Journal of Water Resource and Protection
Vol. 2 No. 9 (2010) , Article ID: 2725 , 12 pages DOI:10.4236/jwarp.2010.29097
Simultaneous Determination of Ten Endocrine Hormone Disrupters in Water Using SPE/LC-(ESI)MS-MS
1Faculty of Pharmacy, Laboratory of Hidrology, Lisbon, Portugal
2EPAL (Empresa Portuguesa das Águas Livres, S.A.), Laboratório Central da EPAL, Lisbon, Portugal
3iMed (The Institute for Medicines and Pharmaceutical Sciences), Faculty of Pharmacy, Lisbon, Portugal
E-mail: vitorcar@epal.pt
Received May 22, 2010; revised June 11, 2010; accepted June 20, 2010
Keywords: Water Analysis, Endocrine Disruptors, Tandem Mass Spectrometry, Solid-phase Extraction, Uncertainty Evaluation
ABSTRACT
The aim of this study was developed and validated an analytical method based on liquid chromatography and tandem mass spectrometry after solid phase extraction to monitorizing ten endocrine hormone disrupters in Lisbon drinking water system. Natural and synthetic hormones (17-β-estradiol, ethinylestradiol, estriol, estrone, progesterone, mestranol and diethylstilbestrol) and some industrial products (4-n-nonylphenol, 4-tert-octylphenol and bisphenol A) were studied. Mass spectrometer detection parameters were optimized, such as the best conditions for the precursor ion formation, namely cone voltage, when applying negative and positive electrospray ionization, and also collision energy for MRM1 and MRM2 transitions. The best conditions of the solid phase extraction (SPE) using Waters Oasis HLB (6 mL, 200 mg) and Isolute C18 (EC) (6 ml, 1000 mg) were also optimized. The method was validated through the application of several statistical tests and the uncertainty estimation of the analytical assay. This method showed a very good linear range for all the studied analytes with determination coefficients (r2) between 0.9962 and 0.9999 and coefficients of variation lower than 4%. There were no significant differences between recoveries obtained with the studied matrices, like groundwater, surface water and water for human consumption. In these matrices, the recovery values varied between 32 and 95%. The limits of method detection were between 0.28 and 22 ng/L. The validated method was applied for the analysis of water samples from the EPAL (Empresa Portuguesa das Águas Livres, S.A.) water supply system including tap water, spring water, groundwater, and river water. Some target compounds (bisphenol A, progesterone, 4-tert-octylphenol, and 4-n-nonylphenol) were found in trace amounts in analysed waters.
1. Introduction
An endocrine disrupter (ED) is an exogenous substance or mixture that alters function(s) of the endocrine system and consequently causes adverse health effects in an intact organism, or its progeny, or (sub)populations [1].
The main evidence suggesting that exposure to environmental chemicals can lead to disruption of endocrine function was first reported in the 1950s, specially in the Great Lakes region (2) and comes from changes seen in a number of wildlife species (molluscs, crustacean, fish, reptiles, birds and mammals) in various parts of the world [2,3]. Their effect on wildlife is well documented and includes the feminization of male foetuses, reproductive disfunctions, behavioral changes and development problems [2,3]. There is also some limited evidence in humans that adverse endocrine-mediated effects have followed either intentional or accidental exposure to high levels of particular chemicals. The main example of an endocrine disrupter in humans is diethylstilbestrol (DES) [4-7].
Potential adverse outcomes in humans are mostly described as effects on reproductive and sexual development and function [8,9], such as decreased sperm quality and increase in subfertility [10-14], changes in sex ratio [15,16], puberty disorders and higher incidence of cryptorchidism and hypospadias [9,12,15,17-19].
Chemicals with hormonal activity, that is, potential endocrine disrupters, include synthetic compounds and natural hormones. Synthetic compounds include a wide variety of products, like pharmaceutical products (including synthetic hormones), pesticides (for example, DDT, dieldrin, lindane and atrazine), industrial products (for example, polychlorinated biphenyl, phthalates, bisphenol A and alkylphenols, such as 4-n-nonylphenol and octylphenol) and their treatment by-products (for example, dioxin and furane) [20]. Some of these industrial products are used in food packaging material, as plasticizers, in water pipes, in water reservoirs as coatings, in personal care products, and in cleaning and disinfecting chemical products.
Regarding natural hormones, they can occur in water due to the natural process of excretion in all species of mammals. Domestic and industrial effluents are the most significant input of these substances in the aquatic environment and these compounds and their metabolites are constantly excreted into the sewers environment and continue to accumulate in the environment. The large variety of compounds with effective or potential endocrine disruptor activity, the diversity of environmental matrices where they can occur, their physical and chemical properties (high to moderate polarity) and the usually low concentrations require an accurate and efficient monitoring of these endocrine disruptors in water sources and water for human consumption Liquidchromatography coupled to tandem mass spectrometry (LC-MS/MS) is being applied with success to the analysis of these compounds [21-26].
However, the analysis of endocrine disruptors in several types of water matrices still require some preliminary sample preparation technique, which is frequently one of the most critical steps in the analytical process. Sample preparation techniques have been largely developed and this allow having the possibility to choose among various efficient techniques, less time consuming and with the advantage of reducing the use of higher volumes of organic solvents. Solid-phase extraction (SPE) is a well-established technique routinely used for the extraction/concentration of these target compounds as well for removing interfering matrices components before chromatographic analysis [25, 27]. The versatility of this technique, due to the variety of solid phases available, is another advantage of this technique.
The aim of this work was to optimize and validate a solid-phase extraction procedure combined with liquid chromatography coupled to tandem mass spectrometry detection with an electrospray ionization source (SPE /LC-(ESI)MS-MS) for the determination of seven natural and synthetic hormones (17-β-estradiol, 17-a-ethinylestradiol, estriol, estrone, progesterone, mestranol and diethylstilbestrol) and three industrial products (4-n-nonylphenol, 4-tert-octylphenol and bisphenol A).
The selection criteria for the analytical methodology were based on the characteristics of the target compounds (medium to high polarity) and also on the trace levels (down to the ng/L level in surface waters and even lower in ground and drinking waters) expected on the environmental matrices [25-27]. Several detection parameters were optimized in the mass spectrometer for each endocrine disruptor studied, such as the best conditions for the precursor ion formation, namely cone voltage, when applying negative and positive electrospray ionization mode. Two transitions were also established, one for quantification (MRM1) and one for qualification (MRM2), after optimization of the triple quadrupole conditions. The selection of two transitions ensures a high degree of selectivity and additional sensitivity to quantify trace levels of organic compounds in water samples.
Several conditions of the SPE were also tested, namely different elution solvents and sorbents as well as sorbent drying conditions, sample flow rate and the eluate concentration step.
The SPE/LC-(ESI)MS-MS was validated through the application of several statistical methods which allow defining the linear range, working ranges, and the method detection limits (MDL). Simultaneously, precision and matrix effect of the analytical assay were studied for the target compounds, according to the requirements of Council Directive 98/83/EC (European Commission, 1998) and International Procedures.
The European publications on this subject appeared first in the early-to-mid 2000s but the studies haven’t included the same endocrine disrupters, different types of waters (natural, residual and water for human consumption) were analysed, different degree of precision and accuracy were applied for the same instrumental analysis, and in same cases the requirements of optimisation and validation were not applied properly.
This study has the opportunity to prove the utility and reliability of the solid-phase extraction method with liquid chromatography coupled to tandem mass spectrometry in detail for the analysis of natural and synthetic hormones, and phenolic compounds in natural (surface or groundwater) and tap waters, which are the main preoccupation of the water distributors.
The majority of publications regarding this issue did not approach the evaluation of the uncertainty of the applied analytical method. In this study an evaluation of the main uncertainty sources of this method were also included, which allowed an estimation of the expanded uncertainties for the target compounds.
