 Advances in Chemical Engi neering and Science , 2011, 1, 176-182 doi:10.4236/aces.2011.14026 Published Online October 2011 (http://www.SciRP.org/journal/aces) Copyright © 2011 SciRes. ACES Study on Co-Effect o f K 2SO4 Deposition and Low Concentration SO2 on Performances of V2O5/AC Catalysts for Low Temperature SCR Xianlong Zhang1,2, Bowen Shi1, Xueping Wu1, Zhanggen Huang2, Zhenyu Liu2*, Baojun Yang1, Cuiping Zhang1 1School of Chemical Engin eering, Hefei University of Technology, Hefei, China 2State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, China E-mail: *zhangxianlong@yahoo.com.cn Recieved August 20, 2011; revised September 13, 2011; accepted Se pt em ber 21, 2011 Abstract Simulated compounds were prepared by loading K2SO4 onto V2O5/AC catalysts. Study the effect of K2SO4 on V1/AC catalysts in the presence of low concentration SO2. Transient response techniques, TPD was car- ried out. The results indicated that the DeNO activity of V1/AC catalysts was decreased seriously in the early period of operation, but the deactivation was gradually diminished with SO2 adsorption and then, it was completely eliminated. For the sulphated catalysts (saturated catalysts by sulphate), their SCR activity were free from existence of gaseous SO2, the loss of activity about 10% caused by K2SO4 was found on them. The deactivation of K2SO4 deposited catalysts was due to the decrease of adsorbed and activated NH3, or some acid sites. Keywords: Vanadium Oxide Catalyst, SCR, K2SO4 Deposition, Deactivation, Sulphated Catalyst 1. Introduction Selective catalytic reduction of NOx (SCR) is one of the most widely used technologies for reducing NOx from stationary sources. The optimum operating temperature of the commercial catalyst (i.e.,V2O5/TiO2 promoted with WO3 or MoO3) is in the range of 300˚C - 500˚C for keep- ing free from SO2 deactivation, which makes it necessary to reheat the flue gas after the electrostatic precip ita tor and desulphuriser (120˚C - 250˚C). Thus the search for new catalysts which are active and stable at low temperatures and in presence of low SO2 is still under way. In recent years, a considerable effort has been focused on some low temperature SCR catalysts su ch as MnOx/NaY zeolite [1], MnOx/activated carbon (MnOx/AC) [2,3] and especially V2O5/AC [4-6]. There are some submicron particles (though very little) which contain alkali and alkali earth metals in coal fired flue gas especially in some coal-biomass co-fired flue gases even after the electrostatic precipitator and desul- phuriser [7-9]. Those metals, mainly potassium, volatize from coal during combustion and then condense to form submicron particles such as K2SO4 or K2O. They are ma- jor poisoning substances for SCR catalyst [10-12]. It is widely believed that the catalyst activity is related to the ammonia adsorbed on Bronsted acid sites associated with V5+-OH [13-15]. When deposited, potassium preferen- tially co-ordinates to the hydroxyl groups on V2O5 and the catalytic activity decrease with the decrease in both the number and the strengt h of the Bronst ed acid si tes [10,12]. Under lower temperature (150˚C - 250˚C), V2O5/AC has been proved to be a promising catalyst for SCR, and also a perfect absorbance of SO2 due to its catalytic oxidation of SO2 to SO3 [5,6,16]. More interestingly, the adsorption of SO2 on V2O5/AC catalyst can remarkably promote the ac- tivity of SCR over it [17] , which was suggestibl y due to t he increased acidity of the catalyst. When the V2O5/AC cata- lyst is used for SCR downstream of desulfurizer, there is also some low concentration of SO2 