 Pharmacology & Pharmacy, 2011, 2, 315-321 doi:10.4236/pp.2011.24040 Published Online October 2011 (http://www.SciRP.org/journal/pp) Copyright © 2011 SciRes. PP 315 Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production Shinichi Takahashi*, Yoshikane Izawa, Norihiro Suzuki Department of Neurology, Keio University School of Medicine, Tokyo, Japan. Email: *takashin@tka.att.ne.jp R Received August 5th, 2011; revised September 13th, 2011; accepted September 25th, 2011. ABSTRACT Cilostazol, a potent inhibitor of type 3 phosphodiesterase (PDE3), has recently been reported to exert neuroprotective effects during acute cerebral ischemic injury. These effects are, at least in part, mediated by the inhibition of oxidative cell death. However, the effects of cilostazol on glucose metabolism in brain cells have not been determined. In the pre- sent study, we examined the effects of cilostazol on the oxidative metabolism of glucose and the resultant formation of reactive oxygen species (ROS) in cultured neurons and astroglia. Cultures of neurons or astroglia were prepared from Sprague-Dawley rats. The cells were treated with cilostazol (0 - 30 μM) for 48 hours prior to the assay. L-[U-14C]lactat e ([14C]lactate) or [1-14C]pyruvate ([14C]pyruvate) oxidation was measured. ROS production was determined using an H2DCFDA assay with a microplate reader. Forty-eight hours of exposure to cilostazol resulted in dose-dependent in- creases in [14C]lactate and [14C]pyruvate oxidation in both the neurons and astroglia. Dibutyryl cyclic AMP (0 - 0.5 mM) also increased [14C]lactate oxidation, indicating cAMP-mediated PDH activation. In contrast, free radical forma- tion was not affected by cilostazol in either the neurons or astroglia. Cilostazol enhanced the oxidative metabolism of glucose in both neurons and astroglia, while it did not augment ROS production. Keywords: Astrocyte, Cilostazol, Glucose, Lactate, Pyruvate Dehydrogenase 1. Introduction Brain function is completely dependent on the oxidative metabolism of glucose for ATP synthesis. The functional activation of local brain lesions triggers ATP consump- tion by Na+, K+-ATPase, which restores the trans-mem- brane Na+ and K+ gradients in the local cells [1]. In con- trast to the brain, the heart muscle is dependent on fatty acid oxidation in mitochondria for its energy production [2]. A type 3 phosphodiesterase (PDE3) inhibitor has been known to enhance cardiac contractile force by enhancing fatty acid metabolism [3]. Accordingly, an in vitro ex- periment has shown that two PDE inhibitors, enoximone and milrinone, suppress glucose oxidation in isolated cardiac myocytes [4] in accordance with Randle’s hy- pothesis [5], implying a reciprocal control of glucose oxidation and fatty acid oxidation in living organisms. Importantly, as fatty acid is not an energy substrate for brain tissue under normal physiological conditions, the effect of PDE inhibitors on fatty acids does not affect brain energy metabolism, and the effect of PDE inhibi- tors on glucose metabolism in neural tissue has not been previously studied, as far as we know. Cilostazol, a potent PDE3 inhibitor [6], has been used for effective secondary prevention of ischemic stroke for the past several years [7-9]. It exerts a potent anti- thrombotic action through the inhibition of PDE3 in both platelets and vascular endothelial cells. Unfortunately, however, the actions of cilostazol on brain parenchymal cells (i.e., neurons and glial cells) have not been exam- ined extensively. Cilostazol also plays a neuroprotective role during acute [10] and subacute [11] phases of cere- bral ischemia. This action is thought to depend on in- creases in the cyclic adenosine 3’,5’-monophosphate (cAMP) concentration by inhibiting PDE, which, in turn, activates