Many factors affect the distribution of the target compounds in different countries or even in different regions of the same country. This work focused on risk contaminants in the Lisbon drinking water supply. For this reason, this method has been applied to the analysis of several samples taken from the main intakes of water (surface and groundwater) used for production of water for human consumption, and from different sampling points of the drinking water distribution system (piping, nets and reservoirs) of EPAL (Empresa Portuguesa das Águas Livres, S.A.). EPAL is the largest drinking water supply company in Portugal and is responsible for the production and supply of drinking water to the city of Lisbon, as well as for the bulk supply to 25 municipalities of the greater Lisbon area, comprising around 2.6 million people. The production system includes two Water Treatment Plants (Asseiceira and Vale da Pedra) which abstract and treat surface water from Castelo de Bode reservoir and Tagus River, respectively. It also has a spring water source in Olhos de Água and 19 groundwater sources. This study allowed us to characterize the quality of different types of water samples, concerning the level of the studied target compounds.
This study was the first research performed to evaluate the occurrence probability of these target compounds in raw water and water supply system of EPAL, and also at national level.
2. Materials and Methods
2.1. Reagents and Standard Solutions
Individual standards of bisphenol A, diethylstilbestrol, 17-β-estradiol, estriol, estrone, 4-n-nonylphenol, 4-tertoctylphenol, mestranol and progesterone were obtained from Tokyo Chemical Industry (TCI) (Boerenveldseweg, Belgium). 17-a-ethinylestradiol was obtained from Sigma-Aldrich (Gillingham, United Kingdom). The purity of these standards was analytical grade.
Stock solutions with 200 mg/L of each compound were prepared in a solution of methanol:water (8:2) + 5 mM of ammonium acetate. These solutions were stored at 5 ± 3˚C in the absence of light.
A standard mixture solution was prepared by diluting each individual standard solution in methanol:water (4:6) + 5 mM of ammonium acetate, with a concentration of 4 mg/L. Appropriate amounts of the standard mixture solution were diluted in methanol:water (4:6) + 5 mM of ammonium acetate in order to get final concentrations between 1 and 400 µg/L.
Methanol (liquid chromatography grade, 99.9%) and ammonium acetate (pro analysis grade, 98%) were provided by Merck (Darmstadt, Germany).
Reagent water was obtained using a Milli-Q device, Academic A-10 model, from Millipore (Molsheim, France).
Acetone (99.5%, gas chromatography grade), methyl tert-butyl ether (gas chromatography grade), n-hexane (gas chromatography grade) all from Merck (Darmstadt, Germany), and dichloromethane (99.8%, High Performance Liquid Chromatography grade) from Sigma-Aldrich (Gillingham, United Kingdom) were used for SPE optimization.
Sodium thiosulphate (Na2S2O3) (99.5%, pro analysis grade) for samples pre-treatment was provided by Merck (Darmstadt, Germany).
2.2. Chromatographic and Mass Spectrometry Analysis
The chromatographic analysis was performed on a Waters Alliance 2795 Separations Module HPLC, equipped with a quaternary pump, automatic injector and a thermostated column compartment (Waters, Mildford, MA, USA) connected to a Quattro micro API triple quadrupole mass spectrometer, equipped with a Z-spray electrospray ionization source, from Micromass (Manchester, UK). The software used for instrumental control and data acquisition was Masslynx version 4.0, 2002 from Micromass. The chromatographic separation was performed with an Atlantis dC18 (2.1 × 150 mm, 5 mm) column from Waters (Mildford, MA, USA), using methanol:water (1:9) + 5 mM of ammonium acetate (solvent A) and methanol:water (9:1) + 5 mM of ammonium acetate (solvent B) as mobile phase. The separation was performed using a gradient profile. The gradient elution conditions were: 100% solvent A (0-15 min), and change to 100% solvent B (15-20 min). At the end of the chromatogram, the mobile phase was changed to its initial composition and the column was equilibrated.
The injection volume was 50 µL, the flow rate was kept constant at 0.3 mL/min under gradient conditions and the temperature of the column compartment was set to 30˚C.
Triple quadrupole operating conditions were studied in order to work in multiple reaction monitoring mode (MRM).
The optimization of the precursor ion formation in the ionization source of the mass spectrometer was performed through the selection of the ionization mode and the optimum cone voltage for each compound in order to achieve the best sensitivity. After setting the capillary voltage at a constant value of 3 kV it was made a comparison of sensitivity between the positive (ESI+) and negative ionization mode (ESI–). Some of the conditions were kept constant, such as: extractor voltage of 2 V, multiplier voltage 650 V, radio frequency (RF) lens at 0.5 V, source temperature at 150˚C and desolvation temperature at 350˚C. The settings of cone voltage varied between 5 and 90 V, in order to get a higher intensity for the signal of the precursor ion. Nitrogen was used as nebulizing, desolvation and cone gas. The flow rate of the desolvation gas was set to 350 L/h and that of the cone gas was set to 60 L/h. Argon was used as collision gas with a pressure of 2.83 × 10-3 mbar. For this purpose, the stock solution of each compound at a concentration around 200 mg/L was injected directly by infusion, in product ion scan.
Two transitions were also established, one for quantification (MRM1) and one for qualification (MRM2), after optimization of the triple quadrupole conditions, using argon as collision gas. This operation was performed through injection of an individual solution of each target compound, with a concentration around 4 mg/L, in the Rheodyne valve of the mass spectrometer, working in MRM mode, and varying the settings of collision energy between 5 and 90 eV, in order to get a higher fragmentation of the precursor ion and choose two products ions with better signal intensity.
2.3. SPE Procedure
A Caliper Life Sciences equipment, model Autotrace (Hopkinton, USA) was used as the automated SPE system. This technique was applied to 500 mL of sample volume. This instrument is equipped with an adjustable nitrogen stream to dry the cartridge before elution. After elution the organic extracts were concentrated to 0.5 mL in a Turbovap evaporation system (Zymark), under a nitrogen stream.
Operational conditions of the SPE were also tested, namely different elution solvents and cartridges as well as cartridge drying conditions under nitrogen stream, sample flow rate, and also the eluate concentration in the Turbovap.
The optimization was performed using 500 mL of reagent water spiked at the following concentrations: bisphenol A, 17-β-estradiol, 4-tert-octylphenol, 17-a-ethinylestradiol and mestranol at 0.40 µg L-1, proge sterone at 0.12 µg/L, estriol and 4-n-nonylphenol at 0.10 µg/L, and diethylstilbestrol and estrone at 0.040 µg/L (SPE optimization solution).
Two different cartridges, namely Waters Oasis HLB (6 mL, 200 mg) and Isolute C18 (EC) (6 ml, 1000 mg) were used in the optimization. These cartridges were washed with 3 mL methanol:acetone (3:2), followed by 6 mL of methanol and 6 mL of reagent water. Afterwards, water sample was drawn into the cartridge connected to a vacuum system at the speed of 30 mL/min and then, the cartridge was ashing with 3 mL of reagent water. After washing, the cartridge was dried under vacuum for 5 min and eluted with 8 mL (4 + 2 +2 mL) of methanol: acetone (3:2). The elution was evaporated to 0.25 mL on Turbovap (40˚C/0.2 bar) and reconstituted in methanol:water (4:6) + 5 mM of ammonium acetate to a final volume of 0.5 mL for further LC-(ESI)MS-MS analysis.
There are many different cartridges commercially available. The chosen cartridges are the best choice as there are many reports analyzing endogenous and synthetic endocrine disrupting chemicals using those cartridges. The several elution solutions used are shown in Table 1.
Other SPE operational conditions such as the sample flow rate (10 and 30 mL/min) and the cartridge drying time (0, 5, 10 min) were also tested.
