Journal of Cancer Therapy
Vol.05 No.13(2014), Article ID:51445,14 pages
10.4236/jct.2014.513125
Dichloroacetic Acid (DCA)-Induced Cytotoxicity in Human Breast Cancer Cells Accompanies Changes in Mitochondrial Membrane Permeability and Production of Reactive Oxygen Species
Zeiyad Alkarakooly1,2, Surya P. Kilaparty2, Qudes A. Al-Anbaky2, Mohammad Saeed Khan3, Nawab Ali2*
1College of Science, University of Diyala, Baquba, Iraq
2Department of Biology, College of Arts, Letters and Sciences, University of Arkansas at Little Rock, Little Rock, USA
3Department of Internal Medicine, Division of Rheumatology, University of Arkansas for Medical Sciences, Little Rock, USA
Email: *nali@ualr.edu
Academic Editor: Sibu P. Saha, University of Kentucky, USA
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 August 2014; revised 25 September 2014; accepted 20 October 2014
ABSTRACT
Cancer cells utilize cytosolic glycolysis for their energy production even in the presence of adequate levels of oxygen (Warbug effect) due to mitochondrial defects. Dichloroacetic acid (DCA) shifts cytosolic glucose metabolism to aerobic oxidation by inhibiting mitochondrial pyruvate dehydrogenase kinase (PDK) and increasing pyruvate uptake. Therefore, DCA has potential in reversing the glycolytic metabolism defect in cancerous cells. DCA is also known to induce apoptosis in a number of cancer cell lines, the mechanism of which is not well understood. In this study, an attempt has been made to investigate the effects of DCA on aggressive human breast cancer (MCF-7) cells as compared with less aggressive mouse osteoblastic (MC3T3) cells. Cell cytotoxicity was determined by MTT, crystal violet and Trypan blue exclusion assays. Western blot was used to detect any changes in the expression of apoptotic markers. Flow cytometry was used to measure apoptotic and necrotic effects of DCA. Mitochondrial integrity was determined by change in mitochondrial membrane potential (Δψm), whereas oxidative damage was determined by production of reactive oxygen species (ROS). DCA caused a concentration-dependent cytotoxicity both in MCF-7 and MC3T3 cell lines. MCF-7 cells were most affected. Flow cytometry results showed a significantly higher apoptosis in MCF-7 even at lower concentrations of DCA. However, higher concentrations of DCA were necrotic. Western blotting showed an increased expression of Mn-SOD-1 upon DCA treatment. Further, DCA decreased Δψm and increased ROS production. The effects of DCA were more pronounced on MCF-7 cells as compared to MC3T3 cells. Our results suggest that DCA-induced cytotoxicity in cancerous cells is mediated via changes in Δψm and production of ROS.
Keywords:
Breast Cancer, Dichloroacetic Acid, DCA, Cancer Therapy, Anticancer Agents, Apoptosis, Mitochondrial Defects, Reactive Oxygen Species (ROS)

1. Introduction
Normally, mammalian cells (non-cancerous) produce their energy by aerobic respiration or oxidative phosphorylation utilizing electron transport chain in mitochondria. It has long been recognized that cancerous cells primarily utilize glycolysis even in the presence of adequate oxygen, a phenomenon termed aerobic glycolysis or “Warburg effect” [1] . This change in cytosolic energy production in malignant cells is associated with reprogramming of mitochondrial function that limits pyruvate uptake for oxidative phosphorylation. This leads to an accumulation of large quantities of cytosolic lactic acid causing lactic acidosis. Accumulating evidence suggests that the persistent activation of aerobic glycolytic pathway in tumor cells plays a crucial role in carcinogenesis. Therefore, the inhibition of the increased glycolytic capacity of malignant cells may provide a key cancer treatment approach.
Dichloroacetic acid (DCA) is a small molecule known to shift cytosolic glucose metabolism to mitochondrial aerobic oxidation [2] [3] by inhibiting mitochondrial pyruvate dehydrogenase kinase (PDK). In recent years, reprogramming of mitochondrial function by DCA has received a great deal of attention for cancer treatment strategy as a result of its effectiveness in killing certain types of tumor cells [4] . Studies have now established that DCA suppresses tumor growth via the inhibition of mitochondrial PDK.
Michelakis and his colleagues reported that DCA had caused cell death in certain types of cancerous cells in vitro by inducing apoptosis. Additionally, they showed that the tumor size in rats was significantly reduced by the administration of DCA [5] . In an independent study, Bonnet and co-workers showed that exposing rats to DCA in drinking water caused regression of their xenografted A549 lung carcinoma cells [2] . In an in vitro analysis, they further showed that DCA only killed cancerous cells; it did not affect normal somatic cells. These studies suggested that DCA could be used as a safer yet effective anticancer treatment agent. In a comprehensive study on cancerous cells, Heshe and colleagues reported that DCA was effective against a panel of 18 immortal tumor cell lines. They also reported that DCA-induced effects were mediated by decreasing mitochondrial membrane potential (Δψm) indicating a role for mitochondria-mediated cell death process [3] . This study further showed that DCA also caused a significant induction of apoptosis in different types of human endometrial cancer cell lines. In other preclinical studies, DCA has been shown to inhibit cell proliferation and induce apoptosis in a number of cancer cell lines including prostate, breast, lung, endometrial and glioblastoma (GBM) cancer cell lines [2] . These studies clearly indicated that the treatments with DCA were linked with reduced rates of cellular proliferation and induction of apoptosis.
Since the major target for DCA was mitochondria, Wong and co-workers studied changes in mitochondrial membrane potential (Δψm) and found that the DCA-induced changes were linked to a reduced Δψm [6] . Recently, in an in vivo study, DCA has been shown to reduce the growth of lung cancer xenografts and significantly diminish lung metastasis in a rat mammary adenocarcinoma [7] . In addition, the growth of a pancreatic tumor xenograft was also found to be reduced perhaps by reversal of the glycolytic phenotype. Thus, selective modulation of the glycolytic phenotype in cancer cells by DCA seems to be a promising approach for an effective and tolerable treatment of cancer in humans, although clinical trials in humans still await confirmation and outcome of the safety data.