cAMP-dependent protein kinase (PKA) and the cAMP-responsive-element-binding protein (CREB) phos- phorylation pathway, resulting in anti-apoptotic effects [12,13]. Moreover, cilostazol is reported to scavenge reactive oxygen species (ROS) [10]. In brain tissue, mi- tochondria are believed to be a main source of ROS be-  Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production 316 cause oxygen consumption in the brain is high and ROS formation is further enhanced during ischemia, leading to cell death [15]. The goal of the present study was to investigate the effect of cilostazol on glucose metabolism in cultured neurons and astroglia. We also examined the effect of cilostazol on ROS production in these two types of cells. 2. Materials and Methods 2.1. Animals Timed-pregnant Sprague-Dawley rats were purchased from Japan SLC, Inc. (Hamamatsu, Japan). All animal procedures were performed in accordance with The Animal Experimentation Guidelines of Keio University School of Medicine and were approved by the Labora- tory Animal Care and Use Committee of Keio Univer- sity. 2.2. Chemicals Chemicals and materials were obtained from the follow- ing sources: L-[U-14C]lactate ([14C]lactate; specific activ- ity, 4.84 GBq/mmol), [1-14C]pyruvate ([14C]pyruvate; specific activity, 0.67 GBq/mmol), Insta-Fluor Plus and hyamine hydroxide 10-X were all obtained from Perkin- Elmer Life Sciences (Boston, MA, USA); Dulbecco’s modified Eagle medium with or without glucose, penicil- lin, and streptomycin were obtained from Life Technolo- gies (Grand Island, NY, USA); defined fetal bovine se- rum was obtained from HyClone Laboratories (Logan, UT, USA); trypsin-EDTA was obtained from (Gibco- BRL/Invitrogen, Grand Island, NY); 2’,7’-dichlorodihy- drofluorescein diacetate (H2DCFDA) was obtained from Molecular Probe Inc. (Eugene, OR, USA); and all other chemicals were obtained from Sigma (St Louis, MO, USA). Cilostazol was a kind gift from Otsuka Pharma- ceutical Co. Ltd. (Tokushima, Japan). Please do not re- vise any of the current designations. 2.3. Preparation of Cells Primary astroglial cultures were prepared from the cere- bral cortex of rat pups 24 to 48 hours after birth [16]. Approximately on day 10, the adherent cells were treated with trypsin-EDTA solution, suspended in fresh high- glucose medium (diluted 1:4), and placed in uncoated 24-well culture plates (Sumitomo Bakelite, Tokyo, Ja- pan), 12-well culture plates (Nalge Nunc International, Rochester, NY, USA), or in 25-cm2 culture flasks (Nalge Nunc International). The culture medium was changed twice a week, and the cells were used once they reached confluence (days 21 to 24). The primary neuronal culture was prepared from the forebrain of fetal rats on embryonic day 16, as described previously [16]. Assays were performed using cultures that were 7 to 10 days old. The nutrient medium re- mained untouched until the experiments were initiated. Forty-eight hours before the assay, cilostazol was ap- plied to cultured cells with or without KT5720, a potent inhibitor of PKA [17]. Cilostazol exposure was typically started on day 5 - 7 for the neurons and on day 19 - 21 for the astroglia. To evaluate the acute effect of cilostazol on ROS generation, some cells were grown without cilostazol; instead, cilostazol was added to the H2DCFDA assay buffer during ROS measurement (see below). 