The effect of the temperature bath in the recovery of the analytes was also optimized in the concentration step of the SPE eluate in the Turbovap system. With this

Table 1. Cartridges and solvents used in SPE optimization.
purpose, a volume of 8 mL of the SPE optimization solution (diluted in methanol) was concentrated to 0.5 mL in the Turbovap at different temperatures 35, 40, 45 and 50˚C. The concentrated solutions were analysed by LC-(ESI)MS-MS method under optimized conditions.
The analytical procedure can be complicated by the contamination of the extract with bisphenol A released from reagents (organic solvents) and plastic materials used in the process. If possible, all plastic materials and/or accessories from sampling and SPE equipment should be replaced by glass materials. Blank samples were regularly tested for contamination control.
The C18 cartridge was used in recovery studies using groundwater, surface and drinking water, and in real water samples analysis.
2.4. Validation Studies
The chromatographic linear range was studied by analysis of twenty one solutions containing all standards at different concentration levels (1 to 400 µg/L - prelimnary working range). For some compounds the range did not start at 1 µg/L due to the absence of MRM2 transition chromatographic signal. Calibration curves were plotted for each compound, applying least-square regression analysis [28], for measured signal values of MRM1 transition.
Linear range was evaluated by the determination coefficient and coefficient of variation of the method, and after application of several statistical tests, such as residual analysis, RIKILT test [29] and Mandel test [28]. The initial results were treated in order to comply with all the defined limits for each statistical test. The final choice of the working range was made after uncertainty evaluation of the calibration [30-32]. For the majority of the compounds the linear dynamic range were around five in order to get a lower component of uncertainty related to the calibration curve, and a more accurate quantification.
Taking into account the linear range, twenty fortified solutions were prepared with all target compounds at the lower (ten solutions) and higher (ten solutions) concentration values. These solutions were analyzed to evaluate the repeatability of the LC-(ESI)MS-MS method. The intermediate precision was also evaluated after analysis of one concentration level of the working range for each compound, on different days.
Matrix effects were evaluated in recovery studies using groundwater, surface and drinking water samples fortified at the following concentrations: 17-a-ethinylestradiol at 80 ng/L; mestranol at 60 ng/L; 17-β-estradiol and 4-tert-octylphenol at 40 ng/L; estriol, bisphenol A, and progesterone at 30 ng/L; 4-n-nonylphenol at 20 ng/L, estrone at 8 ng/L; and diethylstilbestrol at 2 ng/L. In order to eliminate interference from chlorine, drinking water samples were pre-treated with 0.1 g/L of sodium thiosulphate.
The main uncertainty sources of the method were identified and quantified, followed by the determination of the combined standard uncertainty (Uc) using a Gauss propagation model. The last step for uncertainty evaluation of an analytical result was the calculation of the expanded uncertainty (U), using a coverage factor k = 2 (95% of confidence level). Two of the approaches to calculate uncertainty are the “bottom-up” and the “topdown” methods. The “bottom-up” method was proposed by ISO (International Organization for Standardization) in order to quantify uncertainty in physical measurements and was subsequently adopted by Eurachem. On the other hand, the “top-down” method uses validation data and data from proficiency test schemes to estimate the uncertainty of the method [30-32].
Using the “bottom-up” approach we combined the components of uncertainty related with the calibration curve (Ucurve), the standard solution preparation (Ustandard), and the intermediate precision of the method (Uprecision). The standard uncertainty of the standard solution preparation is the combination of other components of uncertainty like the purity of the commercial standard, the volumetric and the weight measurements. The combined uncertainty (Uc) was calculated using the following equation:
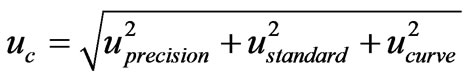 (1)
(1)
Using the ‘top-down’ method we combined the components of uncertainty related with the variability (Uvar) and with the accuracy (Uaccuracy) of the recovery studies (matrix effect). In this approach the combined uncertainty was calculated using another equation:
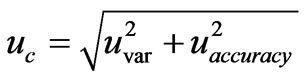 (2)
(2)
The expanded uncertainty (U) was obtained by multiplying Uc by a coverage factor (k) using the following equation:
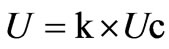 (3)
(3)
In general, the value of the coverage factor k is chosen on the basis of the desired level of confidence to be associated with the interval defined by Uc. Typically, k is in the range 2 to 3. When the normal distribution applies and Uc is a reliable estimate of the standard deviation, U = 2Uc (that is, k = 2) defines an interval having a level of confidence of approximately 95%, and U = 3Uc (that is, k = 3) defines an interval having a level of confidence greater than 99%.
2.5. Water Samples
Between May and June 2008, 43 samples were collected from several sampling points of the EPAL water supply system and also from the main raw water sources of this company (Tagus River, Castelo de Bode reservoir, and Alenquer well). These samples were collected in 1000 mL glass amber bottles with caps and stored at 5 ± 3˚C. For drinking water samples, containing residual chlorine, the sample was pre-treated in water sampling field with 0.1 g/L of sodium thiosulphate to eliminate any chlorine oxidation reaction and subsequent measurement reduce any type of interferences in the final results. The samples were analyzed within 48 hours after sampling. The optimized and validated SPE/LC-(ESI)MS-MS was applied to analyze these samples. The water samples were analyzing during the 48 hours after sampling.
Identification of the analytes was performed by comparing the retention times and the MS signals in the samples with those obtained using standard solutions. Furthermore, for LC-(ESI)MS-MS analysis, the ratio of the two precursor-product ion transitions was compared to that observed for standard analytes (using a tolerance of ± 15%). Quantification was carried out by calculating the response of quantitative ion. Concentrations were determined using linear regression analysis of the peak area.
3. Results and Discussion
3.1. Tandem Mass Spectrometry Conditions
For the majority of the target compounds, the negative ionization was better than positive ionization. Only progesterone and mestranol have a better result using positive ionization mode (Table 2).
The selection of the optimum value of cone voltage for each compound is very important as it has a significant impact in the precursor ion selectivity and subsequently in the signal intensity.
