Journal of Cancer Therapy
Vol.07 No.06(2016), Article ID:67643,16 pages
10.4236/jct.2016.76046
Gene Expression Profiling of Human c-Kit Mutant D816V
Shilpa Sharma, Gurudutta Gangenahalli*
Division of Stem Cell & Gene Therapy Research, Institute of Nuclear Medicine & Allied Sciences (INMAS), New Delhi, India

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 May 2016; accepted 20 June 2016; published 23 June 2016
ABSTRACT
The tyrosine kinase receptor III, c-Kit/stem cell factor receptor and its ligand, human stem cell factor (huSCF) are the predominant regulator of mitogenesis in the hematopoietic stem and progenitor cells. However, gain-of-function mutations alter c-Kit auto-regulatory mechanisms to aberrant c-Kit signaling, leading to the onset or progression of cancerous transformations. The most common mutation of c-Kit is the substitution of aspartic acid residue in position 816 to valine (D816V), which is majorly responsible for its ligand-independent constitutive activation, and is implicated in hematopoietic malignancies. Currently, molecular targeted therapy is increasingly becoming a hot spot due to its specificity and low toxicity. As the molecular mechanisms responsible for D816V-c-Kit mediated tumorogenicity are largely unknown, in this study, we aimed to investigate the D816V-c-Kit signaling mediated downstream molecular targets. Specifically, we created c-Kit active mutant form D816V and performed inducible gene expression of mutant D816V-c-Kit in monomyelocytic cell line U937. Mutant D816V-c-Kit expressing cells revealed significantly enhanced cellular mitogenic activity compared to wild-type c-Kit expressing cells independent of huSCF. To examine the molecular targets regulating tumorogenic proliferation, we evaluated the consequences of mutant D816V-c-Kit expression on downstream gene expression profile by high throughput microarray technology. The levels of some of the relevant genes (PIK3CB, eIF4B, PRKCDBP, MOAP1) were validated by quantitative polymerase chain reaction. SLA, STAT5B, MAP3K2 and MAPK14 emerged as important downstream molecular targets of mutant D816V-c-Kit. Further, by dissecting the signaling pathways, we also demonstrated that the D816V- c-Kit mediated hematopoietic cell proliferation is dependent on molecular target p38 MAP kinase.
Keywords:
c-Kit Mutant, Hematopoietic Cells, Microarray Gene Expression, Proliferation

1. Introduction
c-Kit and huSCF are encoded at the white spotting and steel loci of the mouse, respectively. Mutations at both the W and the Sl locus cause deficiencies in hematopoiesis, gametogenesis and melanogenesis [1] . In humans, c-Kit encodes a transmembrane glycoprotein that belongs to platelet-derived growth factor and macrophage growth factor receptor subfamily [2] . Activation of c-Kit by huSCF binding results in receptor dimerization and activation of intrinsic tyrosine kinase activity. Specific tyrosine residues are autophosphorylated, which results in the activation of downstream signaling pathways, including the Ras/Erk pathway and the phosphoinositide- 3-kinase (PI3K) pathway [3] - [5] .
Though, physiologically regulated tyrosine kinase activity of c-Kit is necessary for the controlled proliferation of hematopoietic stem and progenitor cells, mutational activations disturb c-Kit dynamic regulatory mechanisms, resulting in oncogenic transformation [6] [7] . Gain of independence of external growth stimuli is a crucial step. This can be achieved in several different ways, including mutations that render receptor tyrosine kinases constitutively active in the absence of ligand stimulation [8] [9] . In the case of c-Kit, these mutations most commonly occur either in exon 11 (encoding the juxta-membrane region), predominantly in gastrointestinal stromal tumors or in exon 17 (encoding the activation loop of the kinase domain). This is exemplified by codon 816 mutations found in several human malignancies including acute myeloid leukemia, mastocytosis, germ cell tumors of the seminoma or dysgerminoma types, sinonasal natural killer/T-cell lymphomas, and in intracranial teratomas [10] [11] . D816V mutation leads to the conversion of an aspartic acid residue to a valine, tyrosine, phenylalanine, asparagine or histidine residue [11] [12] . The acquired activating point mutation D816V significantly impairs the efficacy of targeted cancer therapies, thus limiting the treatment options for therapies [13] . Efforts to inhibit mutant c-Kit with tyrosine kinase inhibitors have been unsatisfactory, indicating a need for preclinical approaches to identify alternative molecular targets. Therefore, in this study, we sought to develop a human hematopoietic cell line model expressing D816V-c-Kit as a platform to dissect the molecular mechanisms underlying contributions of mutant D816V-c-Kit to myeloid leukaemia development. To unravel unknown molecular targets driving mutant D816V-c-Kit for leukemogenicity, we used the advanced approach of microarray technology. We used inducible gene expression system for the regulated expression of wild-type c-Kit and mutant D816V-c-Kit in huSCF/c-Kit null U937 cells and assessed the cells’ mitogenic potential. We performed microarray profiling of huSCF activated wild-type c-Kit expressing cells (induced) and mutant D816V-c-Kit expressing cells (induced). To the best of our knowledge, this is the first report that identifies the putative molecular targets triggered by mutant D816V-c-Kit using high throughput microarray in human myeloid leukemic U937 cells. Overall, the results of this study indicate that mutant D816V-c-Kit change the transcript levels of few genes responsible for the cell proliferation. Furthermore, the current data provide novel candidate genes, such as PIK3R1, eIF4B, STAT5B, MAPK14, elicited by mutant D816V-c-Kit in hematopoietic cells. Further, inhibition of proliferative capacity of mutant D816V-c-Kit expressing cells using p38 MAP kinase inhibitor indicates that p38 MAP kinase plays an essential role in D816V-c-Kit mediated tumorogenicity.
2. Materials and Methods
2.1. Creation of Recombinant Wild-Type pc-Kit-TRE2hyg and Its in vitro Site-Directed Mutagenesis
The complete coding sequence of the human c-Kit gene was excised as a BamH1 fragment (1 - 3.2 Kb) from pcDNA3-c-Kit vector (kindly gifted by Dr. Ronnstrand, Department of Experimental Clinical Chemistry, Austria). This fragment was further sub-cloned between BamHI restriction sites in sense orientation of PminCMV promoter using inducible expression vector pTRE2hyg (Clontech, USA). The sub-cloning of c-Kit gene was finally characterized by restriction analysis. The restriction analysis of recombinant wild type c-Kit-pTRE2hyg plasmid DNA using HindIII enzyme (Bioenzyme) generates three characteristic linear fragments of specific size; 4.9 Kb, 2.5 Kb and 1.1 Kb (Figure 1). The c-Kit active mutation as Asp to Val at 816 position (D816V) was introduced by site-directed mutagenesis system (Invitrogen, USA) using wild-type c-Kit-pTRE2hyg plasmid DNA as template and mutagenic primers to create mutation at nucleotide 2467: such as forward Primer 5-’ GAGGTCAAGAAGGTGAAGAGGAAGCTCACCTAC-3’; reverse Primer 5-’CTTCACCTTCTTGACCTCG CCCGTCTTGCCG-3’. The specific mutation of D816V-c-Kit-pTRE2hyg plasmid construct was confirmed by commercial DNA sequencing service (Ocimum Bio-solutions India Pvt. Ltd.) (Figure 2).
