical CO2 Extrac-
tion,” Chemosphere, Vol. 38, No. 3, 1999, pp. 587-599.
doi:10.1016/S0045-6535(98)00213-6
[9] D. R. Banjoo and P. K. Nelson, “Improved Ultrasonic
Extraction Procedure for the Determination of Polycyclic
Aromatic Hydrocarbons in Sediments,” Journal of Chro-
matography A, Vol. 1066, No. 1-2, 2005, pp. 9-18.
doi:10.1016/j.chroma.2005.01.033
[10] H. Budzinski, M. Letellier, P. Garrigues and K. Le Men-
ach, “Optimisation of the Microwave-Assisted Extraction
in Open Cell of Polycyclic Aromatic Hydrocarbons from
Soils and Sediments: Study of Moisture Effect,” Journal
of Chromatography A, Vol. 837, No. 1-2, 1999, pp. 187-
200. doi:10.1016/S0021-9673(99)00067-9
[11] EPA METHOD 3540 C, 1996.
[12] EPA METHOD SW-846 3550C, 2007.
[13] EPA METHOD 3550 B, 1996.
[14] A. Nikolaou, M. Kostopoulou, G. Lofrano and S. Meric,
“Determination of PAHs in Marine Sediments: Analytical
Methods and Environmental Concerns,” Global Nest
Journal, Vol. 11, 2009, pp. 391-405.
[15] C. Anyakora, A. Ogbeche, P. Palmer, H. Coker, G. Ukpo
and C. Ogah, “GC/MS Analysis of Polynuclear Aromatic
Hydrocarbons in Sediment Samples from the Niger Delta
Region,” Chemosphere, Vol. 60, No. 7, 2005, pp. 990-
997. doi:10.1016/j.chemosphere.2004.12.073
[16] R. Boonyatumanond, G. Wattayakorn, A. Togo and H.
Takada, “Distribution and Origins of Polycyclic Aromatic
Hydrocarbons (PAHs) in Riverine, Estuarine and Marine
Sediments in Thailand”, Marine Pollution Bulletin, Vol.
52, No. 8, 2006, pp. 942-956.
doi:10.1016/j.marpolbul.2005.12.015
[17] C. H. Koch, J. S. Khim, D. L. Villeneuve , K. Kannan
and J. P. Giesy, “Characterization of Trace Organic Con-
taminants in Marine Sediment from Yeongil Bay, Korea:
1. Instrumental Analyses,” Environmental Pollution, Vol.
142, No. 1, 2006, pp. 39-47.
doi:10.1016/j.envpol.2005.09.005
[18] A. Filipkowska, L. Lubecki and G. Kowalewska, “Poly-
cyclic Aromatic Hydrocarbon Analysis in Different Ma-
tri- ces of the Marine Environment,” Analytica Chimica
Acta, Vol. 547, No. 2, 2005, pp. 243-254.
doi:10.1016/j.aca.2005.05.023
[19] V. Librando, O. Hutzinger, G. Tringali and M. Aresta,
“Supercritical Fluid Extraction of Polycyclic Aromatic
Hy- drocarbons from Marine Sediments and Soil Sam-
ples,” Chemosphere, Vol. 54, No. 8, 2004, pp. 1189-1197.
doi:10.1016/j.chemosphere.2003.07.008
[20] R. A. Gimeno, E. Comas, R. M. Marcé, J. Ferré, F. X.
Rius and F. Borrull, “Second-Order Bilinear Calibration
for De- termining Polycyclic Aromatic Compounds in
Marine Sediments by Solvent Extraction and Liquid
Chromatography with Diode-Array Detection,” Analytica
Chimica Acta, Vol. 498, No. 1-2, 2003, pp. 47-53.
doi:10.1016/j.aca.2003.08.057
[21] V. Pino, J. H. Ayala, A. M. Afonso and V. Gonzalez,
“Micellar Microwave-Assisted Extraction Combined with
Solid-Phase Microextraction for the Determination of
Polycyclic Aromatic Hydrocarbons in a Certified Marine
Sediment,” Analytica Chimica Acta, Vol. 477, No. 1,
2003, pp. 81-91.
doi:10.1016/S0003-2670(02)01410-1
Copyright © 2011 SciRes. AJAC