The selection of two transitions ensures a high degree of selectivity as well as additional sensitivity to quantify analyte trace levels in water samples. Therefore, it is important to obtain two product ions in good conditions. The optimization of triple quadrupole was performed by
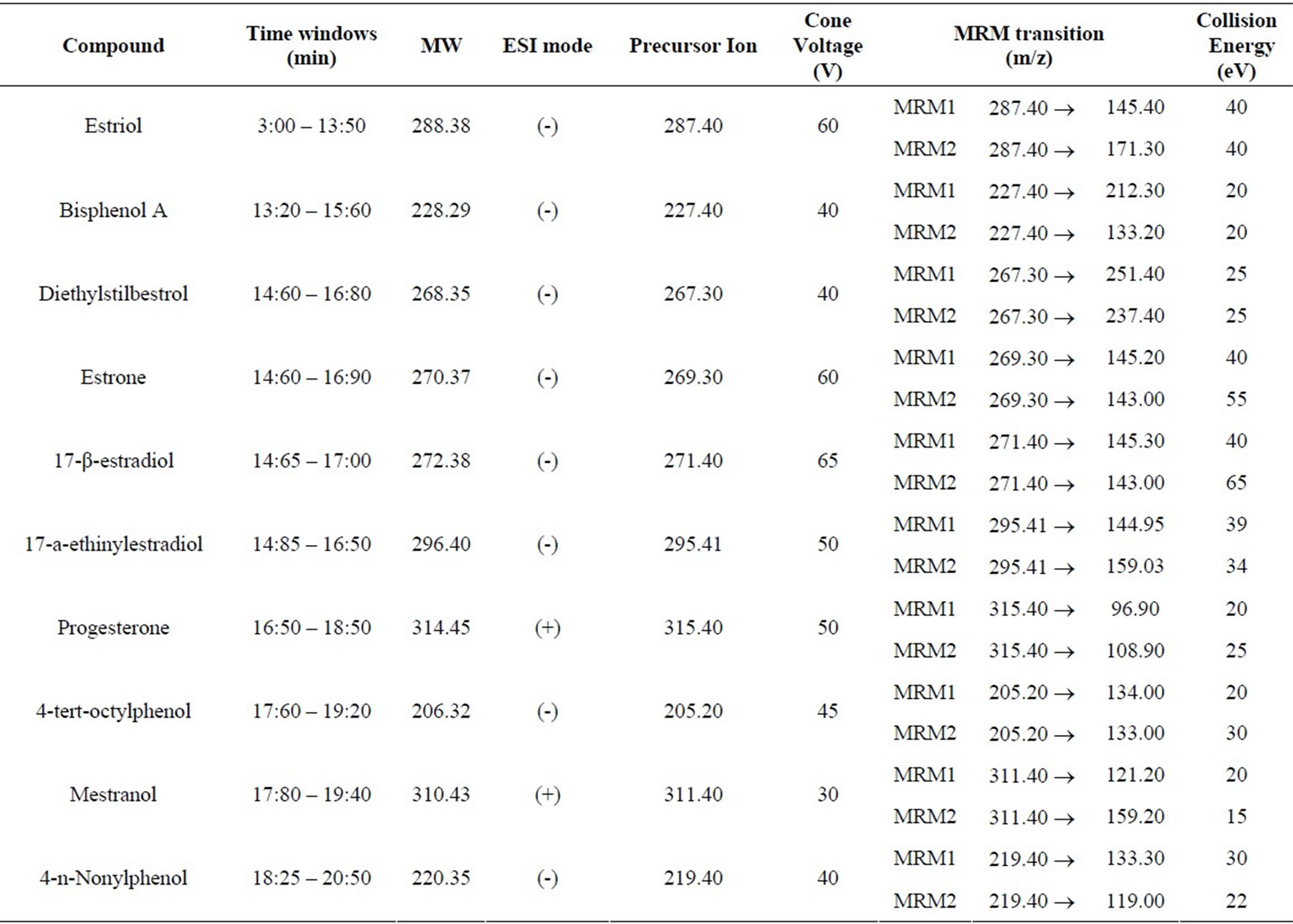
Table 2. Optimized conditions of the massspectrometer for each target compound.
varying collision energy with the objective to select two characteristic product ions (transition MRM1 and MRM2) formed after fragmentation of each precursor ion. The precursor ion fragmentation and the formation of several product ions are influenced by the collision energy. The selection of the product ions is based mainly on the best signal intensity. Transition MRM1 is used for quantification and transition MRM2 for qualification/confirmation, using the ratio MRM1/MRM2.
The best collision energy value was optimized for transitions MRM1 and MRM2 within the same compound. The selected conditions of the triple quadrupole mass spectrometer for the analysis of each target compound are resumed on Table 2.
The elution order of each endocrine disruptor in the chromatographic column was established after optimization of the liquid chromatograph conditions.
3.2. Linear and Working Range
All procedures used for chromatographic method validation requires the study of linear range. Several statistic tests were applied to the initial obtained results in the preliminary defined working range in order to comply with the acceptance criteria of each statistical test [28-29]. One of the established requirements for linear range is r2 ≥ 0.995, which was not accomplished by 4-n-nonylphenol, estrone and diethylstilbestrol. Another statistic test used is the Mandel test (ISO 8466-1) applied in the linear range [28]. The PG values obtained were higher than the F tabulated values for estriol, diethylstilbestrol, estrone, progesterone and 4-n-nonylphenol, for the corresponding degrees of freedom. The coefficient of variation of the method was ≤ 10% for all the compounds, varying from 0.030 to 3.7%. The residual analysis of the calibration data also showed that some compounds, like estriol, bisphenol A, diethylstilbestrol, estrone, 17-β-estradiol, progesterone, 4-tert-octylphenol and 4-n-nonylphenol, did not comply with the criteria of ± 10%. The application of the RIKILT Test [29], with acceptance criteria of 10%, to the same data, showed that mestranol was the only compound to fulfill this requirement.
For this reason, the linear ranges were narrowed until all data met the specified criteria of the statistical tests. The value of the component of uncertainty of the calibration was also taken into account in the definition of the final working range. At the end, r2 varied between 0.9962 and 0.9999; the coefficient of variation was between 0.01 and 3.96%; the residual analysis of the calibration data had a minimum value of –10% and a maximum of 7.6%; the RIKILT values varied between 90 and 114%, and all of the PG values were lower than the F tabulated values, for the corresponding degrees of freedom (Table 3).
The results of the precision study of the chromatographic method using repeatability conditions had RSDs between 2.1 and 17% for the lowest concentration level of the analytes, and between 0.45 and 7.8% for the highest concentration level. The obtained results are presented in Table 4.
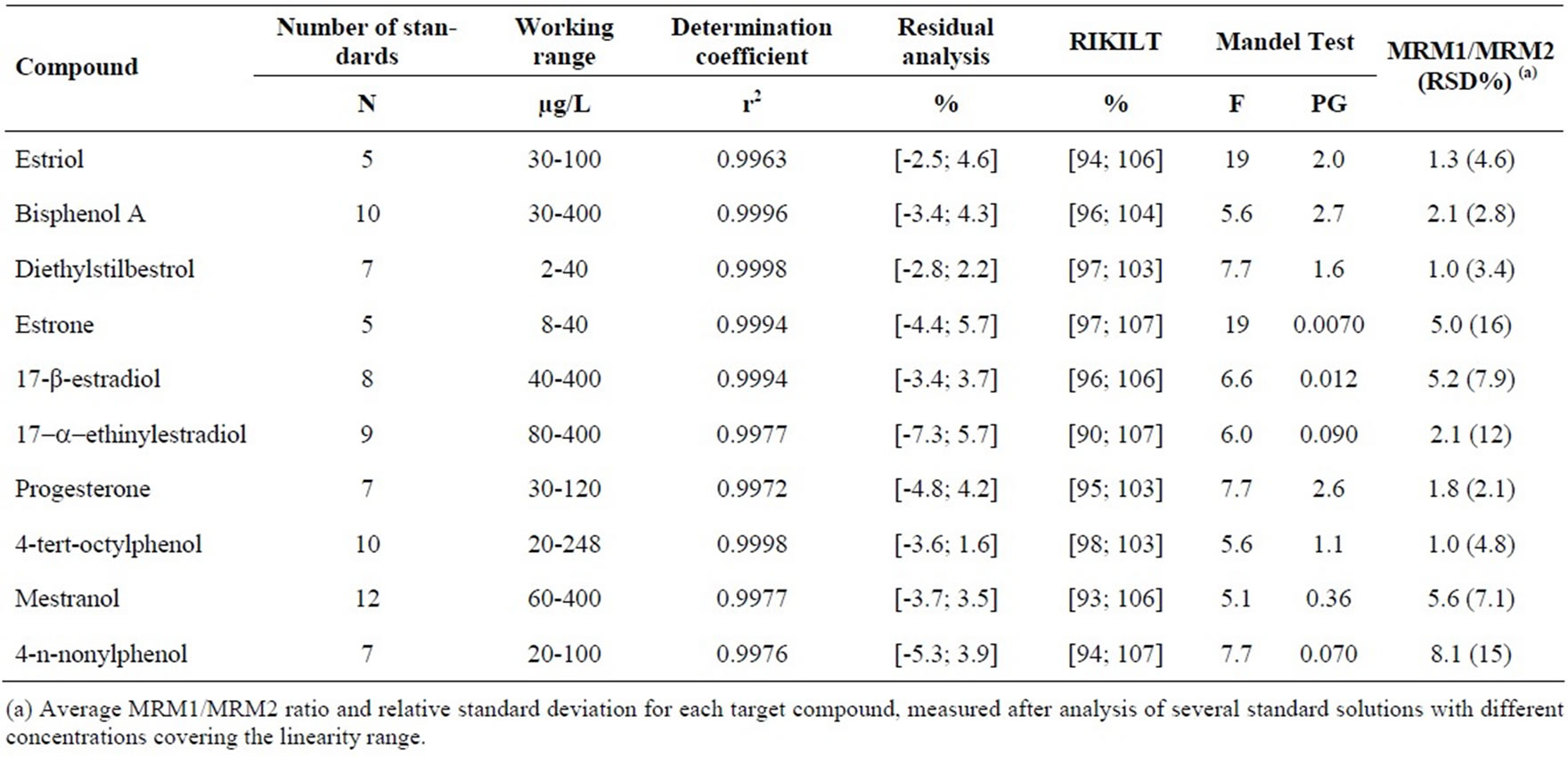
Table 3. Chromatographic working range, determination coefficient, MRM1/MRM2 ratio and Mandel test obtained for each endocrine disruptor by LC-(ESI)MS-MS.

