Journal of Cancer Therapy
Vol.5 No.3(2014), Article ID:44116,9 pages DOI:10.4236/jct.2014.53036
Want to Cure Cancer? Then Revisit the Past; “Warburg Was Correct”, Cancer Is a Metabolic Disease
Robert L. Elliott, Xian P. Jiang, Jonathan F. Head
Elliott-Baucom-Head Breast Cancer Research and Treatment Center, Baton Rouge, USA
Email: akucharchuk@eehbreastca.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 20 February 2014; revised 15 March 2014; accepted 21 March 2014
ABSTRACT
I want to make it very clear at the beginning of this communication; this is a controversial opinion review. However, I believe it is time to rethink our approach to cancer research and therapy. Many cancer researchers, especially those involved in cancer genomic research will disagree. I welcome the disagreement and hope it will stimulate an honest debate and dialog between all disciplines of cancer research and treatment. I am convinced that a vast disconnection exists between those involved in basic research and those in the clinical arena that treat this disease. Cancer researchers in all areas should not ignore the role of cancer metabolism in tumorigenesis, progression and metastasis.
Keywords:Host Immunity; Mitochondrial Dysfunction; Warburg Effect; Aerobic Fermentation; Tumor and Mitochondrial Iron Metabolism

1. Introduction (Why This Review?)
After being involved in cancer research and treating breast cancer patients for over 40 years, I believe that we seriously need to reconsider how we approach and treat the disease. We need to revisit the past and the marvelous work of Otto Warburg. After reviewing the past, I suggest you fast forward to now and study the tremendous work of Thomas Seyfried. He and Shelton have published a great paper on cancer as a metabolic disease [1] , and recently Seyfried had his book on cancer as a Metabolic Disease released by Wiley [2] . The reason we must rethink our approach is because we have not significantly improved the survival of Stage IV cancer patients since President Richard M. Nixon declared war on cancer over 40 years ago.
Cancer is an extremely complex disease, similar but different in each host. There is no solid tumor that is totally homogeneous in regard to tumor biology or genomic mutations. In fact, I believe that we have made a complex disease more complicated by the emphasis on the vast number of genetic mutations discovered in tumors. Though these genomic discoveries are interesting and will assist in some personalized specific therapies, it will in my opinion not be the final answer. Therefore, in order to cure cancer, we need to know exactly what is common to all tumors; not how many genetic mutations are present. Mutations can be numerous and even different from cell to cell in the same tumor. Attacking the predominant mutation may eliminate those cells but allow cells to lack that mutation to remain, grow, metastasize and ultimately kill the patient.
Another important factor ignored in clinical oncology is host immunity. Oncologists treat the disease (tumor biology, mutations, stage) and ignore the immunity of the host harboring that disease. Oncologist must be more aware of the importance of host immunity in cancer. The routine cancer therapy is the “Big Three”: 1) surgery, 2) chemotherapy and 3) radiation—all three of which damage and depress host specific immunity. However, with proper specific individual immunotherapy the “Big Three” can actually be partners in supporting host immunity. Regardless of the type of cancer treatment, host immunity cannot be ignored [3] .
There is now definitely emerging evidence that the main characteristic of nearly all cancers is impaired cellular energy metabolism regardless of cellular or tissue of origin. Normal cells derive their useable energy from oxidative phosphorylation, while most cancer cells become dependent on substrate level phosphorylation for energy demands.
In the 1930s, Otto Warburg described a link between mitochondrial dysfunction and tumorigenesis. He observed a significant increase in glycolysis and lactate production in the presence of oxygen without an increase and an occasional decrease in oxidative phosphorylation [4] [5] . This became known as aerobic fermentation or the “Warburg Effect” and is well documented in tumor cells. Aerobic fermentation causes a marked increase in glucose uptake by cancer cells with an increased lactic acid production. This “Warburg Effect” is the basis for positron emission tomography (PET SCAN) which uses a radioactive labeled glucose analog for tumor imaging.