2.4. Measurement of the Rate of L-[U-14C]Lactate and [1-14C]Pyruvate Oxidation to 14CO2 The rate of [14C]lactate and [14C]pyruvate oxidation to 14CO2 was measured using a modification of a previously described method [18]. After the cells cultured in the 25-cm2 culture flasks were washed twice with phosphate- buffered saline without CaCl2 and MgCl2 (PBS), 2.5 mL of Dulbecco’s balanced salt solution (DBSS: 110 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 0.9 mM NaH2PO4, and 44 mM NaHCO3) supplemented with 2 mM of lactate and 2.5 μL (approximately, 0.85 μM) of [14C]lactate (original concentration: 3.7 MBq/mL) or 2 mM of pyruvate and 2.5 μL (approximately, 20 μM) of [14C]pyruvate (original concentration: 18.5 MBq/mL), which had been pre-warmed at 37˚C and equilibrated with 7% CO2 to adjust the pH to 7.4, was added to the flasks. The culture flasks were then capped with rubber stoppers containing a center well and incubated at 37˚C for 60 minutes. The resulting 14CO2 was trapped by a cotton ball placed in the center well containing 100 μL of hyamine hydroxide 10-X. The reactions were terminated by the injection of 250 μL of 60% perchloric acid through the rubber stopper, and the flasks were kept at 4˚C overnight to trap the 14CO2. The center wells were transferred to 20-mL glass scintillation counter vials, and 500 μL of ethanol and 10 mL of Insta-Fluor Plus were added. The 14C contents of the vials were then evaluated using a liquid scintillation counter. The cell carpets left in the incubation flasks after the removal of the reaction mixtures were then digested with 5 mL of 0.1 M NaOH, and their protein contents were determined using a bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnology, Meridian Road Rock- ford, IL, USA) based on the use of BCA for colorimetric detection [19], according to the manufacturer’s instruct- tions. 2.5. Measurement of ROS Production The production of ROS, mainly H2O2, in the cells was assessed using H2DCFDA [20] and semi-quantitative Copyright © 2011 SciRes. PP 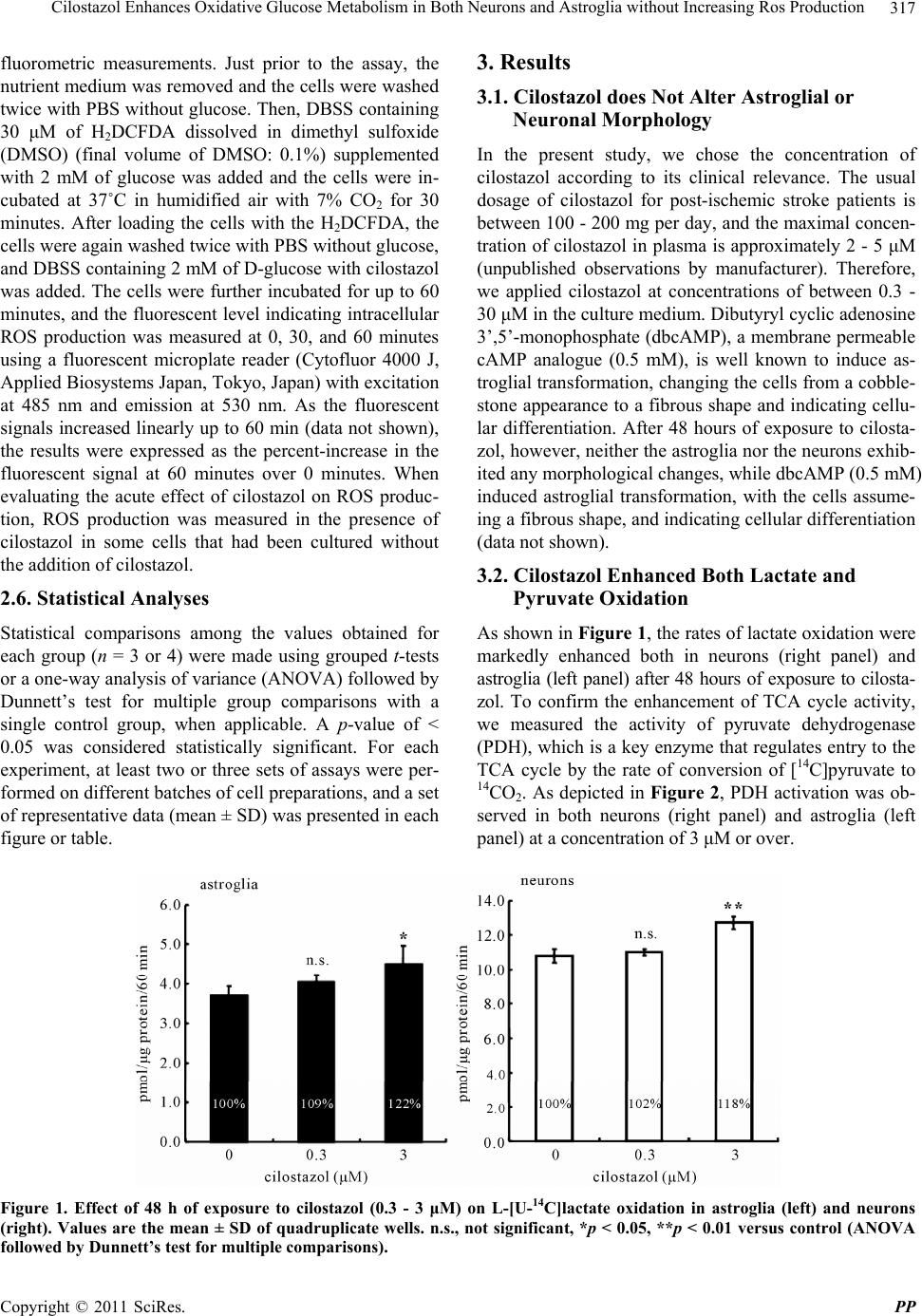 Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production Copyright © 2011 SciRes. PP 317 3. Results fluorometric measurements. Just prior to the assay, the nutrient medium was removed and the cells were washed twice with PBS without glucose. Then, DBSS containing 30 μM of H2DCFDA dissolved in dimethyl sulfoxide (DMSO) (final volume of DMSO: 0.1%) supplemented with 2 mM of glucose was added and the cells were in- cubated at 37˚C in humidified air with 7% CO2 for 30 minutes. After loading the cells with the H2DCFDA, the cells were again washed twice with PBS without glucose, and DBSS containing 2 mM of D-glucose with cilostazol was added. The cells were further incubated for up to 60 minutes, and the fluorescent level indicating intracellular ROS production was measured at 0, 30, and 60 minutes using a fluorescent microplate reader (Cytofluor 4000 J, Applied Biosystems Japan, Tokyo, Japan) with excitation at 485 nm and emission at 530 nm. As the fluorescent signals increased linearly up to 60 min (data not shown), the results were expressed as the percent-increase in the fluorescent signal at 60 minutes over 0 minutes. When evaluating the acute effect of cilostazol on ROS produc- tion, ROS production was measured in the presence of cilostazol in some cells that had been cultured without the addition of cilostazol. 3.1. Cilostazol does Not Alter Astroglial or Neuronal Morphology In the present study, we chose the concentration of cilostazol according to its clinical relevance. The usual dosage of cilostazol for post-ischemic stroke patients is between 100 - 200 mg per day, and the maximal concen- tration of cilostazol in plasma is approximately 2 - 5 μM (unpublished observations by manufacturer). Therefore, we applied cilostazol at concentrations of between 0.3 - 30 μM in the culture medium. Dibutyryl cyclic adenosine 3’,5’-monophosphate (dbcAMP), a membrane permeable cAMP analogue (0.5 mM), is well known to induce as- troglial transformation, changing the cells from a cobble- stone appearance to a fibrous shape and indicating cellu- lar differentiation. After 48 hours of exposure to cilosta- zol, however, neither the astroglia nor the neurons exhib- ited any morphological changes, while dbcAMP (0.5 mM) induced astroglial transformation, with the cells assume- ing a fibrous shape, and indicating cellular differentiation (data not shown). 