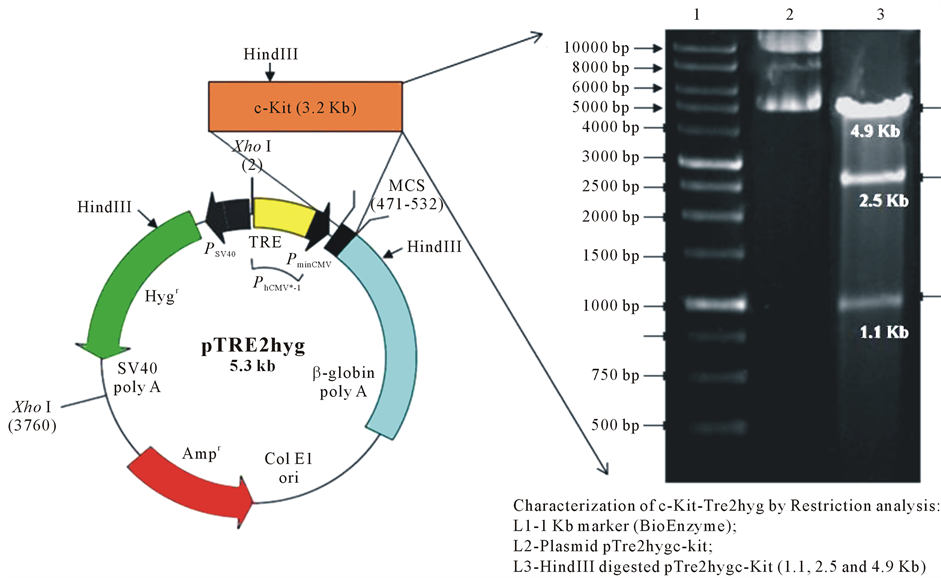
Figure 1. Restriction analysis of subcloned c-Kit. Restriction-digestion analysis of recombinant vector wild-type c-Kit- pTRE2hyg using HindIII give specific fragments of size 4.9 Kb, 2.5 Kb and 1.1 Kb (HindIII has two sites in c-Kit insert and one site in pTRE2hyg).

Figure 2. DNA sequencing of D816V-c-Kit-pTRE2hyg plasmid construct. Electropherogram showing wild type c-Kit and mutant D816V-c-Kit DNA sequence at exon 17. (a) In the wild-type c-Kit cDNA sequence, the codon is GAC; (b) In the mutated D816V-c-Kit sequence, GAC is substituted to codon GTC at nucleotide 2467, the point mutation is shown by the arrowhead.
2.2. Cell Lines and Stable Transfection
Human myelomonocytic leukemic cell line U937 was obtained from National Centre for Cell Science (NCCS, Pune, India). U937 cells were maintained in complete medium (RPMI-1640 medium supplemented with 1.0% sodium pyruvate, 10% FCS, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin) at 37˚C in a fully humidified atmosphere of 95% room air and 5% CO2. The culture was maintained thrice in a week in fresh complete medium. Exponentially growing cells with ≥94% viability were used for transfection. Approximately, 3 × 105/ml number of cells were transfected with p Tet-off plasmid DNA. Transfected cells were selected using puromycin (0.5 ng/µl) containing media. The Tet-off stable U937 cells were maintained by periodically adding doxycycline (1 µg/µl) after every two days. Tet-off transfected U937 cells were subsequently transfected with recombinant vectors-wild type c-Kit-TRE2hyg and D816V-c-Kit-TRE2hyg. Double transfected cells were selected using hygromycin containing culture medium and stable transfected positive clones were screened by limiting dilution. All transfections in U937 cells were performed using hilymax transfection reagent (Dojindo, Japan).
3. Results
3.1. Generation of Inducible Wild-Type and Mutant D816V-c-Kit U937 Cells
3.1.1. Flow Cytometry
Transfection of wild-type and mutant D816V-c-Kit stable transfectants were assessed by flow cytometry. Briefly, one million exponentially growing transfectants were washed and incubated for one hour with 0.5 μg of primary mouse anti-human monoclonal c-Kit antibody (Santa Cruz, USA) at a 1:200 dilution on ice. After washing three times with staining buffer (2% FBS and 0.1% sodium azide in phosphate-buffered saline (PBS), cells were subsequently incubated with secondary FITC-labelled goat anti-mouse antibody (Santa Cruz Biotechnology, USA). These cells were washed to remove unbound secondary antibodies and analyzed by flow cytometry. Non-specific binding was assessed by using isotype-negative controls that were added at the same concentrations. After washing and resuspension, samples were analyzed on a Becton Dickinson (San Jose, CA) FACS Calibur flow cytometer.
3.1.2. Western Blotting
Wild-type and mutant D816V-c-Kit were stimulated with and without huSCF (100 ng/ml) for 24 hrs. Cells were lysed in an appropriate amount of RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM sodium chloride, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA), supplemented with 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, and 1× protease inhibitor. Lysates were incubated for 30 min on ice and then cleared by centrifugation at 14,000 xg for 15 min at 4˚C. Protein content was determined by BCA method (Thermo Scientific, USA). Immunoblotting was performed following a standard protocol. Briefly, samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membrane (Pall Life Science). The membrane was blocked using 3% BSA dissolved in TBS buffer containing 0.1% tween-20 (TBST) for 1hr at room temperature and incubated overnight at 4˚C with primary antibodies: primary anti-human mouse monoclonal p-c-Kit antibody (Santa Cruz Biotechnology, USA), primary anti-human mouse monoclonal p-P38 MAP kinase antibody (Santa Cruz Biotechnology, USA), and anti-β-actin (Santa Cruz Biotechnology, USA). After three 5 min washes with TBST, membranes were incubated with HRP-conjugated secondary antibodies (1:2000) for 1 hr at RT and washed three times with TBST buffer. Blots were developed using TMB stabilized substrate (Promega, Wisconsin). Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, USA) and bands were visualized using enhanced chemiluminescence method (Amersham Pharmacia Biotech Piscataway).
3.2. Proliferation Assay
U937 cells expressing wild-type c-Kit or mutant D816V-c-Kit (2 × 104/well) were re-suspended in serum-free medium and seeded in triplicates in 96-well plates with 100 ng/ml huSCF (Invitrogen, USA) and without huSCF as control. After a 48 hr of stimulation, viable cells were evaluated using the Cell Titer 96 Aqueous One Solution cell proliferation assay kit (Promega, Wisconsin). Data was considered significant if results showed p-value ≤0.05. The effect of p38 MAP kinase inhibitor (Santa Cruz Biotechnology, USA) on cell growth rate was determined by plating cells (2 × 104/well) in a 96-well plate for 24 hrs at the indicated concentrations. Wild-type c-Kit cells and D816V-c-Kit cells were pre-incubated in the presence or absence of p38 MAP kinase inhibitor at concentration of 1 µM for 24 hrs. This was followed by the addition of 100 μl media with or without huSCF at 100 ng/ml.
3.3. Microarray
Wild-type c-Kit expressing cells (induced) stimulated with huSCF (100 ng/ml) for 24 hrs in serum-starved media and mutant D816V-c-Kit expressing cells (induced) were harvested and stored in RNAlater (Ambion Inc., USA). RNA isolation, quality control, and hybridization were performed by Genotypic Technologies Pvt. Ltd (Bangalore, India) according to the manufacturer’s instructions (Affymetrix, Santa Clara, USA). Briefly, RNA was isolated using an RNeasy mini-kit (Qiagen, USA) and quantified in a Nanodrop spectrophotometer. The RNA sample purity ratios were more than 1.9 for ratios of 260/280 nm and 260/230 nm; the RNA Integrity Number (RIN) values were greater than 7.1 as evaluated on a bio-analyzer 2100 (Agilent Technologies, India). Following reverse transcription of RNA in each replicate into respective cDNA, Cy3-labelled cRNA was produced by in vitro transcription and hybridized to a whole genome array chip (human whole gene expression microarray, 8 × 60 K array, Agilent Technologies).