in flue gas. In this case, the possible deactivation of V2O5/AC by deposition of po- tassium compounds is an important factor to be considered for the performance of this catalyst. However, there are few research have been reported to study it so far. This paper is to investigate the effect of potassium in flue gas on activity  X. L. ZHANG ET AL. 177 of V2O5/AC catalyst for SCR in presence of low SO2. K2SO4 was loaded on V2O5/AC catalyst as model com- pound of potassium by method of pore volume impregna- tion. The use of activity measurement, transient response techniques, and temperature programmed techniques (TPD) enable us to probe into the reason for the changes in activ- ity of V2O5/AC under deposition of K2SO4 and sulfa tion in low SO2. 2. Experimental 2.1. Catalyst Preparation The activated carbon (AC) used was a commercial prod- uct from Shanxi Xihua Chem. Co. LTD. The AC was crushed into particles of 0.3 - 0.6 mm in diameter and subjected to proximate and ultimate analyses. V2O5 was supported to the AC by pore volume impregnation using an aqueous solution containing ammonium metavanadate and oxalic acid. The sample was then dried at 50˚C for 12 h and 110˚C for 5 h, followed by calcination in Ar at 500˚C for 8 h and pre-oxidation in air at 250˚C for 5 h. The catalyst used in this work which contains 1wt% V2O5 was termed as V1/AC. K2SO4 was loaded onto V1/AC also through impreg- nation, followed by drying at 50˚C for 12 h and at 110˚C for 5 h. The K2SO4 loaded catalysts were termed as KxV1/AC, where x denoted the weight percentage of K in the catalyst. -S-SCR standed for the catalysts after SCR reaction in presence of low SO2 at 250˚C till being saturated by sulphate and SCR activity keeps steady. 2.2. Catalytic Activity Measurement SCR activity of the catalysts was measured in a fixed bed glass reactor of 15 mm in diameter. The feed rates of NH3/Ar, NO/Ar and SO2/Ar were controlled by mass flow meters and that of Ar and air were controlled by rotameters. H2O vapor was introduced by passing the Ar stream through a heated gas-wash bottle containing de- ionized water. The overall feed contained 600 ppm NO, 600 ppm NH3, 3.4% O2, 2.6% H2O vapor, 300 ppm SO2 (when used) and balanced Ar. The total flow was main- tained at 300 ml/min all the time, corresponding to a space velocity of 22500 - 4500 h–1 in the case of 0.5 - 2.5 g Cat. being used. The reaction temperature was varied between 100˚C - 250˚C. The conc entr ation of NO an d O 2 in the inlet and outlet of the reactor was measured on-line by a flue gas analyzer (KM9006). Conversion of NO, XNO, is defi ned by (1) in out NO in CC X 100% C (1) where Cin and Cout are NO concentrations in the inlet and outlet, respectively. -dXNO, the differences of NO con- version between V1/AC with and with out K2SO4 loaded, is used to denote the deactivation of V1/AC by loading K2SO4. In the experiment, the content of SO2 adsorbed on V1/AC is calculated and labeled as SC (mg SO2/g Cat.). 2.3. Transient-Response Transient-response experiments have been widely used for the SCR study of NO over commercial V2O5/TiO2 catalysts [18-20]. We carried out experiments at 250˚C by stepwise removal-addition one of the reactants SO2, O2 or NH3 from steady-state. Contrarily to steady-state experiments, the dynamics of SCR reaction can be ana- lyzed. Thus these experiments were allowed to unravel the steps involved in the mechanism of reaction. From the steady-state reaction conditions, the same as in 2.2, one reactant was replaced by equal volume of balance Ar, the outlet NO concentration was measured on-line by flue gas