3.2. Cilostazol Enhanced Both Lactate and Pyruvate Oxidation 2.6. Statistical Analyses As shown in Figure 1, the rates of lactate oxidation were markedly enhanced both in neurons (right panel) and astroglia (left panel) after 48 hours of exposure to cilosta- zol. To confirm the enhancement of TCA cycle activity, we measured the activity of pyruvate dehydrogenase (PDH), which is a key enzyme that regulates entry to the TCA cycle by the rate of conversion of [14C]pyruvate to 14CO2. As depicted in Figure 2, PDH activation was ob- served in both neurons (right panel) and astroglia (left panel) at a concentration of 3 μM or over. Statistical comparisons among the values obtained for each group (n = 3 or 4) were made using grouped t-tests or a one-way analysis of variance (ANOVA) followed by Dunnett’s test for multiple group comparisons with a single control group, when applicable. A p-value of < 0.05 was considered statistically significant. For each experiment, at least two or three sets of assays were per- formed on different batches of cell preparations, and a set of representative data (mean ± SD) was presented in each figure or table. Figure 1. Effect of 48 h of exposure to cilostazol (0.3 - 3 μM) on L-[U-14C]lactate oxidation in astroglia (left) and neurons (right). Values are the mean ± SD of quadruplicate wells. n.s., not significant, *p < 0.05, **p < 0.01 versus control (ANOVA followed by Dunnett’s test for multiple comparisons).  Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production 318 Figure 2. Effect of 48 h of exposure to cilostazol (0.3 - 30 μM) on pyruvate dehydrogenase (PDH) activity, as measured by the conversion of [1-14C]pyruvate to 14CO2 in astroglia (left) and neurons (right). Values are the mean ± SD of quadruplicate wells. n.s., not significant, *p < 0.05, **p < 0.01 versus control (ANOVA followed by Dunnett’s test for multiple comparisons). 3.3. Actions of Cilostazol Is Medicated by Protein Kinase a Pathway To explore the downstream pathway that occurs in the presence of the cilostazol-mediated enhancement of glu- cose metabolism, we assessed the effect of dbcAMP. dbcAMP (0 - 0.5 mM) induced astroglial morphological changes (data not shown) and also increased [14C]lactate oxidation in astroglia (Figure 3), implying the involve- ment of cAMP. A highly specific PKA inhibitor, KT5720 (100 nM), abolished the cilostazol-induced (3 μM) in- crease in PDH activity in astroglia (Figure 4, left panel) and diminished that in neurons (Figure 4, right panel), indicating that cilostazol induces the oxidative metabo- lism of glucose through PKA-mediated PDH activation. 3.4. ROS Production in Neurons and Astroglia Was Not Altered by Cilostazol Next, we evaluated whether the cilostazol-induced acti- vation of the oxidative metabolism of glucose is associ- ated with an augmentation of ROS production. Table 1 shows that ROS production in the neurons and astroglia was not affected, irrespective of the increased mitochon- drial activity induced by incubation with cilostazol (0.3 - 30 μM) for 48 hours. As cilostazol is reported to possess radical scavenging effects [10], we also evaluated the acute effect of cilostazol on ROS generation during an ROS assay. Table 1 shows that in neurons, lower con- centrations of cilostazol (0.3 - 3 μM) tended to decrease ROS production while the highest concentration (30 μM) enhanced ROS production (p < 0.01, ANOVA followed by Dunnett’s r test for multiple comparisons). The astro- glia did not exhibit any changes in ROS production when incubated with cilostazol. Figure 3. Effects of 48 h of exposure to dibutyryl cAMP (0.05 - 0.5 mM) on L-[U-14C]lactate oxidation in astroglia. Values are the mean ± SD of quadruplicate wells. n.s., not significant, **p < 0.01 versus control (ANOVA followed by Dunnett’s test for multiple comparisons). 