DCA has recently been regarded as the magic bullet against cancer by the public press, further stimulating the attraction of DCA for cancer treatment [3] . Its potential as anti-cancer agent is more attractive because it is easily available as a water-soluble small molecule, which is cell permeable and effective specifically in cancer cells with no or little toxicity to normal cells. It is also cost-effective. Further, DCA has been used in humans for over 30 years to treat lactic acidosis without any significant adverse side effect being reported. So DCA appears to be a safe drug for human use, although it still needs FDA approval for cancer treatment. The promise that the researchers have found in preclinical studies against adult malignancies and the availability of the limited safety data in adults and children provides a strong rationale to pursue further research on DCA in an effort to understand its mechanism of action in malignant as well as non-malignant cells. Therefore, in this study, an attempt has been made to investigate the effects of DCA on cytotoxicity of cancerous cells and study its mechanism of action. Two representative cell lines, namely MC3T3 mouse osteoblastic cells as less aggressive and MCF-7 human breast cancer cell lines as aggressive cancerous cells were used to compare the effects of DCA.
2. Materials and Methods
2.1. Materials
All cell lines including mouse osteoblastic MC3T3 and human breast adenocarcinoma MCF-7, cell culture media (MEM, DMEM), penicillin, Fetal Bovine Serum (FBS) and streptomycin were obtained from the American Type Culture Collection (ATCC), Manassas, VA. Vybrant apoptosis assay kit # 4 (V-13243, Molecular Probes) for flow cytometry analysis was purchased from Invitrogen, Carlsbad, CA. Dichloroacetic acid (DCA, Na-salt), etoposide, MTT reagents, Crystal Violet, Wortmannin, DMSO, 5’,5’,6’,6’-tetrachloro-1’,1’,3’,3’-iodide (JC-1) dye, 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescence dye, glacial acidic acid, Trypan blue, phosphatase inhibitor cocktail, protease inhibitor cocktail, p-amidinophenyl methanesulfonyl fluoride (PMSF) and other reagents were obtained from Sigma-Aldrich, St. Louis, MO. Manganese dependent super oxide dismutase-1 (Mn-SOD-1) and actin antibodies were obtained from Cell Signaling Technology, Danvers, MA. Secondary antibodies (anti-rabbit IgG or anti-mouse IgG) conjugated to horseradish peroxidase (HRP) were obtained from Sigma-Aldrich, St. Louis, MO. A super Signal West Pico Chemiluminescence kit for detection of immuno-complex on western blot was from Pierce Biotechnology, Rockford, IL. High performance chemiluminescence X-ray film was purchased from Amersham Biosciences, Piscataway, NJ.
2.2. Cell Culture
Cells were grown following instructions from ATCC. A complete medium containing MEM or DMEM with 10% FBS was supplemented with antibiotics penicillin (500 units/ml) and streptomycin (500 units/ml) as per manufacturer recommendation. All cells were grown in a humidified CO2 incubator set at 37˚C with 5% CO2 atmosphere. For sub culturing, cells grown to confluence were washed with PBS (11.9 mM phosphates, pH 7.4, 13.7 mM NaCl, 2.7 mM KCl) and then detached with Trypsin-EDTA (0.05% Trypsin/0.53 mM EDTA in HBSS without sodium bicarbonate, calcium and magnesium) by incubating for 5 minutes or until cells detached. After incubation, cells were collected by centrifugation at 500 g for 5 minutes. The cells were resuspended in 10 ml of the complete medium. Cell density was determined by counting the number of cells using a hemocytometer following staining with Trypan blue dye.
2.3. Cytotoxicity Assays
A number of methods including ethidium bromide/acridine orange (EB/AO) staining, MTT, crystal violet and Trypan blue exclusion assays were used to determine the cytotoxic effects of DCA. For DCA or other drug treatment, cells were seeded at a specific density depending upon the type of experiment and the cell culture plates used. After 24 hrs culture, drugs were added as bolus from stock solutions and mixed immediately to achieve the desired final concentrations. Further, incubations were continued for additional time intervals as required (see figure legends). Cells were then used for various assays, stained using appropriate staining procedures or harvested for biochemical determinations. Any variations in experimental procedures are described under figure legends.
For screening the cytotoxicity of DCA on various cell lines, MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl- tetrazolium bromide (MTT assay) or crystal violet (CV assay) reagents were used. The cells were seeded in 96-well plates (Costar, USA) at a density of 1.0 - 1.5 × 104 cells per well and grown for 24 hrs. Various concentrations of DCA or vehicle controls were then added to the wells and incubated for additional 48 hrs at 37˚C with 5% CO2. In MTT assay, mitochondrial dehydrogenase activity is used as an indicator of cell viability. It was assessed by the mitochondrial-dependent reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan [8] . Briefly, the cells were washed twice with the medium free of FBS. A 10 μl MTT solution (5 mg/ml) was added to 90 μl in cell culture medium free of FBS, and the cells were incubated for an additional 4 hrs in the dark. Thereafter, the medium was removed, and the cells were lysed with 100 μl of dimethylsulfoxide (DMSO) to dissolve the purple insoluble MTT formazan produced by mitochondrial succinate dehydrogenase. An automated microplate reader set at 570 nm wavelength was used to measure the conversion of MTT to formazan by metabolically viable cells. The crystal violet assay is based on the inability of dead cells to adhere to cell culture plastic dish [9] . Cells were washed with PBS to remove dead non-adherent cells. The remaining adherent viable cells were fixed with methanol and stained with 0.1% crystal violet solution for 10 minutes. The plates were thoroughly washed with water, and crystal violet was dissolved in 33% glacial acetic acid. The absorbance of the dissolved dye, corresponding to the number of viable cells, was measured in an automated microplate reader at 570 nm [10] . The results of MTT and crystal violet assays were presented as % of the control values obtained from untreated cells. The cell viability was calculated as follows [11] . The % of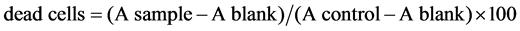 . All determinations were performed in triplicates, and each experiment was repeated at least three times for statistical analysis.
. All determinations were performed in triplicates, and each experiment was repeated at least three times for statistical analysis.
Cell viability was also determined by Trypan blue dye exclusion assay. Briefly, cells were detached by trypsinization and suspended in the medium as described for counting of the cells using hemocytometer. An aliquot of 90 μl of the cell suspension was transferred to an Eppendorf tube and mixed with 10 μl of 0.4% (w/v) Trypan blue dye. An aliquot of 10 μl of this mixture was loaded on each of the two sides of the counting chamber underneath the cover slip of the hemocytometer for cell counting. Cells stained with Trypan blue were counted as dead cells. Cells without Trypan blue stain were counted as live cells. Approximately 200 - 300 cells per treatment were counted for statistical calculations. Each experiment was performed in triplicates and repeated independently at least three times.