analyz er. 2.4. Temperature-Programmed Desorption (TPD) The sulphate species in the catalyst with or without K2SO4 loaded were characterized by temperature- pro- grammed desorption (TPD) with the use of a mass spec- trometer (Balzers QMS422) for the gas stream analysis. TPD experiments were carried out in a quartz micro- reactor. In a typical experiment, 100ml/min of Ar was passed over the catalyst until the signal of the mass spec- trometer was stable. Later the temperature was raised to 700˚C at a rate of 10˚C/min. The following main mass- to-charge (m/e) ratios were used to monitor the outlet gas composition: 17 (NH3), 18 (H2O), 28 (N2/CO), 30 (NO) , 32 (O2), 44 (CO2), and 64 (SO2). 3. Results and Discussion 3.1. Catalytic Activity It is widely believed that vanadium in oxidation state +5 undergoes a redox cycle and oxidizes SO2 to SO3 [16,21-23], and in the case of AC as supporter, the converted SO3 companying with H2O is retained by the AC supporter (sulphation of Cat.). Figure 1 shows the real-time NO conversion along with the outlet SO2 concentration as a function of reaction time on stream over V1/AC catalyst with or without K2SO4 loaded. Firstly, it can be observed that the SO2 concentration describes a typical breakthrough curve. The SO2 breakthrough time on each catalyst follows the order: Copyright © 2011 SciRes. ACES 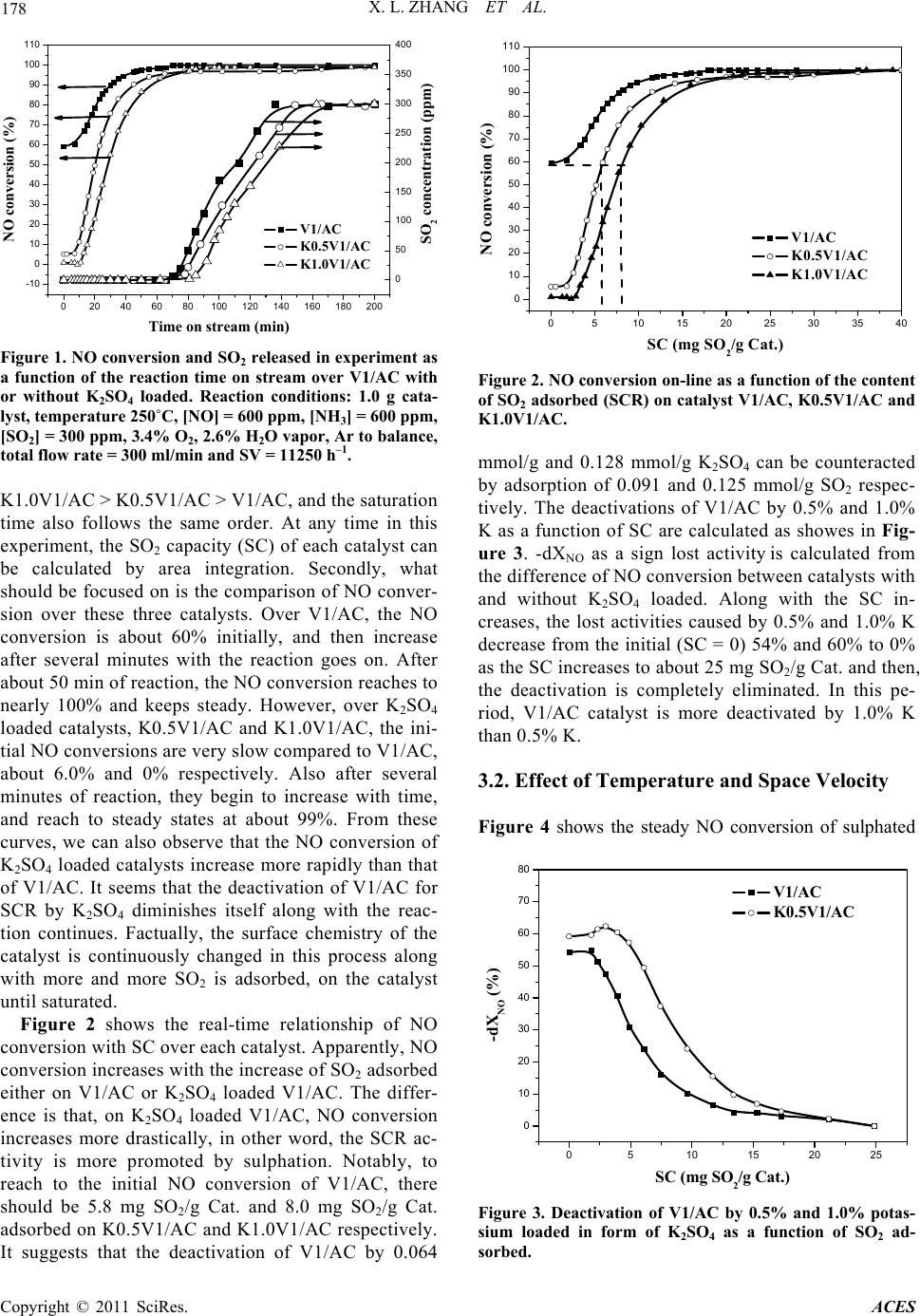 178 X. L. ZHANG ET AL. 020406080100 120 140 160 180 200 -10 0 10 20 30 40 50 60 70 80 90 100 110 SO2 concentration (ppm) NO conversion (%) Time on stream (min) V1/AC K0.5V1/AC K1.0V1/AC 0 50 100 150 200 250 300 350 400 Figure 1. NO conversion and SO 2 released in experiment as a function of the reaction time on stream over V1/AC with or without K2SO4 loaded. Reaction conditions: 1.0 g cata- lyst, temperature 250˚C, [NO] = 600 ppm, [NH3] = 600 ppm, [SO2] = 300 ppm, 3.4% O2, 2.6% H2O vapor, Ar to balance, total flow rate = 300 ml/min and SV = 11250 h–1. K1.0V1/AC > K0.5V1/AC > V1/AC, and the saturat ion time also follows the same order. At any time in this experiment, the SO2 capacity (SC) of each catalyst can be calculated by area integration. Secondly, what should be focused on is the comparison of NO conver- sion over these three catalysts. Over V1/AC, the NO conversion is about 60% initially, and then increase after several minutes with the reaction goes on. After about 50 min of reaction, the NO conversion reaches to nearly 100% and keeps steady. However, over K2SO4 loaded catalysts, K0.5V1/AC and K1.0V1/AC, the ini- tial NO conversions are very slow compared to V1/AC, about 6.0% and 0% respectively. Also after several minutes of reaction, they begin to increase with time, and reach to steady states at about 99%. From these curves, we can also observe that the NO conversion of K2SO4 loaded catalysts increase more rapidly than that of V1/AC. It seems that the deactivation of V1/AC for SCR by K2SO4 diminishes itself along with the reac- tion continues. Factually, the surface chemistry of the catalyst is continuously changed in this process along with more and more SO2 is adsorbed, on the catalyst until satura ted. Figure 2 shows the real-time relationship of NO conversion with SC over each catalyst. Apparently, NO conversion increases with the increase of SO2 adsorbed either on V1/AC or K2SO4 loaded V1/AC. The differ- ence is that, on K2SO4 loaded V1/AC, NO conversion increases more drastically, in other word, the SCR ac- tivity is more promoted by sulphation. Notably, to reach to the initial NO conversion of V1/AC, there should be 5.8 mg SO2/g Cat. and 8.0 mg SO2/g Cat. adsorbed on