4. Discussion The present study revealed that the administration of cilostazol, a PDE3 inhibitor, at clinically relevant con- centrations leads to the enhancement of the oxidative metabolism of glucose both in neurons and astroglia, implying that cilostazol may enhance ATP production in the brain and secure its function. This action of cilostazol seems to be mediated by intracellular cAMP elevation, PKA activation, and enhanced PDH activity. cAMP is reported to enhance Na+, K+-ATPase activity in peripheral nerves [21], and cilostazol prevents Na+, K+- ATPase activity from being reduced in diabetic model animals through the cAMP cmponent [22]. Therefore, o Copyright © 2011 SciRes. PP 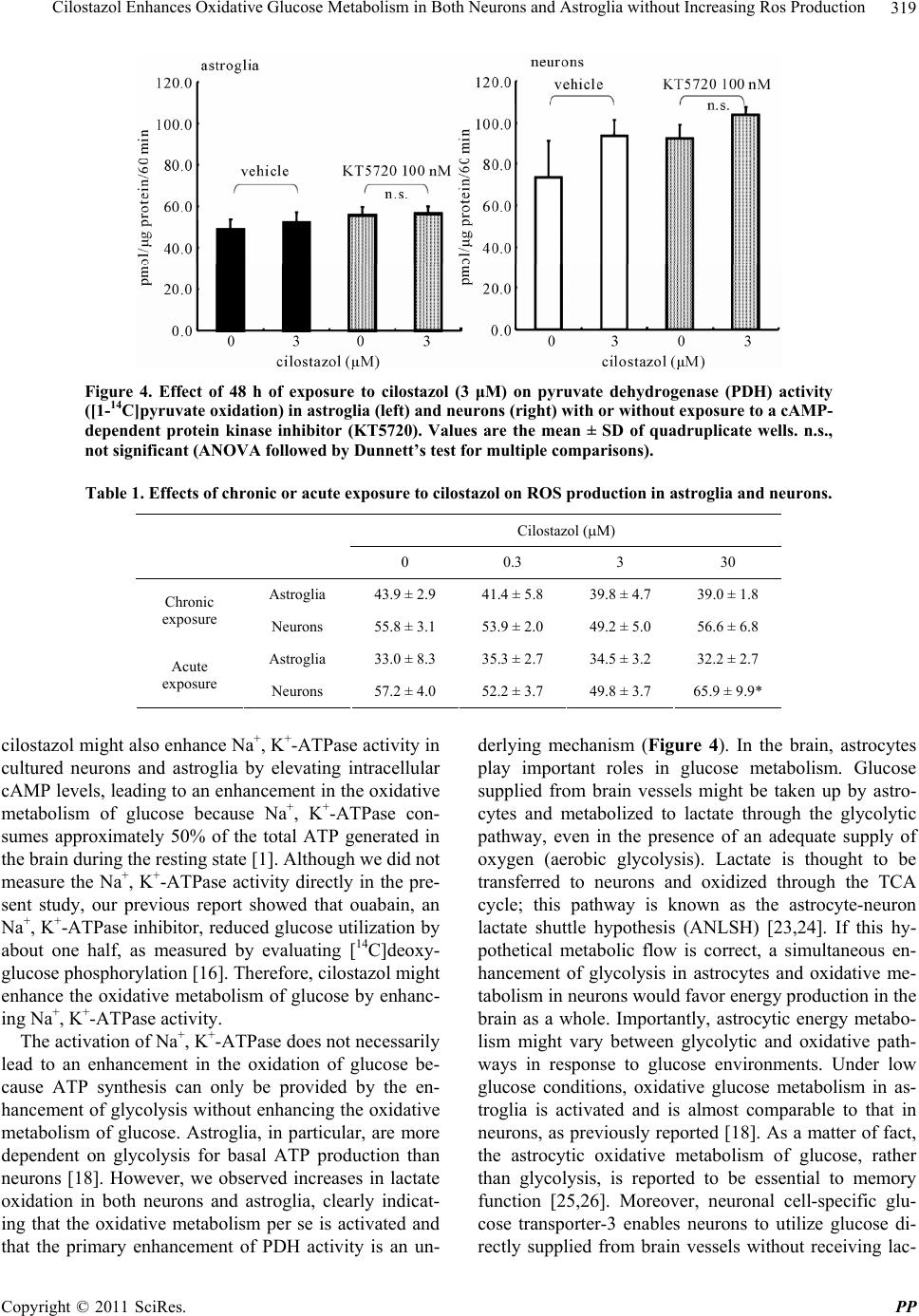 Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production319 Figure 4. Effect of 48 h of exposure to cilostazol (3 μM) on pyruvate dehydrogenase (PDH) activity ([1-14C]pyruvate oxidation) in astroglia (left) and neurons (right) with or without exposure to a cAMP- dependent protein kinase inhibitor (KT5720). Values are the mean ± SD of quadruplicate wells. n.s., not significant (ANOVA followed by Dunnett’s test for multiple comparisons). Table 1. Effects of chronic or acute exposure to cilostazol on ROS production in astroglia and neurons. Cilostazol (M) 0 0.3 3 30 Astroglia 43.9 ± 2.9 41.4 ± 5.8 39.8 ± 4.7 39.0 ± 1.8 Chronic exposure Neurons 55.8 ± 3.1 53.9 ± 2.0 49.2 ± 5.0 56.6 ± 6.8 Astroglia 33.0 ± 8.3 35.3 ± 2.7 34.5 ± 3.2 32.2 ± 2.7 Acute exposure Neurons 57.2 ± 4.0 52.2 ± 3.7 49.8 ± 3.7 65.9 ± 9.9* cilostazol might also enhance Na+, K+-ATPase activity in cultured neurons and astroglia by elevating intracellular cAMP levels, leading to an enhancement in the oxidative metabolism of glucose because Na+, K+-ATPase con- sumes approximately 50% of the total ATP generated in the brain during the resting state [1]. Although we did not measure the Na+, K+-ATPase activity directly in the pre- sent study, our previous report showed that ouabain, an Na+, K+-ATPase inhibitor, reduced glucose utilization by about one half, as measured by evaluating [14C]deoxy- glucose phosphorylation [16]. Therefore, cilostazol might enhance the oxidative metabolism of glucose by enhanc- ing Na+, K+-ATPase activity. The activation of Na+, K+-ATPase does not necessarily lead to an enhancement in the oxidation of glucose be- cause ATP synthesis can only be provided by the en- hancement of glycolysis without enhancing the oxidative metabolism of glucose. Astroglia, in particular, are more dependent on glycolysis for basal ATP production than neurons [18]. However, we observed increases in lactate oxidation in both neurons and astroglia, clearly indicat- ing that the oxidative metabolism per se is activated and that the primary enhancement of PDH activity is an un- derlying mechanism (Figure 4). In the brain, astrocytes play important roles in glucose metabolism. Glucose supplied from brain vessels might be taken up by astro- cytes and metabolized to lactate through the glycolytic pathway, even in the presence of an adequate supply of oxygen (aerobic glycolysis). Lactate is thought to be transferred to neurons and oxidized through the TCA cycle; this pathway is known as the astrocyte-neuron lactate shuttle hypothesis (ANLSH) [23,24]. If this hy- pothetical metabolic flow is correct, a simultaneous en- hancement of glycolysis in astrocytes and oxidative me- tabolism in neurons would favor energy production in the brain as a whole. Importantly, astrocytic energy metabo- lism might vary