EB/AO staining was used as another criterion for cell viability following treatment with DCA. Cells were washed with PBS (10 mM, pH 7.4) and stained with a solution of 100 mg/ml acridine orange and 100 mg/ml ethidium bromide in PBS mixed together in a ratio of 1:1. Cells were then visualized immediately under UV light using a Nikon Labophot fluorescence microscope equipped with a digital camera. Photographs were taken using randomly selected fields. In order to determine the percentage of cells undergoing apoptosis or cell death process, the photographs were used to count the number of live (green) and dead (red) cells. Acridine orange stains live cells green, whereas ethidium bromide stains fragmented nuclear DNA in dead cells red. Approximately 200 - 300 cells per treatment were counted for statistical calculations.
2.4. Flow Cytometry Analysis
For separation of apoptotic and necrotic cells, flow cytometry was performed as described earlier [12] following staining of the cells with YO-PRO-I and PI dyes using Vybrant apoptosis assay kit # 4 (V-13243, Molecular Probes). Briefly, following treatment of cells with DCA to induce apoptosis, cells were harvested by acutase and washed with PBS, pH 7.4. The cell density was adjusted to ≈6 × 105 cells/ ml in PBS, pH 7.4. 1 μl of YO-PRO-I stock solution (component A) and 1 μl of PI stock solution (component B) were mixed per ml of cell suspension. After 30 minutes incubation at 4˚C, the cells were analyzed using BD FasCalibur flow cytometry to sort out cells labelled with each fluorescent probe from a total cell population. Fluorescence emissions were measured at 515 - 545 nm for FITC using a FL-1 PMT detector and 564 - 606 nm for PI using FL-2 PMT detector. In a quadrant (Figure 4), live cells showed little or no fluorescence (lower left), necrotic cells showed red and green fluorescence (upper left and right), and apoptotic cells showed green fluorescence (lower right).
2.5. Determination of Mitochondrial Membrane Potential
For analysis of mitochondrial membrane potential (Δψm), MC3T3 or MCF-7 cells were seeded at a density of 2.5 × 104 cells in 96-well plates and incubated overnight. Cells were then treated with DCA or PBS (control) as described above and maintained in supplemented medium. After 48 hrs, cells were washed with PBS, pH 7.4 and incubated with medium containing 10 μl of 10 mg/ml 5’,5’,6’,6’-tetrachloro-1’,1’,3’,3’-iodide (JC-1dye) for 20 min at 37˚C. In normal cells, the dye concentrates in the mitochondrial matrix due to the electrochemical potential gradient where it forms red fluorescence aggregates. A reaction that affects the mitochondrial membrane potential prevents the accumulation of the JC-1 dye in the mitochondria and thus the dye is dispersed throughout the entire cell leading to a shift from red (590 nm) to green (525 nm) fluorescence. Finally, the cells were washed and resuspended in 100 μl PBS for fluorescence measurements using microplate reader (SYNERGY H4, BioTek, hybrid technology). Mitochondrial depolarization is indicated by a decrease in red/green fluorescence ratio. All mitochondrial membrane potential analyses were performed in triplicates and each experiment was repeated independently at least three times.
2.6. Determination of Reactive Oxygen Species (ROS)
For the assessment of the production of intracellular ROS, MC3T3 and MCF-7 cells were plated in the black clear bottom 96-well plates at a cell density of 1.0 - 1.5 × 104 cells per well and treated with DCA for 48 hrs as described above. A solution of 2’,7’-dihydrochloroflurorescein acetate (DCFH-DA) was added to the medium at a final concentration of 10 μM and the cells were allowed to stain for 90 min in the dark. After DCFH-DA staining, the cells were washed twice and resuspended in 100 μl of PBS. DCFH-DA intensity was examined using fluorescence microplate reader (SYNERGY H4, Bio Tek, hybrid technology) at 485/535 nm. The data presented is a representative experiment performed independently at least three times in triplicate samples.
2.7. Preparation of Cell Lysate and Western Blotting
After appropriate treatments to induce cytotoxicity or apoptosis, the culture media were collected and centrifuged at 1100 g for 5 min to collect floating cells. Cell lysates were prepared by first washing the attached cells once with ice-cold PBS, pH 7.4 and then lysed for 20 min with a lysis buffer (RIPA) containing a protease inhibitor cocktail, a 1 mM phosphatase inhibitor cocktail and 0.1 M PMSF. The floating cells collected were mixed with corresponding cell lysates. Following cell lysis, cell debris was removed by centrifugation at 12,000 g (Eppindroff centrifuge) for 15 min at 4˚C. Protein concentration in cell lysates was determined by using a standard Coomassie Bradford protein assay according to the manufacturer’s instructions (Bio-Rad, Hercules, CA). Western blotting procedures followed were as described by Agarwal et al., 2009. In order to prepare protein samples for SDS-PAGE, cell lysates were mixed with equal volumes of 2× sample buffer (0.5 MTris-HCl, pH = 6.8, 20% glycerol, 4% (w/v) SDS, 0.5% (w/v) bromophenol blue) and boiled for 5 min at 100˚C. Samples containing 100 μg proteins were separated by 10% SDS-PAGE. The proteins from the gels were electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA). The membranes were blocked with 5% skimmed milk prepared in Tris-buffer saline (150 mM NaCl and 20 mM Tris-HCl, pH 7.2) containing 0.05% Tween-20 (TBS-T) for 1 hr at room temperature. The membranes were then incubated with diluted primary antibodies overnight at 4˚C with constant shaking. The primary antibody dilutions were prepared according to the manufacturers’ recommendations. After primary antibody incubation, the membranes were washed three times for 10 min each with TBS-T and were incubated for 2 hrs with a 1:2000 dilution of an appropriate secondary antibody (anti-rabbit IgG for polyclonal or anti-mouse IgG for monoclonal antibodies) conjugated to horseradish peroxidase (HRP) in 2% skimmed milk in TBS-T (Sigma-Aldrich, St. Louis, MO). Signal detection was achieved with a Super Signal West Pico Chemiluminescence kit (Pierce Biotechnology, Rockford, IL) and high performance chemiluminescence film (Amersham Biosciences, Piscataway, NJ).