K0.5V1/AC and K1.0V1/AC respectively. It suggests that the deactivation of V1/AC by 0.064 0510 15 20 25 30 35 40 0 10 20 30 40 50 60 70 80 90 100 110 NO conversion (%) SC (mg SO2/g Cat.) V1/AC K0.5V1/AC K1.0V1/AC Figure 2. NO conversion on-line as a function of the content of SO2 adsorbed (SCR) on catalyst V1/AC, K0.5V1/AC and K1.0V1/AC. mmol/g and 0.128 mmol/g K2SO4 can be counteracted by adsorption of 0.091 and 0.125 mmol/g SO2 respec- tively. The deactivations of V1/AC by 0.5% and 1.0% K as a function of SC are calculated as showes in Fig- ure 3. -dXNO as a sign lost activity is calculated from the difference of NO conversion between catalysts with and without K2SO4 loaded. Along with the SC in- creases, the lost activities caused by 0.5% and 1.0% K decrease from the initial (SC = 0) 54% and 60% to 0% as the SC increases to about 25 mg SO2/g Cat. and then, the deactivation is completely eliminated. In this pe- riod, V1/AC catalyst is more deactivated by 1.0% K than 0.5% K. 3.2. Effect of Temperature and Space Velocity Figure 4 shows the steady NO conversion of sulphated 0510 15 20 25 0 10 20 30 40 50 60 70 80 -dXNO (%) SC (mg SO2/g Cat.) V1/AC K0.5V1/AC Figure 3. Deactivation of V1/AC by 0.5% and 1.0% potas- sium loaded in form of K2SO4 as a function of SO2 ad- sorbed. Copyright © 2011 SciRes. ACES 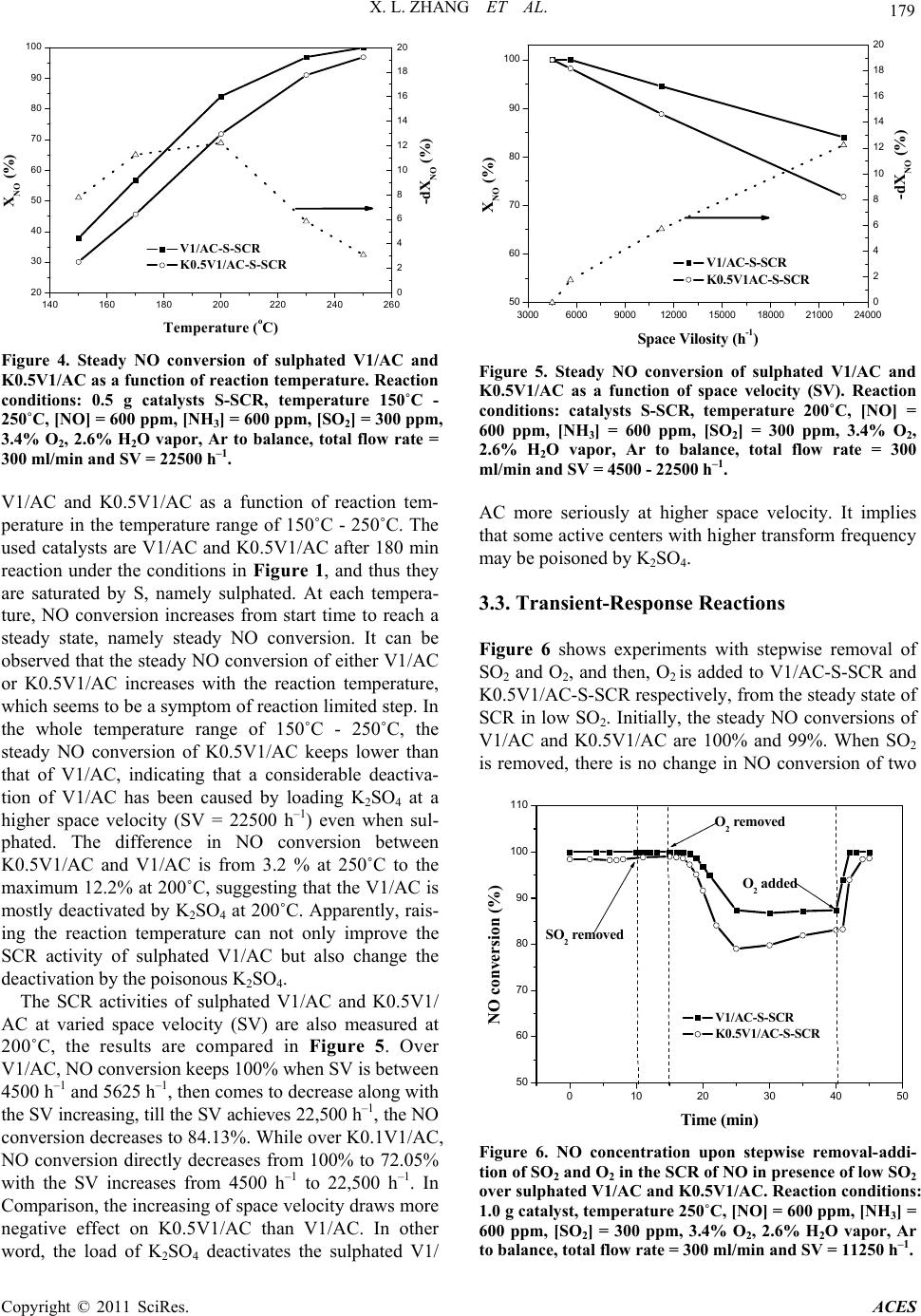 X. L. ZHANG ET AL. 179 140 160 180 200 220 240 260 20 30 40 50 60 70 80 90 100 -dXNO (%) XNO (%) Temperature (oC) V1/AC-S-SCR K0.5V1/AC-S-SCR 0 2 4 6 8 10 12 14 16 18 20 Figure 4. Steady NO conversion of sulphated V1/AC and K0.5V1/AC as a function of reaction temperature. Reaction conditions: 0.5 g catalysts S-SCR, temperature 150˚C - 250˚C, [NO] = 600 ppm, [NH3] = 600 ppm, [SO2] = 300 ppm, 3.4% O2, 2.6% H2O vapor, Ar to balance, total flow rate = 300 ml/min and SV = 22500 h–1. V1/AC and K0.5V1/AC as a function of reaction tem- perature in the temperature range of 150˚C - 250˚C. The used catalysts are V1/AC and K0.5V1/AC after 180 min reaction under the conditions in Figure 1, and thus they are saturated by S, namely sulphated. At each tempera- ture, NO conversion increases from start time to reach a steady state, namely steady NO conversion. It can be observed that the steady NO conversion of eith er V1/AC or K0.5V1/AC increases with the reaction temperature, which seems to be a symptom of reaction limited step. In the whole temperature range of 150˚C - 250˚C, the steady NO conversion of K0.5V1/AC keeps lower than that of V1/AC, indicating that a considerable deactiva- tion of V1/AC has been caused by loading K2SO4 at a higher space velocity (SV = 22500 h–1) even when sul- phated. The difference in NO conversion between K0.5V1/AC and V1/AC is from 3.2 % at 250˚C to the maximum 12.2% at 200˚C, suggesting that the V1/AC is mostly deactivated by K2SO4 at 200˚C. Apparently, rais- ing the reaction temperature can not only improve the SCR activity of sulphated V1/AC but also change the deactivatio n by the poisono us K2SO4. The SCR activities of sulphated V1/AC and K0.5V1/ AC at varied space velocity (SV) are also measured at 200˚C, the results are compared in Figure 5. Over V1/AC, NO conversion keeps 100% when SV is between 4500 h–1 and 5625 h–1, th en co mes to de crease alon g with the SV increasi ng, till the SV achieves 22,500 h –1, the NO conversion decreases to 84.13%. While over K0.1V1/AC, NO conversion directly decreases from 100% to 72.05% with the SV increases from 4500 h–1 to 22,500 h–1. In Comparison, the increasing of space velocity draws more negative effect on K0.5V1/AC than V1/AC. In other word, the