between glycolytic and oxidative path- ways in response to glucose environments. Under low glucose conditions, oxidative glucose metabolism in as- troglia is activated and is almost comparable to that in neurons, as previously reported [18]. As a matter of fact, the astrocytic oxidative metabolism of glucose, rather than glycolysis, is reported to be essential to memory function [25,26]. Moreover, neuronal cell-specific glu- cose transporter-3 enables neurons to utilize glucose di- rectly supplied from brain vessels without receiving lac- Copyright © 2011 SciRes. PP  Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production 320 tate from astrocytes. Collectively, an enhancement in glu- cose oxidation in astrocytes might not necessarily worsen neuronal function. The direct mechanism of the cAMP-dependent en- hancement of PDH activity was not completely eluci- dated in the present study. PKA is a candidate enzyme that might regulate PDH activity because PKA phos- phorylates various enzymes and regulates their action. PDH is regulated by PDH kinase, and the phosphoryla- tion of PDH leads to inactivation [27-29]. As shown in Figure 4, the PKA inhibitor enhanced the basal activity of PDH in both neurons and astroglia. However, the cilostazol-mediated action was not completely abolished in neurons, whereas it completely disappeared in astro- glia. Interestingly, we found that cilostazol did not enhance ROS production in either neurons or astroglia, irrespec- tive of the enhanced oxidative metabolism of glucose. Of course, enhancing oxidative metabolism does not neces- sarily cause an immediate increase in ROS production. Brain tissue possesses an active anti-oxidant system to prevent oxidative stress, and failures in this system lead to various neurodegenerative diseases [30]. Cilostazol reportedly has some action on the ROS scavenging effect [10]. In fact, acute exposure to cilostazol somehow de- creased ROS production in neurons and astroglia (Table 1). The exact mechanism of this effect is unclear. PKA and the productions of ROS are interrelated [31,32]. Ir- respective of the mechanism involved, cilostazol en- hanced brain glucose metabolism without enhancing ROS production, which could have favorable effects on brain function. 5. Acknowledgements The cilostazol used in this study was a kind gift from Otsuka Pharmaceutical Co. Ltd., Tokushima, Japan. REFERENCES [1] D. D. Clarke and L. Sokoloff, “Circulation and Energy Metabolism of the Brain,” In: G. Siegel, B. Agranoff, R. W. Albers and S. Fisher, Ed., Basic Neurochemistry: Mo- lecular, Cellular, and Medical Aspects, 6th Edition, Lip- pincott-Raven, Philadelphia, 1999, pp. 637-669. [2] D. An and B. Rodrigues, “Role of Changes in Cardiac Metabolism in Development of Diabetic Cardiomyopa- thy,” American Journal of Physiology: Heart and Circu- latory Physiology, Vol. 291, No. 4, 2006, pp. H1489- H1506. doi.org/10.1152/ajpheart.00278.2006 [3] S. Abdel-Aleem, M. Badr and C. Frangakis, “Stimulation of Fatty Acid Oxidation in Myocytes by Phosphodi- esterase Inhibitors and Adenosine Analogues,” Life Sci- ences, Vol. 48, No. 18, 1991, pp. PL97-PL102. doi.org/10.1016/0024-3205(91)90219-2 [4] S. Abdel-Aleem, M. K. El Awadi, W. A. Zarouk, D. Taylor and J. E. Lowe, “Effects of Phosphodiesterase In- hibitors on Glucose Utilization in Isolated Cardiac Myo- cytes,” Molecular and Cellular Biochemistry, Vol. 180, No. 1-2, 1998, pp. 129-135. doi.org/10.1023/A:1006803426578 [5] P. J. Randle, P. B. Garland, C. N. Hales and E. A. New- sholme, “The Glucose Fatty-Acid Cycle. Its Role in Insu- lin Sensitivity and the Metabolic Disturbances of Diabe- tes Mellitus,” Lancet, Vol. 1, No. 7285, 1963, pp. 785- 789. [6] Y. Kimura, T. Tani, T. Kanbe and K. Watanabe, “Effect of Cilostazol on Platelet Aggregation and