2.8. Statistical Analysis
The results are expressed as a mean ± SD from three to four independent experiments or as described under figure legends. One-way ANOVA test was used to evaluate statistical significance between control and experimental groups. The p-value of <0.05 was taken as the value with significant difference.
3. Results
3.1. DCA Induces Cytotoxicity in Cancerous Cells
MTT assays are widely used to evaluate the overall cytotoxicity of the cell death-inducing drugs. In our experiments, the ability of DCA to promote cell death or cytotoxicity was evaluated by MTT assays in a number of cancer cell lines. The results are presented in Figure 1. The cytotoxic effects of DCA were observed on all

Figure 1.Effect of DCA on cytotoxicity. Indicated cell lines were grown and seeded at a density of 10 - 15 × 103 cells/well in 96-well plates as described in materials and methods. After 24 hrs, cells were treated with indicated concentrations of DCA. Cell culture continued for additional 48 hrs. Cell viability was determined by MTT assays. The values shown are mean ± SD from at least three independent experiments each performed in triplicates. Experimental values with **p < 0.01 were taken as significantly different as compared with their controls. One-way ANOVA determined statistical significance. *p < 0.05.
cancer cell lines in a dose-dependent manner. MCF-7 breast cancer cell line was the most sensitive to DCA-in- duced cell death. MC3T3 osteoblastic cell line was the least sensitive to DCA-induced cytotoxicity. MC3T3 cell line has been shown to behave like less aggressive or non-aggressive cell line whereas MCF-7 cell line is known to behave as aggressive cancer cell line. In order to compare the effects of DCA on aggressive and non-aggres- sive cancer cells, we selected MCF-7 and MC3T3 as aggressive and non-aggressive cell lines respectively for further studies.
Crystal violet (CV) assays and Trypan blue staining were used as additional criteria to determine DCA-in- duced cytotoxicity in these two selected cell lines. Similar to MTT assays, CV and Trypan blue staining showed that DCA caused more cytotoxicity in MCF-7 breast cancer than MC3T3 osteoblastic cell line (Figure 2). The differential effects of DCA on these two cell lines were determined in a dose dependent manner (25 and 200 mM DCA) for 48 hrs of treatment.
Using a median dose of 50 mM DCA, we have also determined the cytotoxicity of DCA on MCF-7 and MC3T3 cells in a time-dependent manner (Figure 3). Our cytotoxicity assays indicated that DCA caused more cytotoxicity in aggressive cancerous cell line such as MCF-7 as compared to non-aggressive or less aggressive cell line such as MC3T3.
3.2. Cytotoxicity of DCA Is Mediated by Apoptosis
Our goal was to determine whether DCA-induced cytotoxicity was mediated via its apoptotic effects or merely

Figure 2. Comparison of the cytotoxic effect of DCA on MCF-7 and MC3T3 cells. MC3T3 and MCF-7 cells were seeded at a density of 10 - 15 × 103 cells/well in 96-well plates or 10 × 104 cells/plates in 6-well plates as described in materials and methods. After 24 hrs, cells were treated with indicated concentrations of DCA. Cell culture continued for additional 48 hrs. Cell viability was determined by crystal violet (CV) assays (upper panel) or Trypan blue staining (lower panel). The values shown are mean ± SD from at least three independent experiments each performed in triplicates. Experimental values with **p < 0.01 were taken as significantly different as compared with their controls. One-way ANOVA determined statistical significance.

Figure 3. Time-dependent effect of DCA. MC3T3 and MCF-7 cells were seeded at a density of 10 - 15 × 103 cells/well in 96-well plates and grown in normal MEM and DMEM medium respectively with 10% FBS. 50 mM was added to induce apoptosis for 12, 24, 36, 48 and 60 hrs. The percentage of cells undergoing apoptosis was determined by MTT assays. Results shown are the mean values ± SD determined from triplicate samples of three independent experiments. **p < 0.01.
due to non-specific cell death by necrosis. We chose MC3T3 as non-aggressive cell line, which is more resistant to cytotoxicity by DCA, and MCF-7 as aggressive cell line, which is more sensitive to DCA-induced cell death. Acridine orange and ethidium bromide staining was used as a criterion to determine apoptotic cell death. This method is frequently used to study the induction of apoptosis. Characteristic morphological changes due to apoptosis were assessed by fluorescence microscopy using acridine orange and ethidium bromide staining at 25 mM and 100 mM DCA at 48 hrs of treatments. These concentrations were selected because they were close to the IC50 values for more sensitive MCF-7 and more resistant MC3T3 cell lines, respectively. The results showed a significantly higher amount of apoptotic cell death in MCF-7 cells than in MC3T3 cells (Figure 4) complementing our cytotoxicity data. Therefore, it is likely that the DCA-induced cytotoxicity observed earlier in our cytotoxicity assays could be at least partly related to the apoptotic cell death.
To confirm whether DCA-induced cell death was due to apoptosis, we performed flow cytometry analysis. This method distinguishes cells undergoing apoptosis and necrosis from the normal population. Figure 5 shows that at 25 mM DCA, both MCF-7 and MC3T3 cells had a significantly higher population of apoptotic cells as compared with their respective controls. However, necrosis increased with increasing DCA concentration. These results suggest that higher concentrations of DCA cause necrosis, while lower concentrations of DCA induce apoptosis. The overall cell death was, however, more in MCF-7 cells than in MC3T3 cells at all doses of DCA confirming our previous findings on cytotoxicity assays.
3.3. DCA Depolarizes Mitochondrial Membrane in Cancerous Cells
Since mitochondria are the primary target for DCA, we examined whether DCA affects mitochondrial function by measuring changes in its membrane potential. Mitochondrial membrane potential is known to be affected by its oxidative status, which is likely to be impaired in cancerous cells. The JC-1 dye staining to determine change in the ratio of red/green fluorescence is a well-established method to determining mitochondrial membrane integrity. The ratio of red/green fluorescence was decreased drastically in MCF-7 breast cancer aggressive cells following dose-dependent DCA treatment (Figure 6) indicating that DCA depolarizes mitochondrial membrane and decreased the membrane potential in cancerous cells. Such a decrease in mitochondrial membrane potential was not significant in MC3T3 non-aggressive osteoblastic cells.

