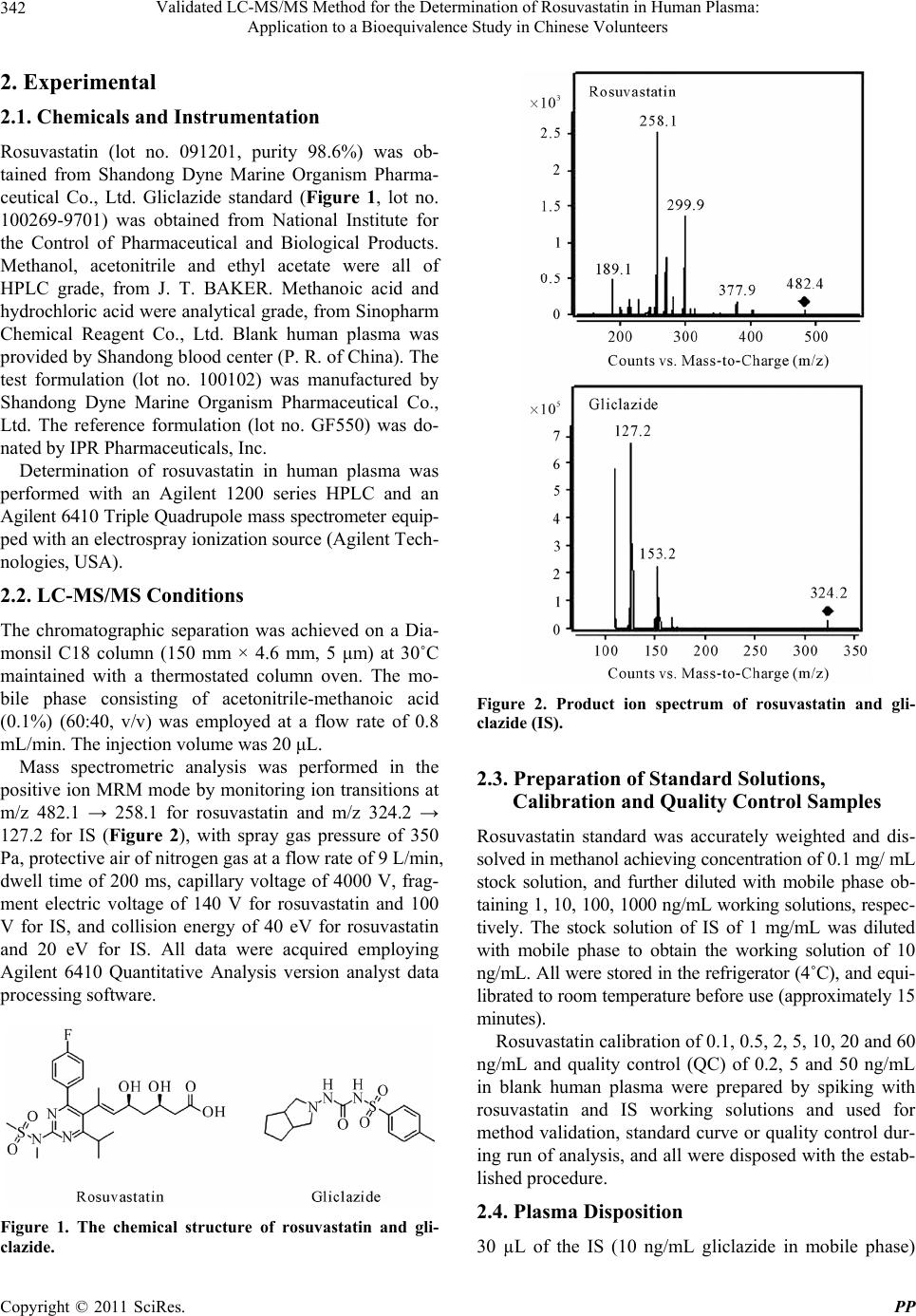
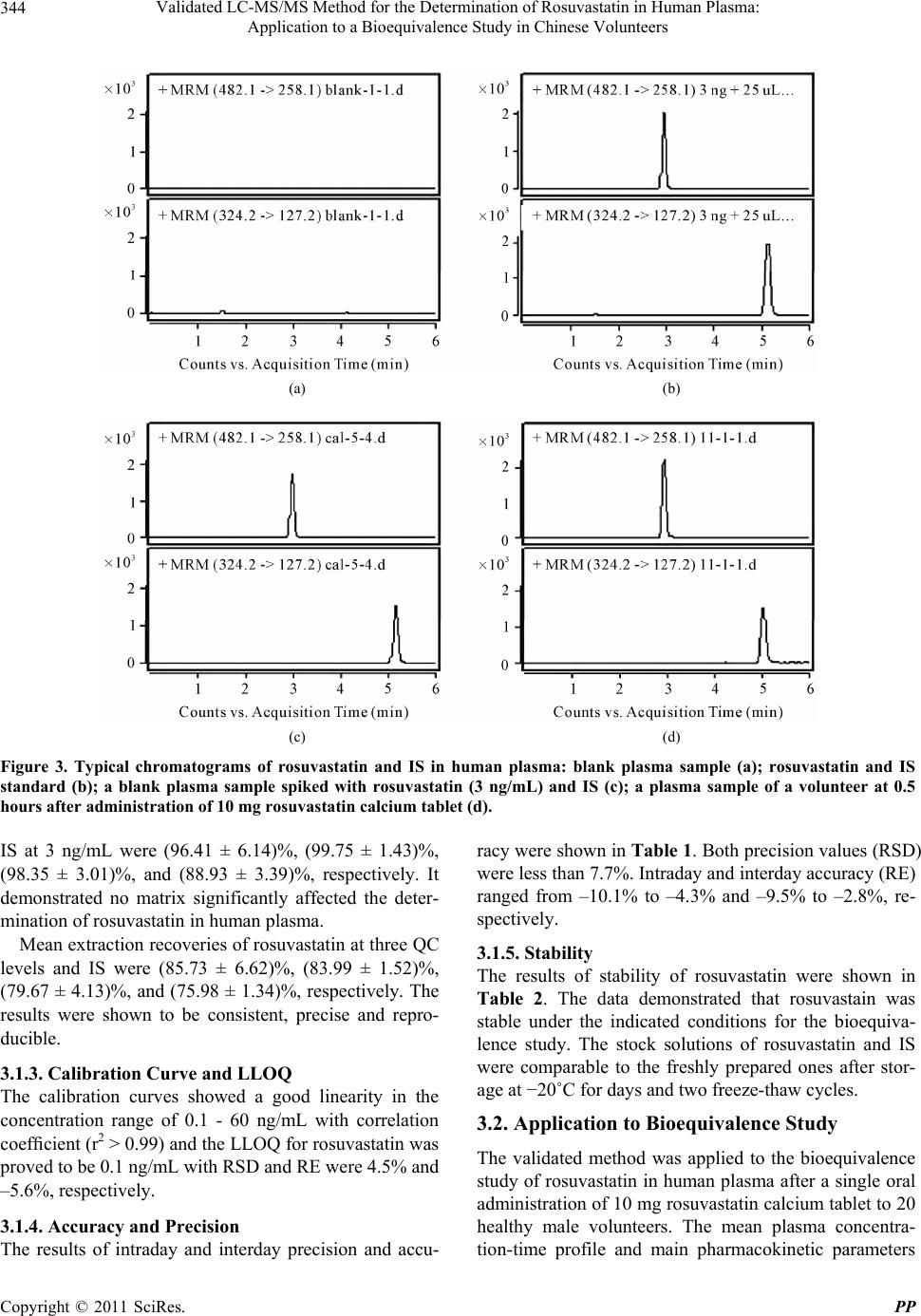
Validated LC-MS/MS Method for the Determination of Rosuvastatin in Human Plasma:
Application to a Bioequivalence Study in Chinese Volunteers
Copyright © 2011 SciRes. PP
346
tan potassium, and gliclazide. Significant interfering peaks
were observed for pravastatin sodium, fluvastatin sodium
and lorazepam during analysis. Different responds were
detected for losartan potassium when spiked with pur-
chased blank plasma and plasma samples from volunteers.
Gliclazide, with suitable retention time, acceptable matrix
effects, and extraction recoveries was selected as internal
standard.
Protein precipitation (PPT), liquid-liquid extraction
(LLE) and solid-phase extraction (SPE) were tried to
obtain a simple and excellent plasma preparation proce-
dure. PPT was easy to dilute the sample and failed to
sufficiently remove endogenous interference. The SPE
column for solid-phase extraction is relatively expensive
for a great quantity of samples. LLE method with various
extraction solvents, including chloroform, ethyl acetate,
n-hexane, dichloromethane, n-hexane-dichloromethane-
isopropanol (20:10:1, v/v/v) was investigated and evalu-
ated for acceptable extraction recoveries and matrix effect,
and ethyl acetate with no-concentration-dependent extrac-
tion recovery and acceptable matrix effect was adopted.
In conclusion, the developed method was simple, spe-
cific, sensitive, and successfully applied to bioequiva-
lence study of rosuvastatin calcium tablets. The two ro-
suvastatin calcium tablets were bioequivalent.
5. Acknowledgements
The authors appreciated Shandong Dyne Marine Organ-
ism Pharmaceutical Co., Ltd, providing financial support
and test and reference products.