load of K2SO4 deactivates the sulphated V1/ 30006000900012000 15000 18000 21000 24000 50 60 70 80 90 100 -dXNO (%) XNO (%) Space Vilosity (h-1) V1/AC- S-SCR K0.5V1AC-S-SCR 0 2 4 6 8 10 12 14 16 18 20 Figure 5. Steady NO conversion of sulphated V1/AC and K0.5V1/AC as a function of space velocity (SV). Reaction conditions: catalysts S-SCR, temperature 200˚C, [NO] = 600 ppm, [NH3] = 600 ppm, [SO2] = 300 ppm, 3.4% O2, 2.6% H2O vapor, Ar to balance, total flow rate = 300 ml/min and SV = 4500 - 22500 h–1. AC more seriously at higher space velocity. It implies that some active centers with higher transform frequency may be poisoned by K2SO4. 3.3. Transient-Response Reactions Figure 6 shows experiments with stepwise removal of SO2 and O2, and then, O2 is added to V1/AC-S-SCR and K0.5V1/AC-S-SCR respectively, from the steady state of SCR in low SO2. Initially, the steady NO conversions of V1/AC and K0.5V1/AC are 100% and 99%. When SO2 is removed, there is no change in NO conversion of two 0 102030405 50 60 70 80 90 100 110 0 O2 added O2 removed SO2 removed NO conversion (%) Time (min) V1/AC-S-SCR K0.5V1 /AC-S-SCR Figure 6. NO concentration upon stepwise removal- addi- tion of SO2 and O2 in the SCR of NO in presence of low SO2 over sulphated V1/AC and K0.5V1/AC. Reaction conditions: 1.0 g catalyst, temperature 250˚C, [NO] = 600 ppm, [NH3] = 600 ppm, [SO2] = 300 ppm, 3.4% O2, 2.6% H2O vapor, Ar to balance, total flow rate = 300 ml/min and SV = 11250 h–1. Copyright © 2011 SciRes. ACES 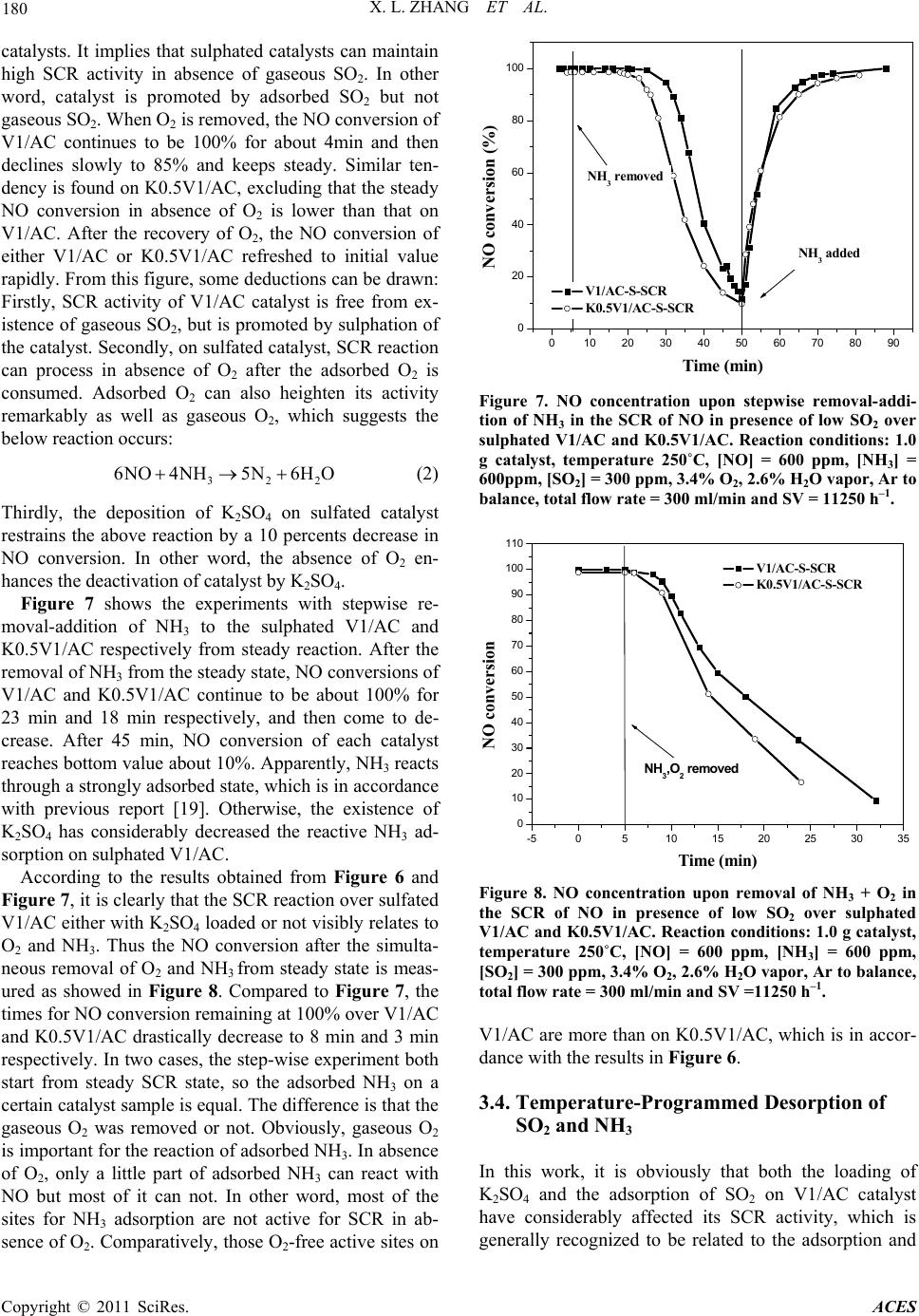 X. L. ZHANG ET AL. 180 2 catalysts. It implies that sulphated catalysts can maintain high SCR activity in absence of gaseous SO2. In other word, catalyst is promoted by adsorbed SO2 but not gaseous SO2. When O2 is removed, the NO conversion of V1/AC continues to be 100% for about 4min and then declines slowly to 85% and keeps steady. Similar ten- dency is found on K0.5V1 /AC, excluding that the steady NO conversion in absence of O2 is lower than that on V1/AC. After the recovery of O2, the NO conversion of either V1/AC or K0.5V1/AC refreshed to initial value rapidly. From this figure, some deductions can be drawn: Firstly, SCR activity of V1/AC catalyst is free from ex- istence of gaseous SO2, but is promoted by sulphation of the catalyst. Secondly, on sulfated catalyst, SCR reaction can process in absence of O2 after the adsorbed O2 is consumed. Adsorbed O2 can also heighten its activity remarkably as well as gaseous O2, which suggests the below reaction occurs: 32 6NO4NH5N6HO (2) Thirdly, the deposition of K2SO4 on sulfated catalyst restrains the above reaction by a 10 percents decrease in NO conversion. In other word, the absence of O2 en- hances the deactivation of catalyst by K2SO4. Figure 7 shows the experiments with stepwise re- moval-addition of NH3 to the sulphated V1/AC and K0.5V1/AC respectively from steady reaction. After the removal of NH3 from the steady state, NO conversions of V1/AC and K0.5V1/AC continue to be about 100% for 23 min and 18 min respectively, and then come to de- crease. After 45 min, NO conversion of each catalyst reaches bottom value about 10%. Apparently, NH3 reacts through a strongly adsorbed state, which is in accordance with previous report [19]. Otherwise, the existence of K2SO4 has considerably decreased the reactive NH3 ad- sorption on sul phat ed V1/AC. According to the results obtained from Figure 6 and Figure 7, it is clearly that the SCR reaction over sulfated V1/AC either with K2SO 4 loaded or not visibly relates to O2 and NH3. Thus the NO conversion after the simulta- neous removal of O2 and