Figure 4. Dose-dependent effect of DCA on apoptosis. MC- 3T3 and MCF-7 cells were seeded at density of 10 × 104 cells/well in six-well plates and grown in normal MEM and DMEM with 10% FBS. Indicated concentrations of DCA were used to induce apoptosis for 48 hours and then cells were stained with acridine orange/ethidium bromide and visualized under UV light using a fluorescence microscope. The percentage apoptosis was determined by counting live (green) and dead (red) cells. Higher doses of treatment lost dead cells during staining. Values in graph are mean ± SD from three experiments performed in triplicates. **p < 0.01.

Figure 5. Flow cytometry analysis. MC3T3 and MCF-7 cells were cultured as described in materials and methods. Cells were then treated with indicated doses of DCA for 48 hours to induce apoptosis. Apoptosis and necrosis was determined by flow cytometry using Vybrant apoptosis assay kit # 4 under optimized conditions. Shown in a quadrant are live cells (lower left), necrotic cells (upper left, upper right), and apoptotic cells (lower right). Flow cytometry results are from a representative experiment repeated three times with similar results.
3.4. DCA Produces Reactive Oxygen Species (ROS) in Cancerous Cells
Production of ROS is another criterion to evaluate oxidative cellular damage and thus cytotoxicity. We examined whether DCA-induced cytotoxicity was linked with ROS production and whether the level of ROS

Figure 6. Determination of mitochondrial membrane potential. MCF-7 and MC3T3 cells were grown as above. Cells were treated with DCA or PBS (control) for 48 hrs. During last 30 min incubation, cells were subjected to media containing JC-1dye (10 mg/ml) for 20 min at 37˚C. The mitochondrial potential was measured using a fluorescence microplate reader. Data are expressed as relative ratio of red to green fluorescence. The level of JC-1 retained by untreated cells was considered to be 100%. Exposure of MC3T3 and MCF-7 cells to DCA was associated with a significant reduction in Δψm compared to control in MCF-7 cells; data shown represent mean ± SD of three independent experiments. **p < 0.01.
production was different in non-aggressive MC3T3 and aggressive MCF-7 cancer cell lines. Our results suggested that DCA significantly increased ROS production in MCF-7 cells in a dose dependent manner (Figure 7). The production of ROS was not significant in MC3T3 non-aggressive cell line suggesting that DCA-induced cytotoxicity observed in aggressive breast cancer cells is, at least in part, mediated by cellular damage due to ROS production.
Manganese super oxide dismutase-1 (Mn-SOD-1), a marker for cytotoxic damage by oxygen free radicals, was also measured in response to DCA treatment. DCA induced the expression of Mn-SOD-1 in MCF-7 cells more than in MC3T3 cells (Figure 8). These results confirm our findings that DCA induces cytotoxicity by inducing oxidative stress in mitochondria due to production of enhanced amounts of ROS in aggressive cancer cells.

Figure 7. Determination of ROS generation. MCF-7 and MC3T3 cells were grown as above. Cells were exposed to different concentrations of DCA or PBS as indicated for 48 hrs. Cells were then incubated in the presence of 10 μM DCFH-DA for 30min for cell staining fluorescence microplate reader. Data shown represent mean ± SD of three independent experiments.

Figure 8. Effect of DCA-induced expression of Mn-SOD1. MC3T3 and MCF-7 Cells were cultured as described in materials and methods. Cells were treated with indicated concentrations of DCA for 48 hrs. 200 μM etoposide was used as a positive control for apoptosis induced expression. After these treatments, cell lysates were prepared in lysis buffer and subjected to SDS-PAGE followed by Western blot analysis, as detailed in materials and methods section. Membranes were probed by specific antibodies to SOD1 followed by horseradish peroxidase conjugated secondary antibody. Actin was used as an internal loading control to account for any variation in protein loading. The blots were visualized by chemiluminescence staining and autoradiography. Data are representative experiments for each antibody repeated multiple times with similar results.
4. Discussion
DCA is a small molecule, which has long been known to act on mitochondria as its primary subcellular target. In cancerous cells, it induces cell death and therefore considered as a molecule of interest in reducing cancer growth both in vitro and in vivo studies [2] [4] [6] [13] -[16] . These beneficial effects of DCA occur without affecting noncancerous cells or causing systemic toxicity. DCA treatment is known to significantly increase glucose oxidation that only takes place in functional mitochondria. Cancerous cells display enormously high amounts of cytosolic glycolysis perhaps due to defect in mitochondrial function. DCA treatment seems to correct this defect by restoring mitochondrial function.
One of the unique bioenergetics features in tumor cells is the observance of “Warburg effect” which has received a great deal of attention as a potential therapeutic target in cancer therapy [17] . Most cancer cells exhibit increased cytosolic glycolysis as a primary metabolic pathway for ATP production, despite the availability of oxygen source. This phenomenon is closely related with the case of apoptotic resistance in cancerous cells. The reversal of this process from cytosolic glycolysis to the mitochondrial oxidation may trigger apoptosis in cancerous cells [2] [5] [6] [18] [19] . The key regulator of glucose oxidation is pyruvate dehydrogenase (PDH), which is inhibited by pyruvate dehydrogenase kinase (PDK) in most tumor cells. Bonnet and co-workers, in 2007, found that DCA could significantly inhibit the PDK activity in tumor cells. DCA was later found to promote apoptosis in lung, breast and glioblastoma cancer cells [20] .
In this study, we have examined the effects of DCA on cell viability of human breast (MCF-7) cancer cell lines and compared with the effects on a non-aggressive MC3T3 cancer cell line. Our study demonstrated that not all cell lines were susceptible to DCA-induced cell death to the same degree of sensitivity. We have also confirmed that DCA induces apoptotic effects at least at lower doses, as previously reported by other researchers [6] [18] [21] . Our data demonstrated that the aggressive cancerous cells MCF-7, was more sensitive to DCA than less aggressive MC3T3. Our findings were also comparable with previous findings [18] , where it was shown that the stimulation of apoptosis or cell death and changes in mitochondrial function were more obvious in highly aggressive and metastatic cancer cells like LoVo than in the less aggressive HT29 and SW480 cells. Our results are in conformity with earlier findings where Wong and co-workers have demonstrated that DCA reduces endometrial cancer cell viability in a dose-dependent manner by inducing apoptosis while having no effect on non-cancerous cells.