Table 4. Precision of LC-(ESI)MS-MS method under repeatability conditions (n = 10).
Mestranol (12%) and estrone (17%) showed a RSD > 10% for the lowest concentration level. Nevertheless, for the majority of the compounds the RSD at the highest concentration level was lower than 5%. These results showed that the chromatographic method had a good precision
3.3. Solid-Phase Extraction
Several recovery studies were performed to evaluate the efficiency of each cartridge in the analysis of the target compounds. The percentage recoveries were between 55-94% and 54-90% for Isolute C18 (EC) and Oasis HLB, respectively. The relative standard deviation was between 7.3-14% and 2.8-12% for Isolute C18 (EC) and Oasis HLB, respectively.
The analysis of variance (ANOVA) showed that statistically there were no significant differences between the percentage recoveries obtained for all compounds, (pvalue = 0.30) using both cartridges. Table 5 lists the results for the two tested cartridges.
The selection of the best elution system for each SPE cartridge was also performed, applying the methodologies described on Table 1. The results obtained for each one are shown in Table 6.
The eluent systems B, C, D and F were immediately rejected due to the lower recoveries of the analytes. On the other hand, the eluent system E gave some problems when applied afterwards in routine, like random decrease of the recovery rates of some analytes, due to the formation of two different phases (MeOH has medium polarity and DCM has low polarity) in the eluate. Consequently, the eluent system A [MeOH:Acetone (3:2)] was selected for the SPE technique applied to the studied compounds.
This system allowed acceptable recovery rates between 55 and 84% with good coefficients of variation (≤ 10%).
The sample flow rates (10 and 30 mL/min) and cartridge drying conditions (0, 5, 10 min) were also tested in order to identify the best SPE conditions. The sample flow rate of 30 mL/min was the optimum value chosen because there are no significant differences between the results obtained using the above sample flow rate. Moreover, the use of a flow rate of 30 mL/min also reduces the time of analysis.
Cartridge drying conditions of 5 minutes with on-line nitrogen stream leads to a lower loss for phenolic compounds. This operation step revealed to be critical to the efficiency of the subsequent eluate evaporation step in the Turbovap system. If the cartridge drying step is not completed, the time for concentration in Turbovap could reach about 5 hours which leads to a higher nitrogen stream exposure of the eluate during the concentration step, and subsequently to a higher loss of the compounds, especially the phenolic compounds. On the other hand, if the cartridge drying time is higher, the eluate concentration step in the Turbovap system can be reduced, but the longer exposure to the nitrogen stream in the SPE equipment can also lead to a loss of compounds. Although most of the target compounds have very low volatility, they can be purged by nitrogen stream and therefore be lost.
The bath temperature of the Turbovap system is another adjustable parameter which needs to be set. The temperature is inversely proportional to the time needed for the concentration of the SPE eluate, and a higher time

Table 5. Comparison of recoveries (R%) of the target hormone disrupters of spiked reagent water by use Isolute C18 (EC) and Oasis HLB cartridges (n = 5).
implies a higher exposure to nitrogen stream. The application of ANOVA to the obtained results indicated that there were no significantly differences (pvalue = 0.81) between them. However, the higher recoveries occurred at 40˚C and 45˚C and the worst results were at 35˚C and 50˚C (higher losses for all the compounds). The optimum water bath temperature of 40˚C was chosen to prevent possible losses of the analytes.
3.4. Matrix Effect and Method Detection Limits (MDL)
The analysis of sample blanks showed no interferences coming from the studied matrices (surface water, groundwater and drinking water), assuring the high selectivity of the analytical method. Table 7 showed the percentage recovery results in different matrices after fortification, and also the variability associated with the entire analytical method. The obtained recovery values were between 32 and 95%. Despite the lower recoveries of some analytes, the variability of the data was satisfactory with a RSD ≤ 15%. There were no significant differences between the recoveries obtained for the analyzed water matrices. Homogeneity of variance analysis was applied to the best (drinking water) and the worst (Tagus river) recovery data from different matrices. However, the results showed that there was not any significant differences between the recoveries obtained for these matrices. All obtained test values (PG) were lower than the tabulated value of Fisher/Snedecor distribution (F (9, 5, 95%) = 10.2) [28].
The method detection limits (MDL) of the SPE/LC- (ESI)MS-MS were between 0.28 and 22 ng/L for progesterone and 17-a-ethinylestradiol, respectively (Table 7). The MDLs were calculated as three times the signal-to-noise level of the chromatographic signal obtained after analysis of the different spiked sample extracts.
These limits are equal or slightly higher than those found by other authors [34-37] because the demands set by validation criteria defined by accreditation process because EPAL laboratory is accredited. For these reason, all routine procedures must be validated according national and international procedures [28-33].
3.5. Uncertainty study
Two approaches were considered for estimation of expanded uncertainty of the analytical method. The first one used the “bottom-up” approach (Method 1) and the second one the “top-bottom” approach (Method 2). Table 8 presents the results concerning these two approaches.
The estimation of uncertainty by method 1 was between 18 and 26%, while by method 2 were between 15 and 30%. In method 1 the calibration component of uncertainty had the highest contribution to the expanded uncertainty. In method 2 there were no statistical significant differences between the contribution of the variability component and the accuracy component to the expanded uncertainty (pvalue = 0.60).