Experimental Th- rombosis,” Arzneimittelforschung, Vol. 35, No. 7A, 1985, pp. 1144-1149. [7] F. Gotoh, H. Tohgi, S. Hirai, A. Terashi, Y. Fukuuchi, E. Otomo, Y. Shinohara, E. Itoh, T. Masuda, T. Sawada, T. Yamaguchi, K. Nishimaru and Y. Ohashi, “Cilostazol Stroke Prevention Study: A Placebo-Controlled Double- Blind Trial for Secondary Prevention of Cerebral Infarc- tion,” Journal of Stroke and Cerebrovascular Diseases, Vol. 9, No. 4, 2000, pp. 147-157. [8] Y. Huang, Y. Cheng, J. Wu, Y. Li, E. Xu, Z. Hong, Z. Li, W. Zhang, M. Ding, X. Gao, D. Fan, J. Zeng, K. Wong, C. Lu, J. Xiao and C. Yao, “Cilostazol as an Alternative to Aspirin after Ischaemic Stroke: A Randomised, Dou- ble-Blind, Pilot Study,” The Lancet Neurology, Vol. 7, No. 6, 2008, pp. 494-499. doi.org/10.1016/S1474-4422(08)70094-2 [9] Y. Shinohara, Y. Katayama, S. Uchiyama, T. Yamaguchi, S. Handa, K. Matsuoka, Y. Ohashi, N. Tanahashi, H. Yamamoto, C. Genka, Y. Kitagawa, H. Kusuoka, K. Ni- shimaru, M. Tsushima, Y. Koretsune, T. Sawada and C. Hamada, “Cilostazol for Prevention of Secondary Stroke (CSPS 2): An Aspirin-Controlled, Double-Blind, Ran- domised Non-Inferiority Trial,” The Lancet Neurology, Vol. 9, No. 10, 2010, pp. 959-968. doi.org/10.1016/S1474-4422(10)70198-8 [10] J. M. Choi, H. K. Shin, K. Y. Kim, J. H. Lee and K. W. Hong, “Neuroprotective Effect of Cilostazol against Focal Cerebral Ischemia via Antiapoptotic Action in Rats,” Journal of Pharmacology and Experimental Therapeutics, Vol. 300, No. 3, 2002, pp. 787-793. doi.org/10.1124/jpet.300.3.787 [11] Y. L. Ye, W. Z. Shi, W. P. Zhang, M. L. Wang, Y. Zhou, S. H. Fang, L. Y. Liu, Q. Zhang, Y. P. Yu and E. Q. Wei, “Cilostazol, a Phosphodiesterase 3 Inhibitor, Protects Mice against Acute and Late Ischemic Brain Injuries,” European Journal of Pharmacology, Vol. 557, No. 1, 2007, pp. 23-31. doi.org/10.1016/j.ejphar.2006.11.003 [12] K. W. Hong, J. H. Lee, K. Y. Kima, S. Y. Park and W. S. Lee, “Cilostazol: Therapeutic Potential against Focal Cere- bral Ischemic Damage,” Current Pharmaceutical Design, Vol. 12, No. 5, 2006, pp. 565-573. doi.org/10.2174/138161206775474323 [13] M. J. Kim, J. H. Lee, S. Y. Park, K. W. Hong, C. D. Kim, K. Y. Kim and W. S. Lee, “Protection from Apoptotic Cell Death by Cilostazol, Phosphodiesterase Type III In- Copyright © 2011 SciRes. PP  Cilostazol Enhances Oxidative Glucose Metabolism in Both Neurons and Astroglia without Increasing Ros Production Copyright © 2011 SciRes. PP 321 hibitor, via cAMP-Dependent Protein Kinase Activation,” Pharmacological Research, Vol. 54, No. 4, 2006, pp. 261- 267. [14] J. H. Lee, S. Y. Park, H. K. Shin, C. D. Kim, W. S. Lee and K. W. Hong, “Protective Effects of Cilostazol against Transient Focal Cerebral Ischemia and Chronic Cerebral Hypoperfusion Injury,” CNS Neuroscience & Therapeu- tics, Vol. 14, No. 2, 2008, pp. 143-152. doi.org/10.1111/j.1527-3458.2008.00042.x [15] A. Y. Abramov, A. Scorziello and M. R. Duchen, “Three Distinct Mechanisms Generate Oxygen Free Radicals in Neurons and Contribute to Cell Death during Anoxia and Reoxygenation,” The Journal of Neuroscience, Vol. 27, No. 5, 2007, pp. 1129-1138. doi.org/10.1523/JNEUROSCI.4468-06.2007 [16] S. Takahashi, B. F. Driscoll, M. J. Law and L. Sokoloff, “Role of Sodium and Potassium Ions in Regulation of Glucose Metabolism in Cultured Astroglia,” Proceedings of the National Academy of Sciences of the USA, Vol. 92, No. 10, 1995, pp. 4616-4620. doi.org/10.1073/pnas.92.10.4616 [17] Y. Y. Huang, K. C. Martin and E. R. Kandel, “Both Pro- tein Kinase A and Mitogen-Activated Protein Kinase Are Required in the Amygdala for the Macromolecular Syn- thesis-Dependent Late Phase of Long-Term Potentiation,” The Journal of Neuroscience, Vol. 20, No. 17, 2000, pp. 6317-6325. [18] T. Abe, S. Takahashi and N. Suzuki, “Oxidative Metabo- lism