MCF-7 cell line originally developed in the Michigan Cancer Foundation (from which it carries its name) in 1973 from a pleural effusion [22] is the most commonly used breast cancer cell line in in vitro research as an aggressive cancer cell line. It is aggressive cancer cell line as it is not derived from the primary breast tumor; it is rather derived from the tumor which was metastasized [23] . On the other hand, the MC3T3 cell line was originally isolated for varying degrees of osteogenic potential and has been widely used as a normal non-cancerous model cell line in bone biology. The MC3T3 cell line used in this study is a MC3T3-E1 sub-line, most frequently and conveniently used as physiologically relevant system for transcriptional control studies [24] . Although it is a non-human cell line, it displays features of a non-cancerous cell line and comparable to other less aggressive human colorectal cell line such as HCT116 used in this study. MC3T3 cell line has also been used as a non-aggressive cancer cell line by other investigators.
We showed that DCA produced differential responses in aggressive and less-aggressive cancer with IC50 value close to 25 mM in MCF-7 cells. This is in agreement with other published studies [18] [25] demonstrating the IC50 values for aggressive cancer cells ranging 20 - 30 mM DCA. The basis for differential effects of DCA on cancerous and non-cancerous cells may reside in its influence on mitochondrial function. DCA inhibits PDK and activates PDH. This leads to an enhanced pyruvate uptake and energy production from mitochondrial oxidative respiration and induction of apoptosis by intrinsic pathway involving caspase-3 and cytochrome c. Although these findings are comparable with other published studies [2] [6] [13] , our results vary in certain respect. In our study, DCA caused cytotoxicity at higher doses; the apoptosis was seen only at lower concentrations of DCA. Similar results are reported by other groups where apoptosis in cancer cells was seen at doses as low as 0.5 - 10 mM [2] [6] [19] . Interestingly, some researchers in their study on breast cancer cells found that DCA inhibited cell proliferation but the induction of apoptosis by DCA was not clear [16] . Therefore, we can safely state that although DCA causes cell death and inhibits growth of some cancer cells but the essential mechanism may be cell-type dependent. Higher doses of DCA cause non-specific cell death or necrosis.
Further to our investigation on the mechanism of action of DCA in inducing cytotoxicity, we looked at changes in mitochondrial membrane potential (Δψm). Treatment with DCA reduced the Δψm in aggressive MCF-7 cells but there was no significant effect on Δψm in less aggressive MC3T3 cells (Figure 6). This suggests that DCA promotes mitochondrial respiration in aggressive cancerous cells leading to the depolarization of mitochondrial membrane and induction of cell death by the proximal pathway of mitochondria as described in previous studies [2] [6] [13] . These results are also in agreement with other findings reported in the literature [18] where changes in mitochondrial function are evidently linked to the invasive nature of LoVo cells than the less invasiveness of HT29 and SW480 cells.
Our study has also provided a link that the cytotoxic effects of DCA are perhaps mediated due to the production of ROS. We have found that the intracellular ROS production was significantly increased in MCF-7 cells treated with DCA (Figure 7). It is well known that cancer cells largely depend on cytosolic glycolysis for their ATP production. Our results indicated that DCA might shift the glucose metabolism of MCF-7 cells back to oxidative phosphorylation. This would lead to an enhanced glucose oxidation by mitochondrial electron transport chain resulting in a sustained production of ROS, which, in turn, could inhibit the mitochondrial H+ efflux and ultimately decreased Δψm. These changes might also lead to the opening of mitochondrial membrane pores (MTP) by DCA and thus allowing the release of cytochrome c and induction of other apoptosis inducing factors with an ultimate result in enhanced cell death [20] .
Most early stage tumors occur in a microenvironment and depend heavily on anaerobic glycolysis for energy needs due to mitochondrial defects [5] [26] . Dysfunction of mitochondria are also a continuous source of ROS in tumor cells. In response to high levels of ROS, cancer cells express high levels of manganese dependent superoxide dismutase (Mn-SOD-1), which converts 2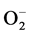 into H2O and O2 [27] . Mn-SOD-1 is an important enzyme responsible for the detoxification of
into H2O and O2 [27] . Mn-SOD-1 is an important enzyme responsible for the detoxification of
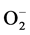 and is considered a key antioxidant in aerobic cells. Deficiency in this enzyme or inhibition of its activity may cause an accumulation of increased amounts of
and is considered a key antioxidant in aerobic cells. Deficiency in this enzyme or inhibition of its activity may cause an accumulation of increased amounts of
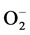 in the cells. This will result in the persistence of the oncogenic phenotype [28] . Consistent with this role of Mn-SOD1, our results showed a significant increase in the expression of Mn-SOD-1 in MCF-7 compared to MC3T3 cells (Figure 8); only high concentrations of DCA increase Mn-SOD-1 expression in MC3T3 cells. This might be due to that MC3T3 cells have less hypoxic environment. Similar to our results, Saed and coworkers [29] have found an increased expression of Mn-SOD-1 during treatment of epithelial ovarian cells (EOC) with DCA. However, in other studies using different cancer cells lines, it was reported that the levels of Mn-SOD-1 decreased. This decrease in Mn-SOD-1 was associated with an increase in apoptosis [30] -[32] . So any disturbance in levels of SOD would affect free radicals, and thus cytotoxicity. Whether ROS promote tumor cell survival or act as antitumorigenic agents depends on the cell and tissue type, the location of ROS production, and the concentration of individual ROS [33] .
in the cells. This will result in the persistence of the oncogenic phenotype [28] . Consistent with this role of Mn-SOD1, our results showed a significant increase in the expression of Mn-SOD-1 in MCF-7 compared to MC3T3 cells (Figure 8); only high concentrations of DCA increase Mn-SOD-1 expression in MC3T3 cells. This might be due to that MC3T3 cells have less hypoxic environment. Similar to our results, Saed and coworkers [29] have found an increased expression of Mn-SOD-1 during treatment of epithelial ovarian cells (EOC) with DCA. However, in other studies using different cancer cells lines, it was reported that the levels of Mn-SOD-1 decreased. This decrease in Mn-SOD-1 was associated with an increase in apoptosis [30] -[32] . So any disturbance in levels of SOD would affect free radicals, and thus cytotoxicity. Whether ROS promote tumor cell survival or act as antitumorigenic agents depends on the cell and tissue type, the location of ROS production, and the concentration of individual ROS [33] .