3.4. Microarray Expression Data Analysis
Transcripts which were reliably detected in all the replicates were considered for subsequent data analysis. The data was normalized by percentile shift normalization method using GeneSpring GX11 (Agilent technologies, India). Following normalization, average signal intensity of the probes showing at least 30% change in expression across the 3 donors were computed and ratios (treated/untreated) were log2 transformed. Statistical analysis of the data was performed using Cyber-T regularized t statistic [14] due to small sample size (n = 3) since it takes into account Bayesian estimate of variance by pooling across genes with similar intensities [15] . The functional annotation tool (DAVID Bioinformatics Resources 6.7) was used to determine the biological relevance of the data and molecular functions represented by differentially regulated genes [16] , enabling us to explore and clarify the biological process, by considering a p-value ≤0.05 as significant. Further, the signaling pathways regulated by significantly altered genes were identified using the Pathway Miner tool [17] , which provides annotation from the Kyoto Encyclopedia of Genes and Genomics (KEGG), Biocarta and GenMAPP, taking Fisher exact p-value ≤0.05 as significant due to the small number of replicates [18] . Genes shown by microarray were classified with a significant change in expression level if they met the criteria of average log2 ratio ≥ ± 0.6 (±1.5 fold) and p-value ≤0.05 in case of mutant D816V-c-Kit expressing cells as compared to wild-type c-Kit expressing cells.
3.5. cDNA Synthesis, PCR and RT-PCR
Specific genes and their expression levels were validated using real-time PCR (RT-PCR) [19] . Total RNA was isolated using GenElute Mammalian Total RNA kit (Sigma, USA). cDNA synthesis was performed using Super Script III cDNA synthesis kit (Invitrogen, USA) as per the manufacturer’s instructions. RT-PCR was done using SYBR green quantitative RT-PCR kit (Sigma, USA) following manufacturer protocol. Gene specific primers were designed by Beacon designer software. Amplification of target genes were performed with thermal cycling condition of 95˚C for 10 min followed by amplification cycles of 95˚C for 30 seconds, 52˚C for 60 seconds, 72˚C for 30 seconds in a spectrofluorometric thermal cycler (Stratagene). Data was analyzed by using the comparative ΔΔCt method [20] by normalizing the ΔCT values for each gene to the ΔCT values of the housekeeping gene, actin. No template control (without cDNA template) was taken as negative control for RT-PCR. Data was normalized by the amount of β-actin mRNA. The primer sets and length of the PCR products are listed in Table 1 .
3.6. Treatment with p38MAPK Inhibitor (SB202190)
The p38MAPK inhibitor (SB202190) was tested for effects on impact-induced changes in viability in U937 cells at indicated concentrations [21] . Effect of p38MAPK inhibitor (SB202190) on proliferation was assessed in wild-type c-Kit and mutant D816V c-Kit expressing cells at a concentration of 1 μM for 24 hrs. Control was incubated with equivalent volumes of solvent (DMSO), which was 0.1% in culture media.
4. Results
4.1. Generation of Stable Mutant D816V-c-Kit Cells Using Doxycycline Regulated Gene Expression
Mutant D816V-c-Kit construct was validated by sequencing across the manipulated region. To study signaling pathways of D816V-c-Kit in progenitor cells, we used U937 cells transfected with wild-type and mutant D816V-c-Kit. Both wild-type c-Kit and the D816V mutant of c-Kit were stably transfected into the hematopoietic cell line U937 using the Tet-off inducible gene expression system [22] - [24] (Figure 3(a)). Flow cytometry analysis showed no change in fluorescence intensity between wild-type c-Kit expressing (induced) and mutant D816V-c-Kit (induced) cells. Induced mutant D816V-c-Kit cells showed significant shift in fluorescence intensity compared to uninduced mutant D816V-c-Kit cells (with doxycycline in media) and isotype control. By western blotting, wild-type c-Kit expressing cells and mutant D816V-c-Kit expressing cells showed receptor expression using c-Kit monoclonal antibody (Figure 3(b)).
4.2. Mutant D816V-c-Kit Mutant Revealed Enhanced Proliferation as Compared to Wild Type c-Kit
The directed proliferation potential of D816V-c-Kit expressing (induced) cells as compared to wild-type c-Kit expressing (induced) cells was analyzed in vitro by MTS assay. As shown Figure 4, D816V-c-Kit expressing cells showed significantly higher proliferation than huSCF (100 ng/ml) stimulated wild-type c-Kit expressing

Table 1. Details of primers used for real-time PCR in induced c-Kit mutant D816V expressing cells.
 (a) (b)
(a) (b)
Figure 3. Establishment of D816V-c-Kit expressing cells. (a) U937 cells expressing wild-type c-Kit and D816V-c-Kit were stained with PE-conjugated antibody or isotype control. The solid red fill shows fluorescence profile of isotype control, black histogram represents uninduced mutant cells (with doxycycline in medium), green panel for (induced) wild type c-Kit expressing cells and blue panel represents (induced) mutant D816V-c-Kit expressing cells. The figure is a representative of three experiments with similar results; (b) Immunoblot showing expression of phosphorylated c-Kit (145 Kda) in wild-type c-Kit and D816V-c-Kit expressing cells.

Figure 4. Mitogenic assay. Wild-type-c-Kit and D816V-c-Kit expressing cells number were assessed by MTS assay and expressed as relative absorbance units. Results are expressed as mean ± SD of triplicate and representative of at least 4 independent experiments (*p < 0.05).
(induced) cells. The wild-type c-Kit expressing cells showed response to growth factor, huSCF by showing their higher proliferation in presence of huSCF. However, mutant D816V-c-Kit expressing cells displayed no significant change in proliferation in the presence or absence of huSCF.
4.3. Transcriptional Response of Mutant D816V-c-Kit in Mutant Cells
Among the differentially regulated genes, genes involved in cell proliferation, PIK3R1 (0.82 fold, p = 0.02), FES (1.06 fold, p = 0.0353), eIF4B (1.11 fold, p = 0.02), ABL2 (0.67 fold, p = 0.0009), MAPK family, MAP2K3 (0.87 fold, p = 0.02), MAPK14 (1.49 fold, p = 0.00), Ras gene family, RAB7B (1.24 fold, p = 0.01), RAB3GAP1 (1.07 fold, p = 0.01), cell adhesion, ABL2 (0.67 fold, p = 0.00), LY9 (0.72 fold, p = 0.03), apoptosis, MOAP1 (−1.42 fold, p = 0.04), transcription factors, TAF5L/PCAF (1.09 fold, p = 0.01), nuclear receptors, U2AF (0.85 fold, p = 0.00), and metabolism, CYP2B6 (0.8 fold, p = 0.02), HMGCS1 (0.87 fold, p = 0.02) were altered in mutant D816V-c-Kit expressing cells. Expression of a number of other signaling molecules involved in mutant D816V-c-Kit mediated ligand-independent proliferation modulated were SLA (1.14 fold, p = 0.155) and STAT5 (2.32 fold, p = 0.103).
4.4. Validation of Microarray data
To verify expression levels of transcripts obtained from microarray, real-time PCR analysis was performed. Up-regulated genes chosen were PIK3R1 (0.82 fold) and eIF4B (1.11 fold); down-regulated genes were MOAP1 (−1.42 fold, p < 0.04) and PRKCDBP (−1.29 fold, p < 0.01). These genes depicted an identical pattern of alteration as seen by microarray expression analysis (Figure 5(a)). Further, we also confirmed expression of few genes which did not qualify selection criteria but are of relevance to the current study such as Src like adaptor, SLA (1.14 fold, p < 0.155) and Signal transducer and activator of transcription 5, STAT5B (2.32 fold, p < 0.103) (Figure 5(b)).