in Cultured Rat Astroglia: Effects of Reducing the Glucose Concentration in the Culture Medium and of D-Aspartate or Potassium Stimulation,” Journal of Cere- bral Blood Flow & Metabolism, Vol. 26, No. 2, 2006, pp. 153-160. doi.org/10.1038/sj.jcbfm.9600175 [19] P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, “ Measurement of Pro- tein Using Bicinchoninic Acid,” Analytical Biochemistry, Vol. 150, No. 1, 1985, pp. 76-85. doi.org/10.1016/0003-2697(85)90442-7 [20] A. Gomes, E. Fernandes and J. L. Lima, “Fluorescence Probes Used for Detection of Reactive Oxygen Species,” Journal of Biochemical and Biophysical Methods, Vol. 65, No. 2-3, 2005, pp. 45-80. doi.org/10.1016/j.jbbm.2005.10.003 [21] H. Shindo, M. Tawata and T. Onaya, “Cyclic Adenosine 3’,5’-Monophosphate Enhances Sodium, Potassium-Ade- nosine Triphosphatase Activity in the Sciatic Nerve of Streptozotocin-Induced Diabetic Rats,” Endocrinology, Vol. 132, No. 2, 1993, pp. 510-516. [22] K. Naka, H. Sasaki, Y. Kishi, M. Furuta, T. Sanke, K. Nanjo and M. Mukoyama, “Effects of Cilostazol on De- velopment of Experimental Diabetic Neuropathy: Func- tional and Structural Studies, and Na+-K+-ATPase Acidity in Peripheral Nerve in Rats with Streptozotocin-Induced Diabetes,” Diabetes Research and Clinical Practice, Vol. 30, No. 3, pp. 153-162. doi.org/10.1016/0168-8227(95)01184-6 [23] L. Pellerin, A. K. Bouzier-Sore, A. Aubert, S. Serres, M. Merle, R. Costalat and P. J. Magistretti, “Activity-De- pendent Regulation of Energy Metabolism by Astrocytes: An Update,” Glia, Vol. 55, No. 12, 2007, pp. 1251-1262. [24] L. Pellerin and P. J. Magistretti, “Glutamate Uptake into Astrocytes Stimulates Aerobic Glycolysis: A Mechanism Coupling Neuronal Activity to Glucose Utilization,” Pro- ceedings of the National Academy of Sciences of the USA, Vol. 91, No. 22, 1994, pp. 10625-10629. doi.org/10.1073/pnas.91.22.10625 [25] M. E. Gibbs, B. S. O’Dowd, E. Hertz and L. Hertz, “As- trocytic Energy Metabolism Consolidates Memory in Young Chicks,” Neuroscience, Vol. 141, No. 1, 2006, pp. 9-13. [26] M. E. Gibbs, D. Hutchinson and L. Hertz, “Astrocytic Involvement in Learning and Memory Consolidation,” Neuroscience & Biobehavioral Reviews, Vol. 32, No. 5, 2008, pp. 927-944. doi.org/10.1016/j.neubiorev.2008.02.001 [27] M. S. Patel and L. G. Korotchkina, “Regulation of Mam- malian Pyruvate Dehydrogenase Complex by Phosphory- lation: Complexity of Multiple Phosphorylation Sites and Kinases,” Experimental and Molecular Medicine, Vol. 33, No. 4, 2001, pp. 191-197. [28] M. S. Patel and L. G. Korotchkina, “Regulation of the Pyruvate Dehydrogenase Complex,” Biochemical Society Transactions, Vol. 34, Part 2, 2006, pp. 217-222. [29] M. C. Sugden and M. J. Holness, “Recent Advances in Mechanisms Regulating Glucose Oxidation at the Level of the Pyruvate Dehydrogenase Complex by PDKs,” Ameri- can Journal of Physiology: Endocrinology and Metabolism, Vol. 284, No. 5, 2003, pp. E855-E862. [30] A. Reynolds, C. Laurie, R. L. Mosley and H. E. Gendel- man, “Oxidative Stress and the Pathogenesis of Neuro- degenerative Disorders,” International Review of Neuro- biology, Vol. 82, 2007, pp. 297-325. doi.org/10.1016/S0074-7742(07)82016-2 [31] N. Koike, T. Takamura and S. Kaneko, “Induction of Re- active Oxygen Species from Isolated Rat Glomeruli by Protein Kinase C Activation and TNF-Alpha Stimulation, and Effects of a Phosphodiesterase Inhibitor,” Life Sciences, Vol. 80, No. 18, 2007, pp. 1721-1728. doi.org/10.1016/j.lfs.2007.02.001 [32] S. Raha, A. T. Myint, L. Johnstone and B. H. Robinson, “Control of Oxygen Free Radical Formation from Mito- chondrial Complex I: Roles for Protein Kinase A and Py- ruvate Dehydrogenase Kinase,” Free Radical Biology & Medicine, Vol. 32, No. 5, 2002, pp. 421-430. doi.org/10.1016/S0891-5849(01)00816-4
|