REFERENCES
[1] C. I. Carswell, G. L. Plosker and B. Jarvis, “Rosuvas-
tatin,” Drugs, Vol. 62, No. 14, 2002, pp. 2075-2085.
doi:10.2165/00003495-200262140-00008
[2] D. J. Betteridge and J. M. Gibson, “Effects of Rosuvas-
tatin on Lipids, Lipoproteins and Apolipoproteins in the
Dyslipidaemia of Diabetes,” Diabetic Medicine, Vol. 24,
No. 5, 2007, pp. 541-549.
doi:10.1111/j.1464-5491.2007.02095.x
[3] F. McTaggart and P. Jones, “Effects of Statins on High-
Density Lipoproteins: A Potential Contribution to Car-
diovascular Benefit,” Cardiovascular Drugs and Therapy,
Vol. 22, No. 4, 2008, pp. 321-338.
doi:10.1007/s10557-008-6113-z
[4] J. R. Crouse III, “An Evaluation of Rosuvastatin: Phar-
macokinetics, Clinical Efficacy and Tolerability,” Expert
Opinion on Drug Metabolism & Toxicology, Vol. 4, No. 3,
2008, pp. 287-304. doi:10.1517/17425255.4.3.287
[5] E. Lee, S. Ryan, B. Birmingham, J. Zalikowski, R. March,
H. Ambrose, R. Moore, C. Lee, Y. Chen and D. Schneck,
“Rosuvastatin Pharmacokinetics and Pharmacogenetics in
White and Asian Subjects Residing in the Same Envi-
ronment,” Clinical Pharmacology & Therapeutics, Vol.
78, No. 4, 2005, pp. 330-341.
doi:10.1016/j.clpt.2005.06.013
[6] T. R. Kumar, N. R. Shitut, P. K. Kumar, M. C. Vinu, V. V.
Kumar, R. Mullangi and N. R. Srinivas, “Determination of
Rosuvastatin in Rat Plasma by HPLC: Validation and Its
Application to Pharmacokinetic Studies,” Biomedi cal Chro-
matography, Vol. 20, No. 9, 2006, pp. 881-887.
doi:10.1002/bmc.611
[7] J. Gao, D. Zhong, X. Duan and X. Chen, “Liquid Chro-
matography/Negative Ion Electrospray Tandem Mass
Spectrometry Method for the Quantification of Rosuvas-
tatin in Human Plasma: Application to a Pharmacokinetic
Study,” Journal of Chromatography B, Vol. 856, No. 1-2,
2007, pp. 35-40. doi:10.1016/j.jchromb.2007.05.012
[8] D. H. Xu, Z. R. Ruan, Q. Zhou, H. Yuan and B. Jiang,
“Quantitative Determination of Rosuvastatin in Human
Plasma by Liquid Chromatography with Electrospray
Ionization Tandem Mass Spectrometry,” Rapid Commu-
nications in Mass Spectrometry, Vol. 20, No. 16, 2006,
pp. 2369-2375. doi:10.1002/rcm.2542
[9] C. K. Hull, A. D. Penman, C. K. Smith and P. D. Martin,
“Quantification of Rosuvastatin in Human Plasma by
Automated Solid-Phase Extraction Using Tandem Mass
Spectrometric Detection,” Journal of Chromatography B,
Vol. 772, No. 2, 2002, pp. 219-228.
doi:10.1016/S1570-0232(02)00088-0
[10] R. K. Trivedi, R. R. Kallem, R. Mullangi and N. R. Srini-
vas, “Simultaneous Determination of Rosuvastatin and
Fenofibric Acid in Human Plasma by LC-MS/MS with
Electrospray Ionization: Assay Development, Validation
and Application to a Clinical Study,” Journal of Phar-
maceutical and Biomedical Analysis, Vol. 39, No. 3-4,
2005, pp. 661-669. doi:10.1016/j.jpba.2005.05.005
[11] S. Vittal, N. R. Shitut, T. R. Kumar, M. C. Vinu, R. Mul-
langi and N. R. Srinivas, “Simultaneous Quantitation of
Rosuvastatin and Gemfibrozil in Human Plasma by High-
Performance Liquid Chromatography and Its Application
to a Pharmacokinetic Study,” Biomedical Chromatography,
Vol. 20, No. 11, 2006, pp. 1252-1259.
doi:10.1002/bmc.692
[12] Y. Shah, Z. Iqbal, L. Ahmad, A. Khan, M. I. Khan, S.
Nazir and F. Nasir, “Simultaneous Determination of ro-
suvastatin and Atorvastatin in Human Serum Using RP-
HPLC/UV Detection: Method Development, Validation
and Optimization of Various Experimental Parameters,”
Journal of Chromatography B, Vol. 879, No. 9-10, 2011,
pp. 557-563. doi:10.1016/j.jchromb.2011.01.004
[13] US Food and Drug Administration, Center for Drug
Evaluation and Research, Guidance for Industry, Bioana-
lytical Method Validation.
http://www.fda.gov/cder/guidance/index.htm
[14] US Food and Drug Administration, “Bioavailability and
Bioequivalence Requirements, Abbreviated Applications,
Proposed Revisions,” Federal Registe r, Vol. 63, No. 223,
1998, pp. 64222-64228.