4.5. Gene Ontology Analysis
Further evaluation of biological processes and functions affected by mutant D816V-cKit expressing cells using DAVID bioinformatics resources identified upregulated genes to be functionally involved in MAPK signaling pathway (MAP2K3, MAPK14), mTOR signaling pathway (PIK3R1, EIF4B), cell cycle (eIF4B, ABL2), cell proliferation (INSIG1, FES, PIK3R1), transcription factors (TAF5L/PCAF, STAT5B) and cell adhesion (LY9). The down-regulated genes consisted of glyceroid metabolism (PPAP2C), basal cell carcinoma (MYCL1), cell differentiation (NDRG4, PDLIM7) and PCLKC (involved in negative regulation for cell growth). Significant (p < 0.04) over represented Gene Ontology terms (molecular function) in down and up-regulated genes is shown in Table 2.

Figure 5. Validation of mRNA levels of genes from microarray data. (a) qPCR assessment of genes showing statistically significant difference on the microarray. mRNA levels of proliferative genes (MOAP1, PI3KCB, eIF4B and PRKCDBP) in mutant D816V-c-Kit expressing U937 cells by Real-Time PCR; (b) qPCR assessment of genes (SLA and STAT5B) not showing statistically significant difference in microarray. Data of 3 independent experiments presented as mean ± SD (*p < 0.05).
Table 2. Gene ontology (biological processes) terms over-represented (p ≤ 0.05) in the set of genes altered by c-Kit mutant form D816V.
4.6. Dose-Dependent Cytotoxic Effect of p38 MAP Kinase in Mutant D816V-c-Kit Cells
Analysis of %viability in U937 cells in the presence of p38 MAP kinase inhibitor (SB202190) at the indicated concentrations showed that p38 MAP kinase inhibitor showed no cytotoxicity at concentration of 1 µM in 24 hrs (Figure 6(a)) but showed reduction in p38 MAP kinase phosphorylation compared to the vehicle, DMSO as shown by immunoblotting using phospho-p38 MAP kinase antibody (Figure 6(b)).
4.7. Generation of Stable Mutant D816V-c-Kit Cells Using Doxycycline Regulated Gene Expression
Mutant D816V-c-Kit construct was validated by sequencing across the manipulated region. To study signaling pathways of D816V-c-Kit in progenitor cells, we used U937 cells transfected with wild-type and mutant D816V-c-Kit. Both wild-type c-Kit and the D816V mutant of c-Kit were stably transfected into the hematopoietic cell line U937 using the Tet-off inducible gene expression system [22] - [24] (Figure 3(a)). Flow cytometry analysis showed no change in fluorescence intensity between wild-type c-Kit expressing (induced) and mutant
 (a) (b)
(a) (b)
Figure 6. Effects of p38MAP kinase inhibitor on cell viability in U937 cells. (a) U937 cells were cultured with various concentrations of p38MAP Kinase inhibitor (0.1 - 7 μM). U937 cells were treated with the indicated concentrations of p38MAP kinase inhibitor for 2 hrs. The cell viability was measured by the metabolic-dye-based MTS assay. Results represent the mean ± SD of three experiments performed in triplicate. The significance was determined by Student’s t-test (*p < 0.05 vs vehicle control); (b) Immunoblot using phospho-p38MAP kinase antibody. Western blot analysis of activated phospho-p38 in the lysates of U937 cells either pretreated with p38MAP kinase inhibitor (0.5 μM and 1 μM) or vehicle control. Blotted proteins were probed with anti-phospho-p38 and then with anti-p38 antibodies, each followed by peroxidase-conjugated secondary antibody. The level of β-actin is shown at the bottom as a loading control. One representative experiment of three independent experiments is shown.
D816V-c-Kit (induced) cells. Induced mutant D816V-c-Kit cells showed significant shift in fluorescence intensity compared to uninduced mutant D816V-c-Kit cells (with doxycycline in media) and isotype control. By western blotting, wild-type c-Kit expressing cells and mutant D816V-c-Kit expressing cells showed receptor expression using c-Kit monoclonal antibody (Figure 3(b)).
4.8. Mutant D816V-c-Kit Mutant Revealed Enhanced Proliferation as Compared to Wild Type c-Kit
The directed proliferation potential of D816V-c-Kit expressing (induced) cells as compared to wild-type c-Kit expressing (induced) cells was analyzed in vitro by MTS assay. As shown Figure 4, D816V-c-Kit expressing cells showed significantly higher proliferation than huSCF (100 ng/ml) stimulated wild-type c-Kit expressing (induced) cells. The wild type c-Kit expressing cells showed response to growth factor, huSCF, by showing their higher proliferation in presence of huSCF. However, mutant D816V-c-Kit expressing cells displayed no significant change in proliferation in the presence or absence of huSCF.
4.9. Transcriptional Response of Mutant D816V-c-Kit in Mutant Cells
Among the differentially regulated genes, genes involved in cell proliferation, PIK3R1 (0.82 fold, p = 0.02), FES (1.06 fold, p = 0.0353), eIF4B (1.11 fold, p = 0.02), ABL2 (0.67 fold, p = 0.0009), MAPK family, MAP2K3 (0.87 fold, p = 0.02), MAPK14 (1.49 fold, p = 0.00), Ras gene family, RAB7B (1.24 fold, p = 0.01), RAB3GAP1 (1.07 fold, p = 0.01), cell adhesion, ABL2 (0.67 fold, p = 0.00), LY9 (0.72 fold, p = 0.03), apoptosis, MOAP1 (−1.42 fold, p = 0.04), transcription factors, TAF5L/PCAF (1.09 fold, p = 0.01), nuclear receptors, U2AF (0.85 fold, p = 0.00), and metabolism, CYP2B6 (0.8 fold, p = 0.02), HMGCS1 (0.87 fold, p = 0.02) were altered in mutant D816V-c-Kit expressing cells. Expression of a number of other signaling molecules involved in mutant D816V-c-Kit mediated ligand-independent proliferation modulated were SLA (1.14 fold, p = 0.155) and STAT5 (2.32 fold, p = 0.103).
4.10. Validation of Microarray Data
To verify expression levels of transcripts obtained from microarray, real-time PCR analysis was performed. Up-regulated genes chosen were PIK3R1 (0.82 fold) and eIF4B (1.11 fold); down-regulated genes were MOAP1 (−1.42 fold, p < 0.04) and PRKCDBP (−1.29 fold, p < 0.01). These genes depicted an identical pattern of alteration as seen by microarray expression analysis (Figure 5(a)). Further, we also confirmed expression of few genes which did not qualify selection criteria but are of relevance to the current study such as Src like adaptor, SLA (1.14 fold, p < 0.155) and Signal transducer and activator of transcription 5, STAT5B (2.32 fold, p < 0.103) (Figure 5(b)).
4.11. Gene Ontology Analysis
Further evaluation of biological processes and functions affected by mutant D816V-cKit expressing cells using DAVID bioinformatics resources identified upregulated genes to be functionally involved in MAPK signaling pathway (MAP2K3, MAPK14), mTOR signaling pathway (PIK3R1, EIF4B), cell cycle (eIF4B, ABL2), cell proliferation (INSIG1, FES, PIK3R1), transcription factors (TAF5L/PCAF, STAT5B) and cell adhesion (LY9). The down-regulated genes consisted of glyceroid metabolism (PPAP2C), basal cell carcinoma (MYCL1), cell differentiation (NDRG4, PDLIM7) and PCLKC (involved in negative regulation for cell growth). Significant (p < 0.04) over represented Gene Ontology terms (molecular function) in down and up-regulated genes is shown in Table 2.