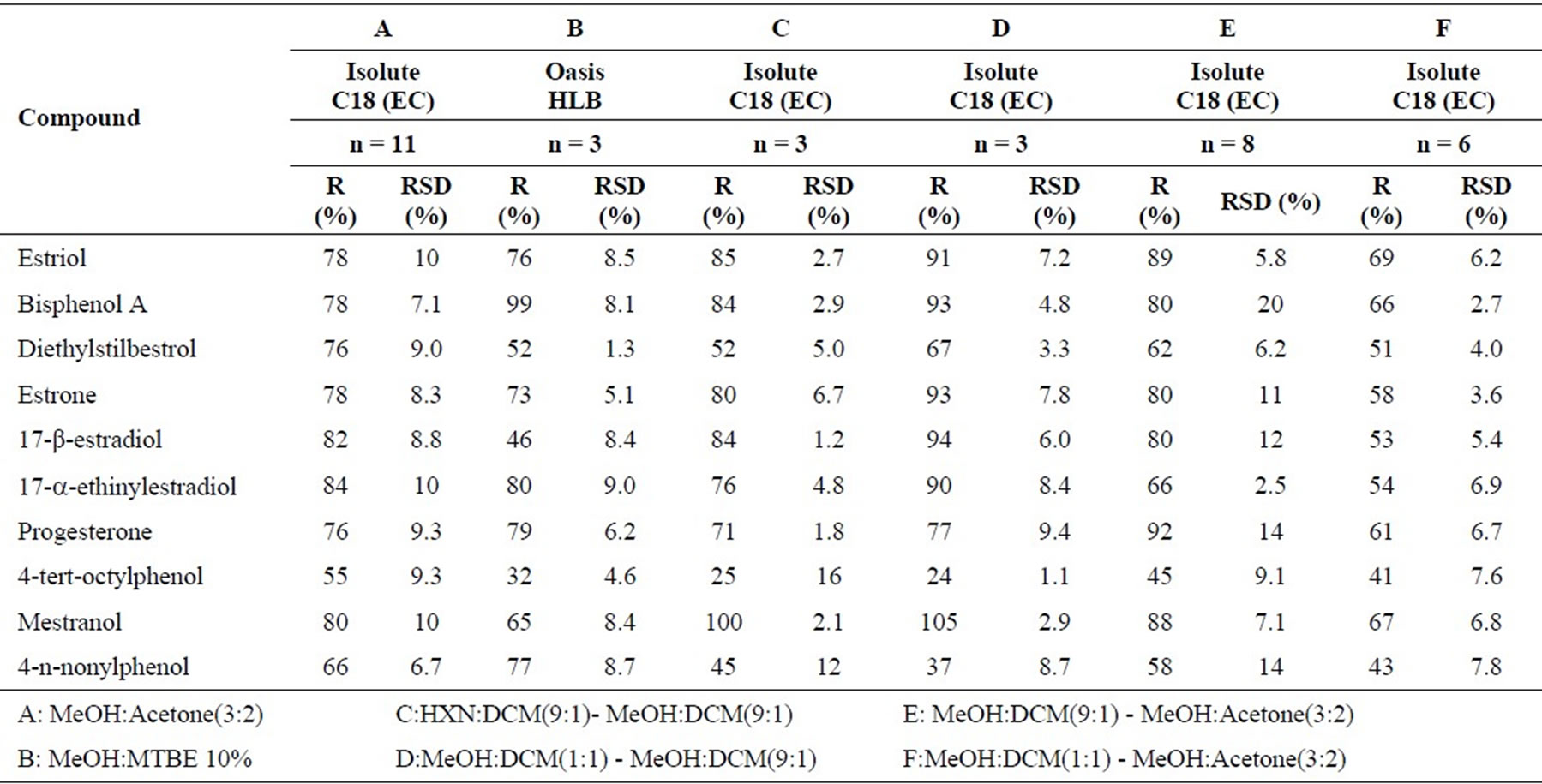
Table 6. Percent recoveries (R%) and relative standard deviation (RSD) for both elution solvents tested.
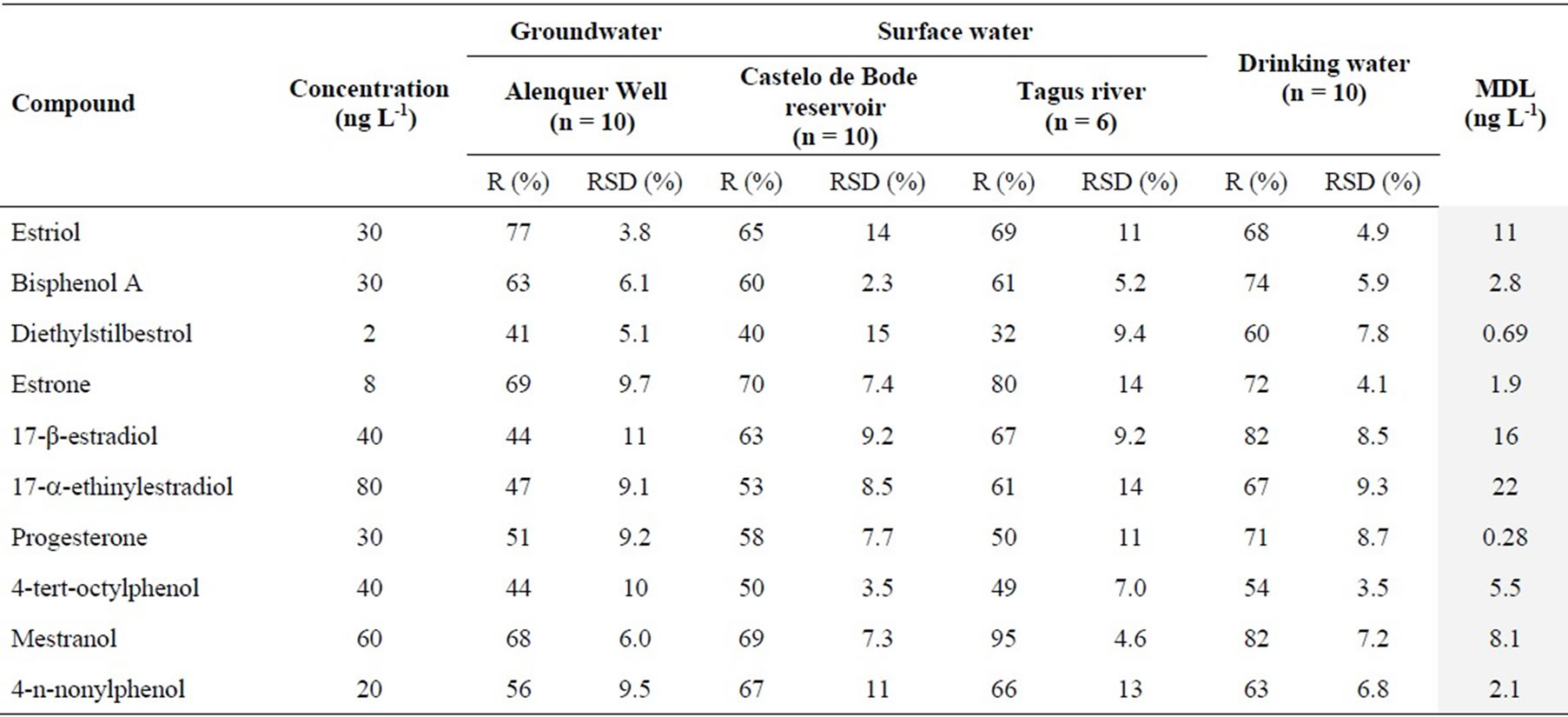
Table 7. Recovery (R%), relative standard deviation (RSD%) and method detection limits (MDL) for each endocrine disruptor and each analyzed matrix by SPE/LC-(ESI)MS-MS.