NH3 from steady state is meas- ured as showed in Figure 8. Compared to Figure 7, the times for NO conv ersion remain ing at 100 % over V1/A C and K0.5V1/AC drastically decrease to 8 min and 3 min respectively. In two cases, the step-wise experiment both start from steady SCR state, so the adsorbed NH3 on a certain catalyst sample is equal. The difference is that the gaseous O2 was removed or not. Obviously, gaseous O2 is important for the reaction of adsorbed NH 3. In absence of O2, only a little part of adsorbed NH3 can react with NO but most of it can not. In other word, most of the sites for NH3 adsorption are not active for SCR in ab- sence of O2. Comparatively, those O2-free active sites on 0 102030405060708090 0 20 40 60 80 100 NH3 added NH3 removed NO conversion (%) Time (min) V1/AC-S- SCR K0.5V1/ AC-S-SCR Figure 7. NO concentration upon stepwise removal- addi- tion of NH3 in the SCR of NO in presence of low SO2 over sulphated V1/AC and K0.5V1/AC. Reaction conditions: 1.0 g catalyst, temperature 250˚C, [NO] = 600 ppm, [NH3] = 600ppm, [SO2] = 300 ppm, 3.4% O2, 2.6% H2O vapor, Ar to balance, total flow rate = 300 ml/min and SV = 11250 h–1. -50510 15 20 25 30 35 0 10 20 30 40 50 60 70 80 90 100 110 NH3,O2 remov e d NO conversion Time (min) V1/AC-S-SCR K0.5V1 / AC-S-SCR Figure 8. NO concentration upon removal of NH3 + O2 in the SCR of NO in presence of low SO2 over sulphated V1/AC and K0.5V1/AC. Reaction conditions: 1.0 g catalyst, temperature 250˚C, [NO] = 600 ppm, [NH3] = 600 ppm, [SO2] = 300 ppm, 3.4% O2, 2.6% H2O vapor, Ar to balance, total flow rate = 300 ml/min and SV =11250 h–1. V1/AC are more than on K0.5V1/AC, which is in accor- dance with the results in Figure 6. 3.4. Temperature-Programmed Desorption of SO2 and NH3 In this work, it is obviously that both the loading of K2SO4 and the adsorption of SO2 on V1/AC catalyst have considerably affected its SCR activity, which is generally recognized to be related to the adsorption and Copyright © 2011 SciRes. ACES  X. L. ZHANG ET AL. 181 activation of NH3 on the catalyst. Thus, TPD experi- ments were carried out on V1/AC-S-SCR and K0.5V1/ AC-S-SCR to examine the desorption of SO2 and NH3 from each sample as shown in Figure 9 and Figure 10 respectively. Figure 9 shows the similar SO2 TPD pro- files of V1/AC-S-SCR and K0.5V1/AC-S-SCR. The desorption of SO2 from each sample starts at about 290˚C and reached the first (and also the main) peak at about 345˚C. Along with the temperature increasing, a should er peak a ppears after 400˚C for both samples. It is apparently that SO2 desorbs from K0.5V1/AC-S-SCR is more than that from V1/AC-S-SCR, which is in accor- dance to the result of Figure 1. In Figure 10, both the V1/AC-S-SCR and K0.5V1/AC-S-SCR appear a main NH3 desorption peak at about 3 15˚C with similar profile. This peak may assign to the NH3 desorption from acid sites which can be independent from alkali metal and adsorbed SO2, so the peak profile and intensity is similar. Also, there aresome notable differences in NH3 TPD profiles of the two samples. Besides the first main peak, 250 300 350 400 450 500 550 600 0.00E+000 5.00E-010 1.00E-009 1.50E-009 2.00E-009 m/e = 64 MS intensity of SO2 (a.u.) Temperature (oC) V1/AC-S-SCR K0.5 V1 / AC-S-SCR Figure 9. SO2 TPD profiles of V1/AC-S-SCR and K0.5V1/ AC-S-SCR. 250 300 350 400 450 500 550 600 0.00E+000 1.50E-010 3.00E-010 4.50E-010 m/e = 15 MS intensity of NH3 (a.u.) Temperature (oC) V1/AC-S-SCR K0.1 V1/AC- S-SCR Figure 10. NH3 TPD profiles of V1/AC-S-SCR and K0.5V1/ AC-S-SCR. another two desorption peaks at about 450˚C and 550˚C are observed on V1/AC-S-SCR,but can’t be found on K0.5V1/AC-S-SCR. These two peaks are related to NH3 desorption from acid sites which inclined to be affected by K ion. When K is addition to the V1/AC-S-SCR catalyst, the acidity of the two acid sites decrease, in other words, NH3 adsorption capacity decrease, so the deactivation take place. More SO2 adsorption on K0.5V1/AC-S-SCR (Figure 2) may enhance the acidity of the two acid sites, so the deactivation can be counter- acted. 