It has been shown that DCA increases PUMA transcripts in endometrial carcinoma cell lines with an apoptotic response suggesting a p53-PUMA-mediated mechanism may be involved in DCA-induced apoptosis [6] . In our studies, we found that DCA increased Akt activation in MCF-7 but not in MC3T3 cells (results not shown). Previous studies [34] showed that DCA when administered on mice reduced the expression of PKB/Akt in liver; though they did not measure the levels of p-Akt. Therefore, it may be possible that alternative pathways are operative in DCA-induced apoptosis [35] [36] .
5. Conclusion
DCA induces more cytotoxicity in aggressive cancerous cells than in non-aggressive cancerous cells. This was established by MTT, crystal violet, Trypan blue and acridine orange/ethidium bromide staining. Flow cytometry was used to confirm and differentiate apoptotic vs. necrotic effects of DCA. At lower concentrations, DCA brings about apoptotic changes but at higher concentrations, most cytotoxic effects of DCA are related to necrosis. Most prominent effects of DCA were seen on changing mitochondrial membrane potential (Δψm) and the production of reactive oxygen species (ROS). The increased expression of Mn-SOD1 is likely a consequence of the increased production of ROS. Taken together, our results suggest that DCA causes significantly higher cytotoxicity in aggressive cancerous cells than in non-aggressive cancerous cells and these effects are primarily mediated by its action on mitochondrial membrane potential and the production of ROS.
Acknowledgements
This study was partly supported from Kathleen Thomsen Hall Charitable Foundation, NASA-EPSCoR, NSF- EPSCoR P3 and Arkansas Space Grant Consortium to Nawab Ali. Zeiyad Alkarakooly is thankful to Iraqi Cultural Office for financial support.
References
- Wu, W. and Zhao, S. (2013) Metabolic Changes in Cancer: Beyond the Warburg Effect. Acta Biochimica et Biophysica Sinica (Shanghai), 45, 18-26. http://dx.doi.org/10.1093/abbs/gms104
- Bonnet, S., Archer, S., Alalunis-Turner, J., Haromy, A., Beaulieu, C., Thompson, R., et al. (2007) A Mitochondria-K Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell, 11, 37-51. http://dx.doi.org/10.1016/j.ccr.2006.10.020
- Heshe, D., Hoogestraat, S., Brauckmann, C., Karst, U., Boos, J. and Lanvers-Kaminsky, C. (2011) Dichloroacetate Metabolically Targeted Therapy Defeats Cytotoxicity of Standard Anticancer Drugs. Cancer Chemotherapy and Pharmacology, 67, 647-655. http://dx.doi.org/10.1007/s00280-010-1361-6
- Papandreou, I., Goliasova, T. and Denko, N.C. (2011) Anticancer Drugs That Target Metabolism: Is Dichloroacetate the New Paradigm? International Journal of Cancer, 128, 1001-1008. http://dx.doi.org/10.1002/ijc.25728
- Michelakis, E., Webster, L. and Mackey, J. (2008) Dichloroacetate (DCA) as a Potential Metabolic-Targeting Therapy for Cancer. British Journal of Cancer, 99, 989-994. http://dx.doi.org/10.1038/sj.bjc.6604554
- Wong, J.Y., Huggins, G.S., Debidda, M., Munshi, N.C. and De Vivo, I. (2008) Dichloroacetate Induces Apoptosis in Endometrial Cancer Cells. Gynecologic Oncology, 109, 394-402. http://dx.doi.org/10.1016/j.ygyno.2008.01.038
- Stacpoole, P.W., Gilbert, L.R., Neiberger, R.E., Carney, P.R., Valenstein, E., Theriaque, D.W. and Shuster, J.J. (2008) Evaluation of Long-Term Treatment of Children with Congenital Lactic Acidosis with Dichloroacetate. Pediatrics, 121, e1223-e1228. http://dx.doi.org/10.1542/peds.2007-2062
- Mosmann, T. (1983) Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. Journal of Immunological Methods, 65, 55-63. http://dx.doi.org/10.1016/0022-1759(83)90303-4
- Flick, D.A. and Gifford, G.E. (1984) Comparison of in Vitro Cell Cytotoxic Assays for Tumor Necrosis Factor. Journal of Immunological Methods, 68, 167-175. http://dx.doi.org/10.1016/0022-1759(84)90147-9
- Isakovic, A., Markovic, Z., Todorovic-Markovic, B., Nikolic, N., Vranjes-Djuric, S., Mirkovic, M., Dramicanin, M., Harhaji, L., Raicevic, N., Nikolic, Z. and Trajkovic, V. (2006) Distinct Cytotoxic Mechanisms of Pristine versus Hydroxylated Fullerene. Toxicological Sciences, 91, 173-183. http://dx.doi.org/10.1093/toxsci/kfj127
- Krmpot, A.J., Janjetovic, K.D., Misirkic, M.S., Vucicevic, L.M., Pantelic, D.V., Vasiljevic, D.M., Popadic, D.M., Jelenkovic, B.M. and Trajkovic, V.S. (2010) Protective Effect of Autophagy in Laser-Induced Glioma Cell Death in Vitro. Lasers in Surgery and Medicine, 42, 338-347. http://dx.doi.org/10.1002/lsm.20911
- Agarwal, R., Mumtaz, H. and Ali, N. (2009) Role of Inositol Polyphosphates in Programmed Cell Death. Molecular and Cellular Biochemistry, 328, 155-165. http://dx.doi.org/10.1007/s11010-009-0085-6
- Cao, W., Yacoub, S., Shiverick, K.T., Namiki, K., Sakai, Y., Porvasnik, S., Urbanek, C. and Rosser, C.J. (2008) Dichloroacetate (DCA) Sensitizes Both Wild-Type and over Expressing Bcl-2 Prostate Cancer Cells in Vitro to Radiation. Prostate, 68, 1223-1231. http://dx.doi.org/10.1002/pros.20788
- Chen, Y., Cairns, R., Papandreou, I., Koong, A. and Denko, N.C. (2009) Oxygen Consumption Can Regulate the Growth of Tumors, a New Perspective on the Warburg Effect. PLoS ONE, 4, e7033. http://dx.doi.org/10.1371/journal.pone.0007033
- Michelakis, E.D., Sutendra, G., Dromparis, P., Webster, L., Haromy, A., Niven, E., Maguire, C., Gammer, T.L., Mackey, J.R., Fulton, D., Abdulkarim, B., McMurtry, M.S. and Petruk, K.C. (2010) Metabolic Modulation of Glioblastoma with Dichloroacetate. Science Translational Medicine, 2, 31ra34. http://dx.doi.org/10.1126/scitranslmed.3000677
- Sun, R.C., Fadia, M., Dahlstrom, J.E., Parish, C.R., Board, P.G. and Blackburn, A.C. (2010) Reversal of the Glycolytic Phenotype by Dichloroacetate Inhibits Metastatic Breast Cancer Cell Growth in Vitro and in Vivo. Breast Cancer Res Treat, 120, 253-260. http://dx.doi.org/10.1007/s10549-009-0435-9
- Hsu, P.P. and Sabatini, D.M. (2008) Cancer Cell Metabolism: Warburg and Beyond. Cell, 134, 703-707. http://dx.doi.org/10.1016/j.cell.2008.08.021
- Madhok, B., Yeluri, S., Perry, S., Hughes, T. and Jayne, D. (2010) Dichloroacetate Induces Apoptosis and Cell-Cycle Arrest in Colorectal Cancer Cells. British Journal of Cancer, 102, 1746-1752. http://dx.doi.org/10.1038/sj.bjc.6605701
- Sun, R.C., Board, P.G. and Blackburn, A.C. (2011) Targeting Metabolism with Arsenic Trioxide and Dichloroacetate in Breast Cancer Cells. Molecular Cancer, 10, 142. http://dx.doi.org/10.1186/1476-4598-10-142
- Xie, J., Wang, B., Yu, D., Lu, Q., Ma, J., Qi, H., Fang, C. and Chen, H. (2011) Dichloroacetate Shifts the Metabolism from Glycolysis to Glucose Oxidation and Exhibits Synergistic Growth Inhibition with Cisplatin in Hela Cells. International Journal of Oncology, 38, 409-417.