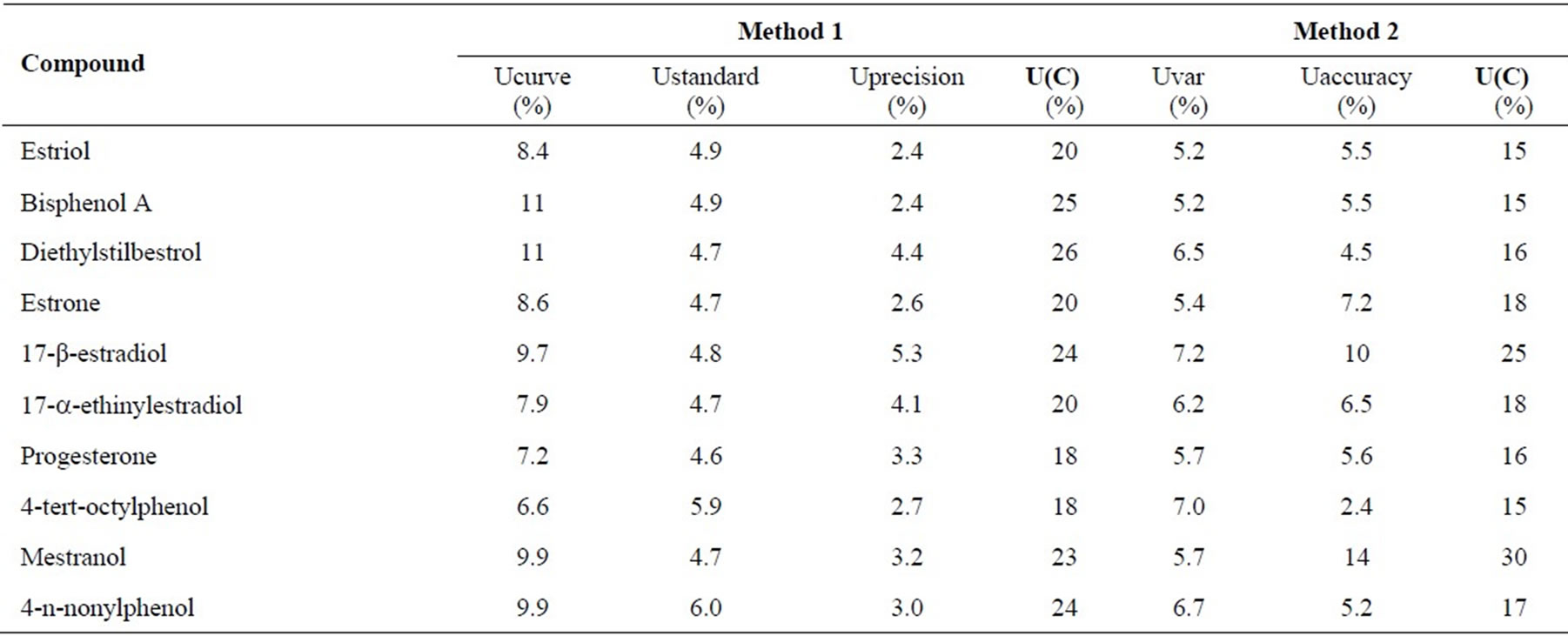
Table 8. Individual uncertainty components and respective expanded uncertainty estimated in the analysis of the target compounds by SPE-LC-ESI-MS/MS.
The value of the uncertainty component of the calibration data of each compound was also used to define its working range. In some cases (estriol and estrone) the number of standards (N) was reduced from 21 to 5 after applying linear range tests and uncertainty estimation (Table 3).
3.6. Sample Analysis
The validated SPE/LC-(ESI)MS-MS method was successfully applied to the analysis of the ten endocrine hormone disrupters in 43 samples from EPAL water supply system including tap water, spring water, groundwater, and river water. Estriol, estrone, 17-a-ethinylestradiol, diethylstilbestrol, mestranol and 17-β-estradiol were not detected in all samples. Some raw waters showed trace amounts of bisphenol A (six samples, 14% positive samples), progesterone (fourty samples, 93% positive samples), 4-tert-octylphenol (three samples, 6.9% positive samples), and 4-n-nonylphenol (six samples, 14% positive samples). The concentrations in these positive samples were between 2.9-4.8 ng/L for bisphenol A, 0.45-1.8 ng/L for progesterone, 27-29 ng/L for 4-tert-octylphenol, and 2.7-5.7 for 4-n-nonylphenol.
The level of detection of the majority of these compounds is well below the method quantification limit, and the estimated concentration levels had a high degree of uncertainty. On the other hand, they are also far from the proposal value of 0.1 mg/L, established by the Drinking Water Directive for compounds without guideline values, taking into account the precautionary principle. This study should be continued with a large number of samples collected in the same sampling points, during a longer time schedule.
4. Conclusions
Tandem mass spectrometry provides a highest degree of certainty in the identification of target compounds. The selection of specific fragment ions for multiple reaction monitoring (MRM) in tandem MS, with optimization of cone voltage and collision energy, guarantee a high degree of selectivity as well as additional sensitivity to quantify trace levels of endocrine disruptors.
The optimized and validated SPE/LC-(ESI)MS-MS method allowed the detection and quantification of all the studied compounds in water samples: estriol, bisphenol A, diethylstilbestrol, estrone, 17-β-estradiol, 17-a-ethinylestradiol, progesterone, mestranol, 4-tert-octylphenol and 4 -n-nonylphenol.
The LC-(ESI)MS-MS method showed a very good linear range for all the studied analytes with determination coefficients (r2) between 0.9963 and 0.9998, and coefficients of variation lower than 4%. The method showed a good precision under repeatability conditions for the majority of the compounds. The confirmation criteria set for each compound (MRM1/MRM2 ratio) showed a good stability over the defined linear range with a RSD ≤ 15%.
Both Isolute C18 (EC) and Oasis HLB cartridges can be used in the SPE procedure because there is no significant difference between them on the recovery tests performed. The best results were obtained using methanol:acetone (3:2) elution system, and a sample flow rate of 30 mL/min. The recovery data obtained in several water matrices were between 32 and 77%. Despite the lower recoveries of some analytes, the variability of the data had a RSD ≤ 15%. The method detection limits (MDL) of the SPE/LC-(ESI)MS-MS were between 0.28 and 22 ng/L The estimation of uncertainty was between 15 and 30%, and the calibration component had the highest contribution for the expanded uncertainty.
Estriol, estrone, 17-a-ethinylestradiol, mestranol, diethylstilbestrol, and 17-β-estradiol were not detected in all samples. Some raw waters showed trace amounts of bisphenol A, progesterone, 4-tert-octylphenol, and 4-nnonylphenol. On the other hand, they are also far from the proposal value of 0.1 mg/L, established by the Drinking Water Directive for compounds without guideline values, taking into account the precautionary principle. As a final conclusion we state that the optimized and validated method can be implemented in routine for the studied endocrine disruptor compounds monitoring with a high level of confidence.
REFERENCES
- Commission of the European Communities, “On Implementation of the Community Strategy for Endocrine Disrupters-A Range of Substances Suspected of Interfering with the Hormone Systems of Humans and Wildlife,” Progress Report, COM 706, 1999.
- T. Colborn, D. Dumanoski and J. P. Myers, “Our Stolen Future,” 1st Edition, Plume, Penguin Books USA inc, New York, Copyright, 1996.
- M. Gilbertson, T. Kubiak, J. Ludwig and G. Fox, “Great Lakes Embryo Mortality, Edema, and Deformities Syndrome (GLEMEDS) in Colonial Fish-eating Birds: Similarity to Chick-edema Disease,” Journal of Toxicology and Environmental Health, Vol. 33, No. 4, 1991, pp. 455- 520.
- R. Meyers, “D.E.S., the Bitter Pill,” 1st Editon, Seaview /Putnam, New York, 1983.
- A. L. Herbst, H. Ulfelder and D.C. Poskanzer, “Adenocarcinoma of the Vagina. Association of Maternal Stilbestrol Therapy with Tumor Appearance in Young Women,” The New England Journal of Medicine, Vol. 284, No. 15, 1971, pp. 878-881.
- H. Klip, J. Verloop, J. D. van Gool, M. E. T. A. Koster, C. W. Burger and F. E. van Leeuwen, “Hypospadias in Sons of Women Exposed to Diethylstilbestrol in Utero: A Cohort Study,” The Lancet, Vol. 359, No. 9312, 2002, pp. 1081-1082.
- M. M. Brouwers, W. F. J. Feitz, et al, “Hypospadias: A Transgenerational Effect of Diethylstilbestrol?” Human Reproduction, Vol. 21, No. 3, 2006, pp. 666-669.