4. Conclusions The deposited K2SO4 decrease the SCR activity of V2O5/AC catalyst, but the deactivation caused by K2SO4 can be counteracted by SO2 adsorption. The SCR activity of both V1/AC and KxV1 /AC at low temperature (150˚C - 250˚C) can be promoted by SO2 adsorption especially the latter. The deactivation caused by K2SO4 can be en- hanced with space velocity increasing but being coun- teracted with temperature increasing. When the catalysts are saturated and steady, their SCR activity is no longer affected by the gaseous SO2. And their SCR process can still process without gaseous or adsorbed O2, but the ab- sence of O2 enhances the deactivation of catalyst by K2SO4. The deactivation of K2SO4 deposited catalysts is due to the decrease of adsorbed and activated NH3, or some acid sites. 5. Acknowledgements The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (40902020, 51002042), Ph.D. Programs Foundation of Ministry of Education of China (20090111120019). 6. References [1] M. Richter, A. Trunschke, U. Bentrup, K.Brzezinka, E. Schreier, M. Schneider, M. Pohl and R. Fricke, “ Selec- tive Catalytic Reduction of Nitric Oxide by Ammonia over Egg-Shell MnOx/NaY Composite Catalysts,” Jour- nal of Catalysis, Vol. 206, No. 1, 2002, pp. 98-113. doi:10.1006/jcat.2001.3468 [2] G. Marban, T.Valdes-Solis and A. B. Fuertes, “Mecha- nism of Low-Temperature Selective Catalytic Reduction of NO with NH3 over Carbon-Supported Mn3O4. Active Phase and Role of Surface NO Species,” Physical Che- mistry Chemical Physics, Vol. 6, No. 2, 2004, pp. 453- 464. doi:10.1039/b313752j [3] T. Grzybek, J. Pasel and H. Papp, “Supported Manganese Catalysts for the Selective Catalytic Reduction of Nitro- gen Oxides with Ammonia Part II. Catalytic Experi- ments,” Physical Chemistry Chemical Physics, Vol. 1, No. Copyright © 2011 SciRes. ACES  X. L. ZHANG ET AL. Copyright © 2011 SciRes. ACES 182 2, 1999, pp. 341-348. doi:10.1039/a806913a [4] T. Valdes-Solis, G. Marban and A. B. Fuertes, “Low- Temperature SCR of NOx with NH3 over Carbon- Ceramic Supported Catalysts,” Applied Catalysis B: En- vironmental, Vol. 46, No. 2, 2003, pp. 261-271. doi:10.1016/S0926-3373(03)00217-0 [5] Z. Zhu, Z. Liu, H. Niu and S.Liu, “Promoting Effect of SO2 on Activated Carbon-Supported Vanadia Catalyst for NO Reduction by NH3 at Low Temperatures,” Journal of Catalysis, Vol. 187, No.1, 1999, pp. 245-248. doi:10.1006/jcat.1999.2605 [6] Z. Zhu, Z. Liu, H. Niu, T. Hu, T. Liu and Y. Xie, “Mechanism of SO2 Promotion for NO Reduction with NH3 over Activated Carbon-Supported Vanadium Oxide Catalyst,” Journal of Catalysis, Vol. 197, No. 1, 2001, pp. 6-16. doi:10.1006/jcat.2000.3052 [7] T. Valmari, T. Lind and E. I. Kauppinen, “Field Study on Ash Behavior during Circulating Fluidizied-Bed Com- bustion of Biomass. 