- Shahrzad, S., Lacombe, K., Adamcic, U., Minhas, K. and Coomber, B.L. (2010) Sodium Dichloroacetate (DCA) Reduces Apoptosis in Colorectal Tumor Hypoxia. Cancer Letters, 297, 75-83. http://dx.doi.org/10.1016/j.canlet.2010.04.027
- Soule, H.D., Vazguez, J., Long, A., Albert, S. and Brennan, M. (1973) A Human Cell Line from a Pleural Effusion Derived from a Breast Carcinoma. Journal of the National Cancer Institute, 51, 1409-1416.
- Burdall, S.E., Hanby, A.M., Lansdown, M. and Speirs, V. (2003) Breast Cancer Cell Lines: Friend or Foe? Breast Cancer Research, 5, 89-95. http://dx.doi.org/10.1186/bcr577
- Bilezikian, J.P., Raisz, L.G. and Martin, T.J. (2008) Principles of Bone Biology: Two-Volume Set. Academic Press, Waltham.
- Ko, L. and Allalunis-Turner, J. (2009) Investigation on the Mechanism of Dichloroacetate (DCA) Induced Apoptosis in Breast Cancer. Journal of Clinical Oncology, 27, e14637.
- Gatenby, R.A. and Gillies, R.J. (2004) Why Do Cancers Have High Aerobic Glycolysis? Nature Reviews Cancer, 4, 891-899. http://dx.doi.org/10.1038/nrc1478
- Kinnula, V.L. and Crapo, J.D. (2004) Superoxide Dismutases in Malignant Cells and Human Tumors. Free Radical Biology and Medicine, 36, 718-744. http://dx.doi.org/10.1016/j.freeradbiomed.2003.12.010
- Tandon, V.R., Sharma, S., Mahajan, A. and Bardi, G.H. (2005) Oxidative Stress: A Novel Strategy in Cancer Treatment. JK Science, 7, 1-3.
- Saed, G.M., Fletcher, N.M., Jiang, Z.L., Abu-Soud, H.M. and Diamond, M.P. (2011) Dichloroacetate Induces Apoptosis of Epithelial Ovarian Cancer Cells through a Mechanism Involving Modulation of Oxidative Stress. Reproductive Sciences, 18, 1253-1261. http://dx.doi.org/10.1177/1933719111411731
- Hileman, E.O., Liu, J., Albitar, M., Keating, M.J. and Huang, P. (2004) Intrinsic Oxidative Stress in Cancer Cells: A Biochemical Basis for Therapeutic Selectivity. Cancer Chemotherapy and Pharmacology, 53, 209-219. http://dx.doi.org/10.1007/s00280-003-0726-5
- Bhosle, S., Pandey, B., Huilgol, N. and Mishra, K. (2003) Membrane Oxidative Damage and Apoptosis in Cervical Carcinoma Cells of Patients after Radiation Therapy. In: Advanced Flow Cytometry: Applications in Biological Research, Springer, Berlin, 65-68.
- Huang, P., Feng, L., Oldham, E.A., Keating, M.J. and Plunkett, W. (2000) Superoxide Dismutase as a Target for the Selective Killing of Cancer Cells. Nature, 407, 390-395. http://dx.doi.org/10.1038/35030140
- Reuter, S., Gupta, S.C., Chaturvedi, M.M. and Aggarwal, B.B. (2010) Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radical Biology and Medicine, 49, 1603-1616. http://dx.doi.org/10.1016/j.freeradbiomed.2010.09.006
- Lingohr, M.K., Thrall, B.D. and Bull, R.J. (2001) Effects of Dichloroacetate (DCA) on Serum Insulin Levels and Insulin-Controlled Signaling Proteins in Livers of Male B6C3F1 Mice. The Journal of Toxicological Sciences, 59, 178-184. http://dx.doi.org/10.1093/toxsci/59.1.178
- Bates, R.C., Edwards, N.S., Burns, G.F. and Fisher, D.E. (2001) A CD44 Survival Pathway Triggers Chemoresistance via Lyn Kinase and Phosphoinositide 3-Kinase/Akt in Colon Carcinoma Cells. Cancer Research, 61, 5275-5283.
- Pelicano, H., Xu, R.H., Du, M., Feng, L., Sasaki, R., Carew, J.S., Hu, Y., Ramdas, L., Hu, L., Keating, M.J., Zhang, W., Plunkett, W. and Huang, P. (2006) Mitochondrial Respiration Defects in Cancer Cells Cause Activation of Akt Survival Pathway through a Redox-Mediated Mechanism. The Journal of Cell Biology, 175, 913-923. http://dx.doi.org/10.1083/jcb.200512100
NOTES

*Corresponding author.