Hanahan and Weinberg described six essential alterations in cell physiology that might underlie malignant cell growth [6] . These six alterations were described as the hallmarks of cancer. These six alterations were: 1) self-sufficiency in growth signals, 2) insensitivity to inhibitory growth signals, 3) evasion of apoptosis, 4) limitless replicative potential, 5) sustained vascularity (angiogenesis), and 6) tissue invasion and metastasis. Seyfried, others and I believe that in addition to these six recognized hallmarks of cancer, the “Warburg Effect” or aerobic fermentation is also a very robust metabolic hallmark of most tumors [7] -[12] .
Another abnormal metabolic defect common to all cancers is dysfunction in tumor cellular iron metabolism. I discussed this in detail in a recent article [12] and will address it again later in this communication. I am convinced that mitochondrial dysfunction (impaired cellular respiration) leading to aerobic fermentation is also related to tumor iron metabolism and stabilization of the transcription factor Hypoxia Inducible factor-1alpha (HIF1-a), which plays a major role in both.
2. Warburg Effect in Tumor Cells
This review is to emphasize the point that cancer is a metabolic disease rather than the now popular belief that it is genetic. For those readers interested in more science and detail on the subject, I highly recommend Seyfried’s book “Cancer as a Metabolic Disease (on the origin, management, and prevention of cancer)” [2] . I will attempt to discuss and summarize some of his very important points supporting cancer as a metabolic disease. However, I will not get into the hard science and mechanisms of the abnormal metabolic defects. It is stated that there are no specific gene mutations or chromosomal abnormality common to all cancers [13] -[15] , but nearly all cancers regardless of their cellular or tissue of origin express aerobic fermentation. This means that cancer cells have increased glucose uptake with increased lactic acid production in the presence of oxygen. The majority of cancers overexpress the genes for glycolysis [7] -[18] .
There is no doubt that the origin of the Warburg effect in cancer cells is controversial. Warburg himself purposed that aerobic fermentation was a manifestation of a problem in cancer cell physiology, which was impaired cellular respiration [5] [19] . Seyfried [1] points out that only those body cells able to increase glycolysis during intermittent respiratory damage are capable of promoting tumorigenesis. The cells unable to induce glycolysis in response to respiratory damage will perish due to energy failure. Therefore, a most common phenotype found in cancer is aerobic fermentation, arising from damaged respiration.
After reviewing the metabolic data collected from many human and animal samples, Warburg postulated with confident certainty that irreversible respiratory damage was the main cause of cancer [5] [19] [20] . However, the scientific community attacked Warburg’s theory as too simplistic and not explaining the evidence of apparent normal respiration in some cancer cells [21] -[29] . They stated that his theory did not address the role of tumorassociated mutations or the progression of metastasis. They complained that it also did not link the molecular mechanisms of uninhibited cell growth directly to damaged respiration. Therefore, the concept of cancer as a metabolic disease was replaced with the view that cancer was a genetic disease. The concept that cancer is a genetic disease has dominated cancer research for the past several decades. The genomic researchers though aware of the metabolic defects felt these defects in cancer cells arose primarily from genomic mutability during tumor progression [30] -[33] . There is now much emerging evidence that questions the genetic origin of cancer and supports data that cancer is a metabolic disease. Our recent paper on transplantation of isolated normal mitochondria to cancer cells support the evidence that cancer is probably a metabolic disease caused by mitochondrial dysfunction [34] . This study demonstrated that isolated normal mammary mitochondria easily enter breast cancer cells. This organelle transplantation into cancer cells inhibits proliferation, increases drug sensitivity and decreases the expression of glucose transporter III (Glut III) (Figures 1-4).