2. Ash Deposition and Alkali Vapor Condensation,” Energy & Guels, Vol. 13, No. 2, 1999, pp. 390-395. [8] L. Zhang and Y. Ninomiya, “Emission of Suspended PM10 from Laboratory-Scale Coal Combustion and Its Correlation with Coal Mineral Properties,” Fuel, Vol. 85, No. 2, 2006, pp. 194-203. doi:10.1016/j.fuel.2005.03.034 [9] L. Zhang, M. Ito, A. Sato and Y. Ninomiya, “Fate of Al- kali Elements during Pyrolysis and Combustion of Chi- nese Coals,” Journal of Chemical Engineering of Japan, Vol. 36, No. 3, 2004, pp. 759-768. [10] J. P. Chen and R. T. Yang, “Mechanism of Poisoning of the V2O5/TiO2 Catalyst for the Reduction of NO by NH3,” Journal of Catalysis, Vol. 125, No. 2, 1990, pp. 411-420. doi:10.1016/0021-9517(90)90314-A [11] R. Khodayari, “Selective Catalytic Reduction of NOx: Experimental and Theoretical Studies of Deactivatin,” Li- centiate Thesis, University of Lund, Lund, 1998. [12] H. Kamata, K. Takahashi and C. U. I. Odenbrand, “The Role of K2O in the Selective Reduction of NO with NH3 over a V2O5 (WO3)/TiO2 Commercial Selective Catalytic Reduction Catalyst,” Journal of Molecular Catalysis A: Chemical, Vol. 139, No. 5, 1999, pp. 189-198. doi:10.1016/S1381-1169(98)00177-0 [13] N. Y. Topsoe, H. Topsoe and J. A. Dumesic, “Vanadia/ Titania Catalysts for Selective Catalytic Reduction (SCR) of Nitric-Oxide by Ammonia: I. Combined Temperature- Programmed in-Situ FTIR and On-line Mass-Spectro- scopy Studies,” Journal of Catalysis, Vol. 151, No. 1, 1995, pp. 226-240. doi:10.1006/jcat.1995.1024 [14] N. Y. Topspe, J. A. Dumesic and H. Topsoe, “Vana- dia-Titania Catalysts for Selective Catalytic Reduction of Nitric-Oxide by Ammonia:ⅡStudies of Active Sites and Formulation of Catalytic Cycles,” Journal of Catalysis, Vol. 151, No. 1, 1995, pp. 241-252. doi:10.1006/jcat.1995.1025 [15] J. A. Dumesic, N. Y. Topsoe, H. Topsoe, Y. Chen and T. Slabisk, “Kinetics of Selective Catalytic Reduction of Ni- tric Oxide by Ammonia over Vanadia/Titania,” Journal of Catalysis, Vol. 163, No. 2, 1996, pp. 409-417. doi:10.1006/jcat.1996.0342 [16] P. Davini, “Adsorption of Sulphur Dioxide on Thermally Treated Active Carbon,” Fuel, Vol. 68, No. 2, 1989, pp. 145-148. doi:10.1016/0016-2361(89)90314-1 [17] E. Garcia-Bordeje, J. L. Pinilla, M. J. Lazaro, R. Moliner and J. L. G. Fierro, “Role of Sulphates on the Mechanism of NH3-SCR of NO at Low Temperatures over Presul- phated Vanadium Supported on Carbon-Coated Mono- liths,” Journal of Catalysis, Vol. 233, No. 1, 2005, pp. 166-175. doi:10.1016/j.jcat.2005.04.032 [18] L. Lietti, G. Ramis, F. Berti, G. P. Toledo, D. Rbba, G. Busca and P. Forzatti, “Chemical, Structural and Mecha- nistic Aspects on NOx SCR over Commercial and Model Oxide Catalysts,” Catalysis Today, Vol. 42, No. 18, 1998, pp. 101-116. doi:10.1016/S0920-5861(98)00081-9 [19] L. Lietti, I. Nova, S. Camurri, E. Tronconi and P. Forzatti, “Dynamics of the SCR-DeNOx Reaction by the Tran- sient-Response Method,” AIChE Journal, Vol. 43, No.17, 2004, pp. 2559-2570. [20] E. Tronconi, L. Lietti, P. Forzatti and S. Malloggi, “Ex- perimental and Theoretical Investigation of the Dynamics of the SCR-DeNOx Reaction,” Chemical Engineering Science, Vol. 51, No. 11, 1996, pp. 2965-2970. doi:10.1016/0009-2509(96)00182-0 [21] M. Amiridis, I. Wachs, G. Deo, J. Jehng and D. Kim, “Reactivity of V2O5Catalysts for the Selective Catalytic Reduction of NO by NH3: Influence of Vanadia Loading, H2O, and SO2,” Journal of Catalysis, Vol. 161, No. 1, 1996, pp. 247-253. doi:10.1006/jcat.1996.0182 [22] J. Dunn, P. Koppula, H. Stenger and I. Wachs, “Influence of Pt Concentration on Activity and Combustion of Coke on Pt/Al2O3,” Applied Catalysis, Vol. 19, No. 1, 1985, pp. 203-206. doi:10.1016/S0166-9834(00)82681-0 [23] E. Tronconi, A. O. C. Cavanna and P. Forzatti, “Transient Kinetics of SO2 Oxidation Over SCR-DeNOx Monolith Catalysts,” Industrial and Engineering Chemistry Resear- ch, Vol. 38, No. 7, 1999, pp. 2593-2598. doi:10.1021/ie980673e
|