4.12. Dose-Dependent Cytotoxic Effect of p38 MAP Kinase in Mutant D816V-c-Kit Cells
Analysis of %viability in U937 cells in the presence of p38 MAP kinase inhibitor (SB202190) at the indicated concentrations showed that p38 MAP kinase inhibitor showed no cytotoxicity at concentration of 1 µM in 24 hrs (Figure 6(a)) but showed reduction in p38 MAP kinase phosphorylation compared to the vehicle, DMSO as shown by immunoblotting using phospho-p38 MAP kinase antibody (Figure 6(b)).
4.13. p38 MAP Kinase Inhibitor Mediated Reduction of Proliferation in Mutant D816V-c-Kit Cells
The vehicle, DMSO did not modify any investigated parameter in comparison with control culture. Investigation of the effect of p38 MAP kinase inhibitor in mutant D816V-c-Kit cells revealed significant reduction in cell proliferation. Mutant D816V-c-Kit expressing cells without huSCF showed more inhibition as compared to huSCF induced growth of mutant cells. Wild-type c-Kit expressing cells showed no effect on proliferation by p38 MAP kinase inhibitor either in presence or absence of huSCF. U937 cells showed no significant reduction in overall survival in the presence or absence of inhibitor, suggesting no cytotoxic effects on cell survival at dose of 1 µM at 24 hrs (Figure 7).

Figure 7. Growth inhibition by p38MAPK inhibitor in D816V-c-Kit expressing U937 cells. Wild-type c-Kit and D816V-c-Kit expressing cells were treated with indicated concentration of 1 µM of p38MAPK inhibitor for 24 hrs and then cultured for 24 hrs in growth media alone or supplemented with 100 ng/mL huSCF. Viable cells were detected by proliferation assay using MTS assay.
5. Discussion
Receptor tyrosine kinases are one of the most important targets underlying oncogenesis as their deregulation increases cellular proliferation in hematological malignancies [5] . D816V-c-Kit mutation is considered to be the most common gain-of-function mutation in case of mastocytosis, a myelo-proliferative neoplasm, and is involved to a lesser extent in germ-cell tumors, acute myeloid leukemia and mucosal melanoma [11] . Though, c-Kit inhibitor, imatinib is widely used in treatment of these diseases, however, imatinib fails to inhibit cells that exhibit the D816V mutation [12] [13] . New paradigm of cancer therapy involves the molecular characteristics of tumor to guide the therapeutic regimens. Biological significance of D816V mutation of c-Kit needs to be defined at molecular level to target causative kinase proteins for anti-cancer therapies.
Therefore, in the present study, we tried to understand the molecular response of mutant form of c-Kit at D816V residue c-Kit for the following reasons: 1) c-Kit mutation at residue D816V significantly impairs the efficacy of cancer therapies, limiting the treatment options for therapies; 2) the signaling pathways activated by D816V-c-Kit remain largely unidentified and 3) this will aid in developing novel molecular inhibitors with the potential to overcome resistance mutations. We investigated the downstream signaling events in D816V-c-Kit expressing cells compared to wild-type c-Kit using high-throughput gene expression profiling. Specifically, we developed a stable mutant D816V-c-Kit expressing cellular system and studied its correlation with proliferation and altered signal transduction compared to wild-type c-Kit expressing cells. We selected human hematopoietic progenitor U937 cells as this cell population is huSCF and c-Kit null at mRNA transcript level, which would eliminate the interference of host endogenous c-Kit gene expression [25] . Also, human U937 cells resource offers the opportunity to explore the study of c-Kit mutant expression and its function in the complex human conditions, thus would enable support for translational medical research in future. Expression of c-Kit gene could be done through viral transduction; however, transduction with viral vectors involves the risk of malignant transformation [26] . Furthermore, it has been reported that low efficiency of viral transduction of HSCs in human clinical trials severely decrease the level of chimerism or long-term repopulating ability of hematopoietic cells [27] . Further, to avoid undesirable oncogenic effects by constitutive activated signaling, we first made an effort to develop a system for transgene expression of c-Kit (wild-type and mutant) in a regulated manner using Tet-off inducible gene expression vector system. Additionally, this also offers the opportunity to precisely explore and study the transgene expression and its function in mammalian cell system. In this system, transacti- vator (tTA) is prevented from interacting with its binding site on DNA (tetO) by the effector substance, doxycycline, at low non-toxic concentrations [28] . Consequently, induced mutant D816V-c-Kit expressing cells (without doxycycline) showed much higher expression than uninduced mutant D816V-c-Kit cells (with doxycycline in media) by flow cytometry. Unexpectedly, mutant D816V-c-Kit expressing cells showed similar expression to wild type c-Kit expressing cells instead of showing higher receptor expression. This is due to increased rate of degradation of surface receptor mutant D816V-c-Kit and subsequently rapid receptor turnover [29] .
After achieving the doxycycline inducible regulated transgene surface expression of c-Kit genetic construct in the hematopoietic progenitor cell line U937, we further analyzed the proliferation of these cells towards huSCF in vitro. We used serum-free media to avoid non-specific activation of c-Kit by other serum proteins, thus to reduce signal-to-noise ratio. Our results revealed significantly enhancedproliferation of the mutant D816V-c-Kit expressing U937 cells as compared to wild-type c-Kit expressing cells. D816V-c-Kit expressing cells showed similar proliferation in presence or absence of huSCF (ligand-independent) whereas wild-type c-Kit cells showed proliferation in presence of huSCF only (ligand-dependent). huSCF showed no contribution for D816V mediated leukemogenicity in D816V-c-Kit expressing U937 cells unlike in mast cells [30] . This signifies for difference in mutant D816V-c-Kit downstream signaling compared to wild-type c-Kit, which ultimately regulates the cell proliferation.
Further, we attempted here to dissect downstream molecular targets of mutant D816V-c-Kit by microarray approach using an exhaustive 44,000 probe array. Our study provides a comprehensive analysis of transcriptome of D816V-c-Kit expressing cells. We employed three independent D816V-c-Kit transfection experiments and routinely cultured flasks to account for biological variance. Interesting set of genes responsible for factor independent proliferation is reported here for the first time. A search for the genes regulated by D816V-c-Kit revealed up-regulation of genes involved in cellular proliferation including PIK3R1, FES, INSIG1, eIF4B; and mitogen-activated protein kinases (MAPK) signaling pathway genes, MAP2K3 and MAPK14. The constitutively activated D816V-c-Kit mutation leads to the recruitment of major pro-oncogenic signaling cascades, such as the PI3K signaling, STAT, or RAS/MAPK pathway [31] . Quantitative PCR validated upregulation of PIK3R1. Earlier studies have also shown that PIK3R1 encoded protein, p85 subunit ofPI3K, is downstream target of D816V-c-Kit [32] . Rather, PI3K is constitutively associated with D816V-c-Kit receptor [32] . PI3K has been earlier shown to be implicated as participant in signaling pathways regulating multiple cellular functions, including proliferation, differentiation, anti-apoptosis and tumorigenesis [33] [34] . Another proliferative gene, FES, which encodes protein with tyrosine kinase activity, was also discerned up-regulated in mutant D816V-c- Kit expressing cells. This is in accordance with previous studies which have shown that FES is an essential effector of D816V-c-Kit proliferation signal. Reduction of FES expression leads to decrease in proliferation of D816V-c-Kit expressing cells [35] . Another up-regulated gene, eukaryotic translation initiation factor 4B (EIF4B) encode for proteins that are involved in the early initiation of protein synthesis. PI3K signaling pathway has also been shown to be related to EIF4B [36] .