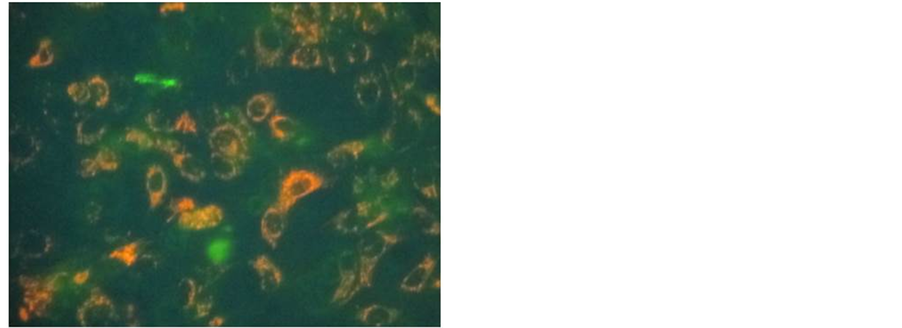
Figure 1. Fluorescent micrograph of MCF-7 cells co-cultured with isolated JC-1 stained MCF-12A mitochondria.
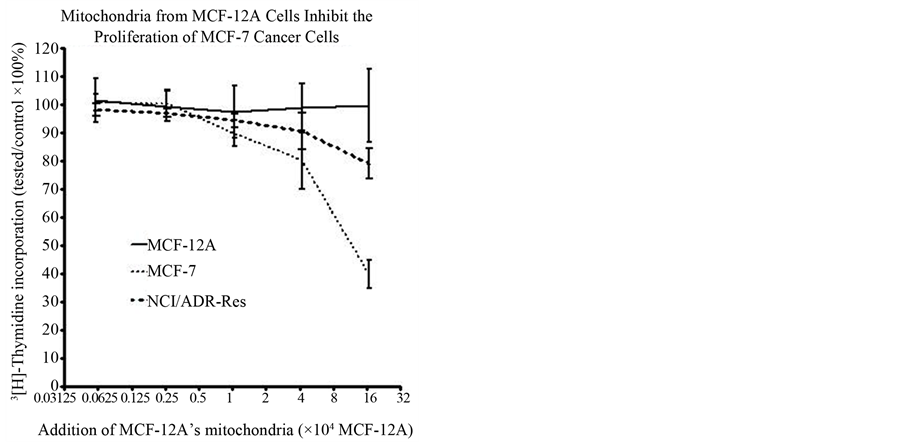
Figure 2. Graph of how the mitochondria of MCF- 12A inhibit the proliferation when added to the cancer cell lines MCF-7 and NCI/ADR-Res.
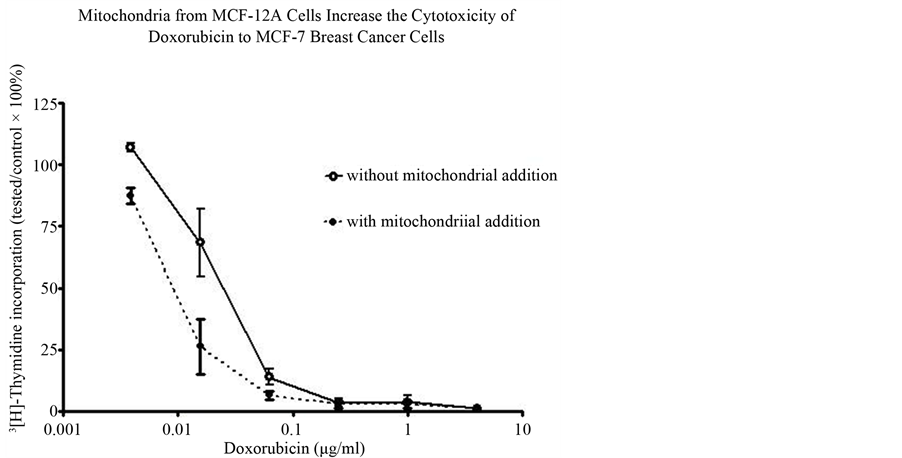
Figure 3. Graph depicting the addition of MCF-12A mitochondria to the MCF-7 breast cancer cell line and the resulting increase in the cytotoxicity of doxorubicin.