- R. L. Cooper and R. J. Kavlock, “Endocrine Disruptors and Reproductive Development: A Weight-of-evidence Overview,” Journal of Endocrinology, Vol. 152, No. 2, 1997, pp. 159-166.
- O. Per-Erik, B. Bertil, B. Brunström, H. Hakansson and E. Klasson-Wehler, “Endocrine Disrupting Substances Impairment of Reproduction and Development,” Swedish Environmental Protection Agency, Stockholm, Sweden, 1998.
- D. S. Irvine, “Falling Sperm Quality,” British Medical Journal, Vol. 309, No. 6952, 1994, p. 476.
- L. Multigner and O. A. Luc, “Secular Variations in Sperm Quality: Fact or Science Fiction?” Cadernos de Saúde Pública, Vol. 18, No. 2, 2002, pp. 403-412.
- J. Toppari, A. Haavisto and M. Alanen, “Changes in Male Reproductive Health and Effects of Endocrine Disruptors in Scandinavian Countries,” Cadernos de Saúde Pública, Vol. 18, No. 2, 2002, pp. 413-420.
- J. W. Dallinga, E. J. C. Moonen, J. C. M. Dumoulin, J. L. Evers, L. H. Johannes, J. P. M. Geraedts and J. C. S. Kleinjans.Decreased, “Human Semen Quality and Organochlorine Compounds in Blood,” Human Reproduction, Vol. 17, No. 8, 2002, pp. 1973-1979.
- P. Mocarelli, P. M. Gerthoux, D. G. Patterson, S. Milani, G. Limonta, M. Bertona, S. Signorini, P. Tramacere, L. Colombo, C. Crespi, P. Brambilla, C. Sarto, V. Carreri, E. J. Sampson, W. E. Turner and L. L. Needham, “Dioxin Exposure, from Infancy through Puberty, Produces Endocrine Disruption and Affects Human Semen Quality,” Environmental Health Perspectives, Vol. 116, No. 1, 2008, pp. 70-77.
- M. Rittler and E. E. Castilla, “Endocrine Disruptors and Congenital Anomalies,” Cadernos de Saúde Pública Pública, Vol. 18, No. 2, 2002, pp. 421-428.
- J. R. Barret, “Shift in the Sexes: Are Endocrine Disruptors Changing Birth Ratios?” Environmental Health Perspectives, Vol. 115, No. 6, 2007, pp. 941-946.
- M. H. Hsieh, B. N. Breyer, M. L. Eisenberg and L. S. Baskin, “Associations among Hypospadias, Cryptorchidism, Anogenital Distance, and Endocrine Disruption,” Current Urology Reports, Vol. 9, No. 2, 2008, pp. 137-142.
- F. H. Pierik, A. Burdorf, J. A. Deddens, R. E. Juttmann and R. F. A. Weber, “Maternal and Paternal Risk Factors for Cryptorchidism and Hypospadias: A Case-control Study in Newborn Boys,” Environmental Health Perspectives, Vol. 112, No. 15, 2004, pp. 1570-1576.
- L. J. Paulozzi, “International Trends in Rates of Hypospadias and Cryptorchidism,” Environmental Health Perspectives, Vol. 107, No. 4, 1999, pp. 297-302.
- Commission of the European Communities, “On Implementation of the Community Strategy for Endocrine Disrupters - a Range of Substances Suspected of Interfering with the Hormone Systems of Humans and Wildlife,” Communication from the Commission to the European Parliament and Council, Progress Report, COM 262, 2001.
- E. S. Chiron and R. J. Analusis, “Determination of Nonionic Polyethoxylate Surfactants in Wastewater and Sludge Samples of Sewage Treatment Plants by Liquid Chromatography-Mass Spectrometry,” EDP Sciences, Vol. 28, No. 6, 2000, pp. 535-542.
- T. Isobe, H. Shiraihi, M. Yasuda, A. Shinoda, H. Suzuki and M. Morita, “Determination of Estrogens and their Conjugates in Water Using Solid-Phase Extraction Followed by Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 984, No. 2, 2003, pp. 195-202.
- M. S. Díaz-Cruz, M. L. Alda, R. López and D. Barceló, “Determination of Estrogens and Progestrogens by Mass Spectrometric Techniques (GC/MS, LC/MS and LC/MS /MS),” Journal of Mass Spectrometry, Vol. 38, No. 9, 2003, pp. 917-923.
- Y. K. K. Koh, T. Y. Chiu, A. Boobis, E. Cartmell, J. N. Lester, and M. D. Schrimshaw, “Determination of Steroid Estrogens in Wastewater by High Performance Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1173, No. 1-2, 2007, pp. 81-87.
- M. J. L. Alda, S. Díaz -Cruz, M. Petrović and D. Barceló, “Analysis of Selected Emerging Pollutants (Steroid Sex Hormones, Drugs and Alkilphenolic Surfactants) in the Aquatic Environmental by LC-MS and LC-MS-MS,” Journal of Chromatography A, Vol. 1000, No. 1-2, 2003, pp 503- 526.
- B. Nicholson, “Organic Chemical Issues in Wastewater Quality: A Review of Current Analytical Methods,” Australian Water Quality Centre for Water Quality and Treatment, September 2006.
- S. D. Richardson and T. A. Ternes, “Water Analysis: Emerging Contaminants and Current Issues,” Analytical Chemistry, Vol. 77, No. 12, 2005, pp. 3807-3838.
- International Standard Organization, “ISO 8466-1 Water quality-Calibration and Evaluation of Analytical Methods and Estimation of Performance Characteristics; Part I: Statistical Evaluation of the Linear Calibration Function,” 1990.
- J. M. P. van Trijp and A. H. Roos, “RIKILT-DLO, Model for the Calculation of Calibration Curves,” RIKILT report 91.02, Wageningen, Netherlands, 1991.
- EURACHEM/CITAC, “Quantifying Uncertainty in Analytical Measurement,” Guide CG 4, 2nd Edition, 2000.
- VAM Project 3.2.1., “Development and Harmonization of Measurement Uncertainty Principles,” Part (d): Protocol for Uncertainty Evaluation from Validation Data, 2000.
- EA (European co-operation for Accreditation), “EA Guidelines on the Expression of Uncertainty in Quantitative Testing,” EA-4/16, 2003.
- D. T. Burns, K Danzer and A. Townsend, “Use of the Terms Recovery and Apparent Recovery in Analytical Procedures (IUPAC Recommendations 2002),” Pure and Applied Chemistry, Vol. 74, No. 11, 2002, pp. 2201-2205.
- L. Sun, W. Yong, X. Chu and J. Lin, “Simultaneous Determination of 15 Steroidal Oral Contraceptives in Water Using Solid-Phase Disk Extraction Followed by High Performance Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Chromatography A, Vol. 1216, No. 28, 2009, pp. 5416-5423.
- D. Matějíčeka and V. Kubáň, “High Performance Liquid Chromatography/Ion-Trap Mass Spectrometry for Separation and Simultaneous Determination of Ethynylestradiol, Gestodene, Levonorgestrel, Cyproterone Acetate and Desogestrel,” Analytica Chimica Acta, Vol. 588, No. 2, 2007, pp. 304-315.
- D. P. Grover, Z. L. Zhang, J. W. Readman and J. L. Zhou, “A Comparison of Three Analytical Techniques for the Measurement of Steroidal Estrogens in Environmental Water Sample,” Talanta, Vol. 78, No. 3, 2009, pp. 1204 -1210.
- M. Pedrouzo, F. Borrull, E. Pocurull and R. M.Marcé, “Estrogens and their Conjugates: Determination in Water Samples by Solid-Phase Extraction and Liquid Chromatography-Tandem Mass Spectrometry,” Talanta, Vol. 78, No. 4-5, 2009, pp. 1327-1331.