MOAP1 and PRKCDBP down-regulation in mutant D816V-c-Kit expressing cells compared to wild-type c-Kit expressing cells were also validated by quantitative PCR. MOAP1 gene encodes protein which functionally mediates caspase-dependent apoptosis by its interaction with apoptosis regulator protein [37] . The gene encoding protein kinase C delta binding protein, PRKCDBP expression has been shown to induce the G1 cell-cycle arrest and increased cellular sensitivity to various apoptotic stresses [38] . The expression of this protein was found to be down-regulated in various cancer cell lines, suggesting the possible tumor suppressor function of this protein.
Few up-regulated microarray outcome genes, selectively, SLA (1.14 fold, p < 0.15) and STAT5B (2.32 fold, p < 0.10), did not meet the selection criteria, however, exhibit statistically significant change in expression when assessed by quantitative RT-PCR. This difference arises because of the sensitivities of the two techniques and use of different statistical analysis methods. It has been reported that SLA recruits ubiquitin ligases, which tag mutant D816V-c-Kit for degradation, contributing to its lower surface expression compared to wild-type c-Kit [39] . This is the most common mechanism through which SLA protein regulate receptor stability as well as downstream signaling [40] . STAT5 up-regulation by mutant D816V-c-Kit expressing cells as downstream target is consistent with STAT5 up-regulation as downstream target in D816V-c-Kit expressing HMC-1 cells important for cell proliferation. STAT activation can be aberrant as it is involved with other pathological situations [31] [41] . The up-regulation of BCR/ABL in D816V-c-Kit expressing cells, which encodes for kinase active fusion protein, opens the possibility that mutant D816V phosphorylates additional substrate molecules that are not phosphorylated by wild-type c-Kit and is expected to constitute novel targets for selective therapy. Earlier studies reported that Bcr/Abl kinase is an oncogenic fusion which protects hematopoietic progenitor cells from spontaneous apoptosis [42] .
Genes known to be regulated by MAPK signaling (MAP2K3 and MAPK14) were also shown to be modulated by D816V-c-Kit expressing cells. Growing evidence suggests that p38 MAP kinase subgroup of MAPKs is involved in cell proliferation, cell transformation and tumor progression [43] . We examined the effect of MAPK14 gene product, p38 MAP kinase on proliferative ability of mutant D816V-c-Kit expressing cells compared to wild-type c-Kit expressing cells using inhibitor of p38 MAP kinase. Mutant D816V-c-Kit expressing cells showed significant reduction in cell proliferation in the presence of p38MAP kinase inhibitor whereas wild-type c-Kit expressing cells showed p38 MAP kinase independent proliferation. This suggests that MAPK14 is implicated as important participant in signaling pathways regulating proliferation of mutant D816V- c-Kit cells. Comparative higher proliferation of huSCF treated mutant D816V-c-Kit expressing cells in presence of p38 MAP kinase inhibitor as compared to huSCF independent mutant D816V-c-Kit expressing cells treated with p38 MAP kinase inhibitor; suggest that factor (huSCF) independent proliferation of D816V-c-Kit expressing U937 cells is more sensitive for inhibition by p38 MAP kinase inhibitor than huSCF treated D816V-c-Kit expressing cells. This indicates that mutant D816V-c-Kit expressing cells without huSCF are more dependent on p38 MAP kinase signaling for proliferation as compared to mutant cells with huSCF. huSCF is a survival factor which rescue cells from growth inhibition to some extent. Also, both factor-dependent and -independent growth of D816V-c-Kit expressing cells was inhibited to a greater extent than huSCF induced growth of wild-type c-Kit cells. These results highlight the likely activation of p38 MAPK signaling pathway during proliferation mediated by D816V-c-Kit expressing cells. Though, p38 MAPK pathway becomes activated in a wide variety of cancers, the relative importance of p38 MAPK leading to mitogenesis is variable and appears to differ among different cell types [44] - [46] . Thus, inhibition of p38 MAPK can decrease cell survival and enhance the effects of chemotherapeutic drugs in many types of cancer cells. It seems that p38 MAP kinase signaling pathways represent a potential target for therapeutic intervention.
Overall, the results of this study indicate that mutant D816V-c-Kit expressing cells show upregulation of gene expression of BCR-ABL, PI3K, SLA, p38 MAPK and STAT5 pathways for cellular proliferation (Figure 8). Further in-depth broadband research of mutant D816V-c-Kit receptor would unveil an approach to manipulate HSCs with molecular targeted drugs to maximize efficacy with minimal dose.

Figure 8. Schematic diagram of mutant D816V-c-Kit mediated signal transduction responsible for cellular proliferation. STAT5 can be directly activated by the mutant c-Kit receptor in the absence of JAK proteins. SLA and BCR-ABL phosphorylate and localize STAT5. PI3K interact with the Ras GDP/GTP exchange protein. Recent evidence suggests that a significant amount of cross-talk occurs between the PI3K and MAPK pathways. The MAP kinase signal transduction pathways regulate the proliferation in a manner inextricable from other signal transduction system. Genes responsible for mediating apoptosis (MOAP1) was found downregulated. Upregulated genes are shown in red arrows and downregulated genes in green arrows (SLA: Src like adaptor; BCR/ABL: B-cell receptor/abelson murine leukemia virus; STAT5B: signal transducer and activated protein; PI3K: phosphatidyl inositol-3-kinase; eIF4B: elongation initiation factor 4B; RAS: rat sarcoma; MAP2K3: Mitogen activated protein kinase; CBP: CREB binding protein; MOAP1: Modulator of apoptosis1).
6. Conclusion
Present study demonstrated that D816V-c-Kit mutant is a key molecular player displaying efficacy for factor- independent hematopoietic cellular hyper-proliferation. Genes (PIK3R1, eIF4B, SLA, MAPK14, STAT5) modulated upon mutant D816V-c-Kit induction reveals transformation of huSCF-dependent cells to factor independent proliferation. A better understanding of the relationship between MAP kinase signal transduction system and the regulation of cell proliferation is essential for the rational design of novel pharmaco-therapeutic approaches [47] . Study provides a better fundamental understanding for cellular and molecular mechanism of mutant D816V-c-Kit, by revealing the up-regulation of group of genes which are known as crucial regulators of hematopoietic proliferation and survival. Deciphering molecular targets and functional role of dozens of uncharacterized genes identified needs to be undertaken along with identification of signaling pathways that would help pharmacologists and molecular biologists to design novel drugs targeting at site responsible for mutation D816V mediated tumorous transformation of c-Kit.
Acknowledgements
We are thankful to Dr. R.P. Tripathi, Director of INMAS for providing us necessary facilities. Authors also thank the Ministry of Defence, Department of Defence Research and Development for research grant. We thank Mrs. Namita Kalra for help during data acquisition by flow cytometry. Shilpa Sharma in particular thanks Indian Council of Medical Research (ICMR) for the award of Senior Research Fellow and Research Associateship.