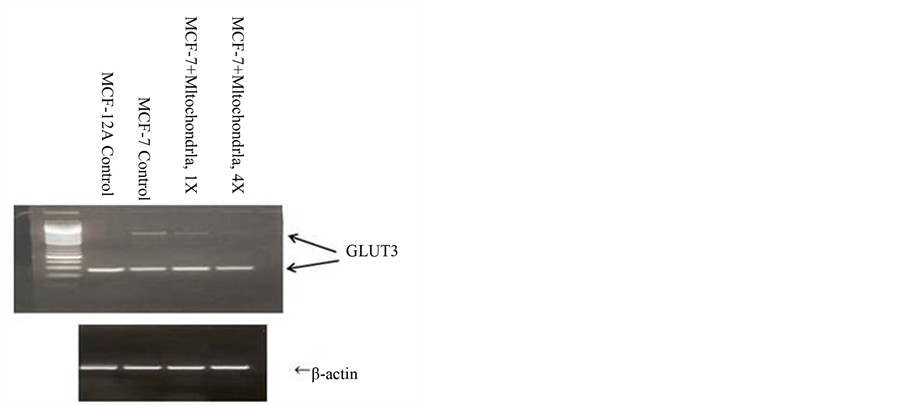
Figure 4. Effect of exogenous mitochondria of MCF- 12A cells on Mrna expression of glucose transporter 3 in breast cancer MCF-7 cells (RT-PCR).
A recent paper by Sebastian, Zwaans, Silberman and Gymrek et al. supports the concept that cancer is a metabolic disease [35] . They present strong evidence that the Histone Deacetylase SIRT6 is a tumor suppressor that controls metabolism. SIRT6 deficient cells are tumorigenic and SIRT6 immortalized knockout (KO) mouse embryonic fibroblast (MEFs) showed increased glucose uptake and lactate production (aerobic glycolysis). They performed experiments in SIRT6 (KO) and wild type (WT) (MEFs) and confirmed that tumorigenesis in SIRT6 (KO) cells is oncogene independent. Pyruvate dehyrogenase kinase (PDK)-1 and lactate dehydrogenase (LDH)- A protein levels are upregulated in SIRT6 (KO) cells showing that these cells are highly glycolytic. They concluded that the enhanced glycolysis rather than oncogene activation was probably the driving force for tumorigenesis in SIRT6 deficient cells. Their work is just more evidence that genomic instability is downstream of the metabolic changes in cancer cells, such as, aerobic fermentation (the glycolytic phenotype) [35] .
Seyfried’s goal is to resolve the conflict and provide evidence that genomic mutability and the hallmarks of cancer, including the Warburg effect (aerobic fermentation) can be linked to energy metabolism and damaged respiration. He believes that impaired cellular respiration precedes the genome instability that is associated with tumorigenesis. The genome instability that is associated with tumorigenesis is downstream of impaired cellular respiration and mitochondrial dysfunction. The genome instability contributes to further respiratory damage, increased genome mutability and tumor progression (effects become causes). Seyfried bases his hypothesis on evidence that nuclear genome integrity is mostly dependent on homeostasis of mitochondrial energy and that all cells require a continuous level of useable energy to maintain viability. I believe Warburg was definitely correct, though he did not link his effect to what we now recognize as the hallmarks of cancer. Seyfried does a great job reviewing the evidence and making the connection and expanding Warburg’s ideas of how impaired energy metabolism might be exploited for tumor therapy and prevention. I believe that Warburg and Seyfried are correct, and that Cancer is a Metabolic Disease. ARE YOU HERE? IF NOT WAKE UP!
3. Tumor and Mitochondrial Iron Metabolism
Now I am going to discuss briefly tumor and mitochondrial iron metabolism. I believe that some type of defect in iron sulfur (FeS) cluster biosynthesis in the mitochondria might be the defect that initiates mitochondrial respiratory damage and abnormal energy production. In other words, it might be the promoter of aerobic fermentation. We have been working on tumor iron metabolism for over 30 years. Hopefully, this discussion of tumor and mitochondrial iron metabolism will stimulate great researchers like Seyfried to attempt to link it to aerobic fermentation and cancer metabolism.