Cite this paper
Shilpa Sharma,Gurudutta Gangenahalli, (2016) Gene Expression Profiling of Human c-Kit Mutant D816V. Journal of Cancer Therapy,07,439-454. doi: 10.4236/jct.2016.76046
References
- 1. Yarden, Y., Kuang, W.J., Yang, F.T., Coussens, L., Munemitsu, S., Dull, T.J., et al. (1987) Human Proto-Oncogene c-Kit: A New Cell Surface Receptor Tyrosine Kinase for an Unidentified Ligand. EMBO Journal, 6, 3341-3351.
- 2. Qiu, F., Ray, P., Brown, K., Barker, P.E., Jhanwar, S., Ruddle, F.H. and Besmer, P. (1988) Primary Structure of c-Kit: Relationship with the CSF-1/PDGF Receptor Kinase Family Oncogenic Activation of v-Kit Involves Deletion of Extracellular Domain and C Terminus. EMBO Journal, 7, 1003-1011.
- 3. Ashman, L.K., Cambareri, A.C., To, L.B., Levinsky, R.J. and Juttner, C.A. (1991) Expression of the YB5.B8 Antigen (c-Kit Proto-Oncogene Product) in Normal Human Bone Marrow. Blood, 78, 30-37.
- 4. Papayannopoulou, T., Brice, M., Broudy, V.C. and Zsebo, K.M. (1991) Isolation of c-Kit Receptor-Expressing Cells from Bone Marrow, Peripheral Blood, and Fetal Liver: Functional Properties and Composite Antigenic Profile. Blood, 78, 1403-1412.
- 5. Sharma, S., Gurudutta, G.U., Satija, N.K., Pati, S, Afrin, F., Gupta, P., et al. (2006) Stem Cell c-KIT and HOXB4 Genes: Critical Roles and Mechanisms in Self-Renewal, Proliferation, and Differentiation. Stem Cells and Development, 15, 755-778.
http://dx.doi.org/10.1089/scd.2006.15.755 - 6. Ashman, L.K. (1999) The Biology of Stem Cell Factor and Its Receptor c-Kit. International Journal of Biochemistry & Cell Biology, 31, 1037-1051.
http://dx.doi.org/10.1016/S1357-2725(99)00076-X - 7. Linnekin, D. (1999) Early Signaling Pathways Activated by c-Kit in Haematopoietic Cells. International Journal of Biochemistry & Cell Biology, 31, 1053-1074.
http://dx.doi.org/10.1016/S1357-2725(99)00078-3 - 8. Longley, B.J., Reguera, M.J. and Ma, Y. (2001) Classes of c-Kit Activating Mutations: Proposed Mechanisms of Action and Implications for Disease Classification and Therapy. Leukemia Research, 25, 571-576.
http://dx.doi.org/10.1016/S0145-2126(01)00028-5 - 9. Rodrigues, G.A. and Park, M. (1994) Oncogenic Activation of Tyrosine Kinases. Current Opinion in Genetics & Development, 4, 15-24.
http://dx.doi.org/10.1016/0959-437X(94)90086-8 - 10. Moriyama, Y., Tsujimura, T., Hashimoto, K., Morimoto, M., Kitayama, H., Matsuzawa, Y., et al. (1996) Role of Aspartic Acid 814 in the Function and Expression of c-Kit Receptor Tyrosine Kinase. Journal of Biological Chemistry, 271, 3347-3350.
http://dx.doi.org/10.1074/jbc.271.7.3347 - 11. Agarwal, S., Kazi, J.U., Mohlin, S., Pahlman, S. and Ronnstrand, L. (2015) The Activation Loop Tyrosine 823 Is Essential for the Transforming Capacity of the c-Kit Oncogenic Mutant D816V. Oncogene, 34, 4581-4590.
http://dx.doi.org/10.1038/onc.2014.383 - 12. Tsujimura, T., Hashimoto, K., Kitayama, H., Ikeda, H., Sugahara, H., Matsumura, I., et al. (1999) Activating Mutation in the Catalytic Domain of c-Kit Elicits Hematopoietic Transformation by Receptor Self-Association Not at the Ligand-Induced Dimerization Site. Blood, 93, 1319-1329.
- 13. Lee, S., Lee, H., Kim, J., Lee, S., Kim, S.J., Choi, B.S., et al. (2014) Development and Biological Evaluation of Potent and Selective c-KIT (D816V) Inhibitors. Journal of Medicinal Chemistry, 57, 6428-6443.
http://dx.doi.org/10.1021/jm500413g - 14. Baldi, P. and Long, A.D. (2001) A Bayesian Framework for the Analysis of Microarray Expression Data: Regularized t-Test and Statistical Inferences of Gene Changes. Bioinformatics, 17, 506-519.
http://dx.doi.org/10.1093/bioinformatics/17.6.509 - 15. Murie, C., Woody, O., Lee, A.Y. and Nadon, R. (2009) Comparison of Small n Statistical Tests of Differential Expression Applied to Microarrays. BMC Bioinformatics, 10, 45.
http://dx.doi.org/10.1186/1471-2105-10-45 - 16. Huang, D.W., Sherman, B.T. and Lempicki, R.A. (2009) Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nature Protocols, 4, 44-57.
http://dx.doi.org/10.1038/nprot.2008.211 - 17. Pandey, R., Guru, R.K. and Mount, D.W. (2004) Pathway Miner: Extracting Gene Association Networks from Molecular Pathways for Predicting the Biological Significance of Gene Expression Microarray Data. Bioinformatics, 20, 2156-2158.
http://dx.doi.org/10.1093/bioinformatics/bth215 - 18. Curtis, R.K., Oresic, M. and Vidal-Puig, A. (2005) Pathways to the Analysis of Microarray Data. Trends in Biotechnology, 23, 429-435.
http://dx.doi.org/10.1016/j.tibtech.2005.05.011 - 19. Heid, C.A., Stevens, J., Livak, K.J. and Williams, P.M. (1996) Real Time Quantitative PCR. Genome Research, 6, 986-994.
http://dx.doi.org/10.1101/gr.6.10.986 - 20. Livak, K.J. and Schmittgen, T.D. (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods, 25, 402-408.
http://dx.doi.org/10.1006/meth.2001.1262 - 21. Warrior, U., Chiou, X.G., Sheets, M.P., Sciotti, R.J., Parry, J.M., Simmer, R.L., et al. (1999) Development of a p38 Kinase Binding Assay for High Throughput Screening. Journal of Biomolecular Screening, 4, 129-135.
http://dx.doi.org/10.1177/108705719900400306 - 22. Gossen, M., Freundlieb, S., Bender, G., Müller, G., Hillen, W. and Bujard, H. (1995) Transcriptional Activation by Tetracyclines in Mammalian Cells. Science, 268, 1766-1769.
http://dx.doi.org/10.1126/science.7792603 - 23. Gossen, M., Bonin, A.L., Freundlieb, S. and Bujard, H. (1994) Inducible Gene Expression Systems for Higher Eukaryotic Cells. Current Opinion in Biotechnology, 5, 516-520.
http://dx.doi.org/10.1016/0958-1669(94)90067-1 - 24. Mayerhofer, M., Gleixner, K.V., Hoelbl, A., Florian, S., Hoermann, G., Aichberger, K.J., et al. (2008) Unique Effects of KIT D816V in BaF3 Cells: Induction of Cluster Formation, Histamine Synthesis, and Early Mast Cell Differentiation Antigens. The Journal of Immunology, 180, 5466-5476.
http://dx.doi.org/10.4049/jimmunol.180.8.5466 - 25. Chandrashekran, A., Gordon, M.Y. and Casimir, C. (2004) Targeted Retroviral Transduction of c-Kit+ Hematopoietic Cells Using Novel Ligand Display Technology. Blood, 104, 2697-2703.
http://dx.doi.org/10.1182/blood-2003-10-3717 - 26. Sadelain, M. (2004) Insertional Oncogenesis in Gene Therapy: How Much of a Risk? Gene Therapy, 11, 569-573.
http://dx.doi.org/10.1038/sj.gt.3302243 - 27. Schuster, J.A., Stupnikov, M.R., Ma, G., Liao, W., Lai, R., Ma, Y. and Aguila, J.R. (2012) Expansion of Hematopoietic Stem Cells for Transplantation: Current Perspectives. Experimental Hematology & Oncology, 1, 12.
http://dx.doi.org/10.1186/2162-3619-1-12 - 28. Woods, S.L. and Bishop, J.M. (2011) A New Transgenic Mouse Line for Tetracycline Inducible Transgene Expression in Mature Melanocytes and the Melanocyte Stem Cells Using Dopachrome tautomerase Promoter. Transgenic Research, 20, 421-428.
http://dx.doi.org/10.1007/s11248-010-9421-6 - 29. Sun, J., Pedersen, M. and Ronnstrand, L. (2009) The D816V Mutation of c-Kit Circumvents a Requirement for Src Family Kinases in c-Kit Signal Transduction. c-Kit Expression in Human Megakaryoblastic Leukemia Cell Lines, The Journal of Biological Chemistry, 284, 11039-11047.
http://dx.doi.org/10.1074/jbc.M808058200 - 30. Amagai, Y., Tanaka, A., Matsuda, A., Jung, K., Ohmori, K. and Matsuda, H. (2013) Stem Cell Factor Contributes to Tumorigenesis of Mast Cells via an Autocrine/Paracrine Mechanism. Journal of Leukocyte Biology, 93, 245-250.
http://dx.doi.org/10.1189/jlb.0512245 - 31. Chaix, A., Lopez, S., Voisset, E., Gros, L., Dubreuil, P. and De Sepulveda, P. (2011) Mechanisms of STAT Protein Activation by Oncogenic KIT Mutants in Neoplastic Mast Cells. The Journal of Biological Chemistry, 286, 5956-5966.
http://dx.doi.org/10.1074/jbc.M110.182642 - 32. Chian, R., Young, S., Danilkovitch-Miagkova, A., Ronnstrand, L., Leonard, E., Ferrao, P., Ashman, L. and Linnekin, D. (2001) Phosphatidylinositol 3 Kinase Contributes to the Transformation of Hematopoietic Cells by the D816V c-Kit Mutant. Blood, 98, 1365-1373.
http://dx.doi.org/10.1182/blood.V98.5.1365 - 33. Blume-Jensen, P., Janknecht, R. and Hunter, T. (1998) The Kit Receptor Promotes Cell Survival via Activation of PI 3-Kinase and Subsequent Akt-Mediated Phosphorylation of Bad on Ser136. Current Biology, 8, 779-782.
http://dx.doi.org/10.1016/s0960-9822(98)70302-1 - 34. Arcaro, A. and Guerreiro, A.S. (2007) The Phosphoinositide 3-Kinase Pathway in Human Cancer: Genetic Alterations and Therapeutic Implications. Current Genomics, 8, 271-306.
http://dx.doi.org/10.2174/138920207782446160 - 35. Voisset, E., Lopez, S., Dubreuil, P. and De Sepulveda, P. (2007) The Tyrosine Kinase FES Is an Essential Effector of KITD816V Proliferation Signal. Blood, 110, 2593-2599.
http://dx.doi.org/10.1182/blood-2007-02-076471 - 36. Gabillot-Carre, M., Lepelletier, Y., Humbert, M., de Sepuvelda, P., Hamouda, N.B., Zappulla, J.P., et al. (2006) Rapamycin Inhibits Growth and Survival of D816V-Mutated c-Kit Mast Cells. Blood, 108, 1065-1072.
http://dx.doi.org/10.1182/blood-2005-06-2433 - 37. Foley, C.J., Freedman, H., Choo, S.L., Onyskiw, C., Fu, N.Y., Yu, V.C., et al. (2008) Dynamics of RASSF1A/MOAP-1 Association with Death Receptors. Molecular and Cellular Biology, 28, 4520-4535.
http://dx.doi.org/10.1128/MCB.02011-07 - 38. Lee, J.H., Kang, M.J., Han, H.Y., Lee, M.G., Jeong, S., Ryu, B.K., et al. (2011) Epigenetic alteration of PRKCDBP in Colorectal Cancers and Its Implication in Tumor Cell Resistance to TNFα-Induced Apoptosis. Clinical Cancer Research, 17, 7551-7562.
http://dx.doi.org/10.1158/1078-0432.CCR-11-1026 - 39. Kazi, J.U., Agarwal, S., Sun, J., Bracco, E. and Ronnstrand, L. (2014) Src-Like-Adaptor Protein (SLAP) Differentially Regulates Normal and Oncogenic c-Kit Signaling. Journal of Cell Science, 127, 653-662.
http://dx.doi.org/10.1242/jcs.140590 - 40. Dragone, L.L., Shaw, L.A., Myers, M.D. and Weiss, A. (2009) SLAP, a Regulator of Immunoreceptor Ubiquitination, Signaling, and Trafficking. Immunological Reviews, 232, 218-228.
http://dx.doi.org/10.1111/j.1600-065X.2009.00827.x - 41. Bibi, S., Arslanhan, M.D., Langenfeld, F., Jeanningros, S., Cerny-Reiterer, S., Hadzijusufovic, E., et al. (2014) Cooperating STAT5 and AKT Signaling Pathways in Chronic Myeloid Leukemia and Mastocytosis: Possible New Targets of Therapy. Haematologica, 99, 417-429.
http://dx.doi.org/10.3324/haematol.2013.098442 - 42. Yu, C., Krystal, G., Varticovksi, L., McKinstry, R., Rahmani, M., Dent, P., et al. (2002) Pharmacologic Mitogen-Activated Protein/Extracellular Signal Regulated Kinase Kinase/ Mitogen Activated Protein Kinase Inhibitors Interact Synergistically with STI571 to Induce Apoptosis in Bcr/Abl-Expressing Human Leulemia Cells. Cancer Research, 62, 188-199.
- 43. Koul, H.K., Pal, M. and Koul, S. (2013) Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes & Cancer, 4, 342-359.
http://dx.doi.org/10.1177/1947601913507951 - 44. Maher, P. (1999) p38 Mitogen-Activated Protein Kinase Activation Is Required for Fibroblast Growth Factor-2-Stimulated Cell Proliferation but Not Differentiation. The Journal of Biological Chemistry, 274, 17491-17498.
http://dx.doi.org/10.1074/jbc.274.25.17491 - 45. Kanda, Y., Nishio, E., Kuroki, Y., Mizuno, K. and Watanabe, Y. (2001) Thrombin Activates p38 Mitogen-Activated Protein Kinase in Vascular Smooth Muscle Cells. Life Sciences, 68, 1989-2000.
http://dx.doi.org/10.1016/S0024-3205(01)00990-0 - 46. Adam, R.M., Roth, J.A., Cheng, H.L., Rice, D.C., Khoury, J., Bauer, S.B., et al. (2003) Signaling Through Pi3k/Akt Mediates Stretch and PDGF-Bb-Dependent DNA Synthesis in Bladder Smooth Muscle Cells. Journal of Urology, 169, 2388-2393.
http://dx.doi.org/10.1097/01.ju.0000063980.99368.35 - 47. Zhang, W. and Liu, H.T. (2002) MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Research, 12, 1989-2000.
http://dx.doi.org/10.1038/sj.cr.7290105
NOTES

*Corresponding author.