4. Tumor Iron Metabolism
Abnormal iron metabolism could be the main culprit in tumorigenesis as it is involved in tumor metabolism, mitochondrial dysfunction and tumor immuno-suppression [12] . Iron (Fe) is an essential metal vital for living cells [36] -[38] . It is required by heme and—heme enzymes and proteins, which are essential for oxygen transport and oxidative phosphorylation [39] . Iron is a cofactor for ribonucleotide reductase, an enzyme that converts ribonucleotides to deoxyribonucelotides, and thus a key enzyme in DNA synthesis. This requires a continuous supply of iron for ribonucleotide reductase to maintain its activity [40] [41] . Therefore, iron is directly associated with cell proliferation.
Transferrin (TF), a bilobed glycoprotein is the chief iron transport protein in mammalian blood. It transports iron from sites of absorption and storage to sites of iron utilization [42] . Cancer cells markedly overexpress on their cell surface transferrin receptors. Iron bound (TF) specifically interacts with the cell surface transferrin receptor (TFR1) that promotes the transport of iron across the cell membrane by endocytosis. The entire (TF-TFR1) complex is internalized and the pH within the endosome decreases due to a proton pump in the endosomal membrane. The acidic environment allows ferric (Fe3+) atoms to release from the complex [43] . The ferric is reduced to the ferrous state by a ferrireductase and then transported out of the endosome into the cytoplasm by the divalent metal transporter (DMT1). The (apo-TF-TFR1) complex is then recycled to the cell surface and apotransferrin is released into the bloodstream [44] .
In 1993, we reported on the role of iron metabolism in breast carcinoma [45] . This was an extensive cytochemical, tissue culture, and ultrastructural study. It demonstrated that the iron storage protein ferritin was increased and located in the cytoplasm. It also confirmed that transferrin could be used as a carrier to target toxic therapy selectively to tumor tissue. We had previously reported preliminary evaluation of a platinum transferrin complex (MPTC-63) as a potential nontoxic therapy for breast cancer [46] . It is undeniable that iron plays a significant role in the proliferation, growth and progression of cancer. Mitochondrial iron metabolism is the cradle of all iron metabolism and now we will briefly address mitochondrial iron metabolism.
5. Mitochondrial Iron Metabolism
Mitochondria are essential for iron metabolism and a site for iron sulfur (FeS) cluster biosynthesis and the only site of heme synthesis. Richardson, Lane and Becker et al, have done a great job reporting on mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and the cytosol [47] . It is somewhere in the integration of iron metabolism between the mitochondrion and the cytosol that I believe a defect takes place that initiates respiratory damage contributing to aerobic fermentation and tumorigenesis. Why might I think that; because the mitochondrion is well known for its key role in energy production, electron transport, oxygen transport, deoxynucleotide synthesis, reactive oxygen species, and apoptosis. However, little is appreciated that it is a focal point of iron metabolism. There is very little known about the regulation of iron uptake by the mitochondrion and how this is merged with iron metabolism in other organelles and the cytosol. The discovery of proteins involved in mitochondrial iron transport (mitoferrin 1 and 2) and storage (mitochondrial ferritin) has revealed a definite communication between iron metabolism in the mitochondrion and the cytosol [47] . I believe that any minor defect in the communication of iron between the mitochondrion and the cytosol, mitochondrial iron import and iron metabolism could lead to mitochondrial respiratory damage and genomic instability and thus contribute to tumorigenesis.
A great paper by Veatch, McMurray, Nelson and Gottschling support this concept [48] . They showed that mitochondrial dysfunction leads to nuclear genome instability via an iron an iron-sulphur (FeS) cluster defect. The loss of mtDNA leads to nuclear genome instability which causes a cellular crisis. The crisis is not mediated by absence of respiration, but correlates with a reduction in the mitochondrial membrane potential. They identified a defect in (FeS) cluster biogenesis in cells undergoing this crisis. Therefore, genomic instability (mutations) arises as a downstream epiphenomenon of disturbed iron metabolism in mitochondria. Their results suggest mitochondrial dysfunction stimulates nuclear genome instability by inhibiting the production of (FeS) cluster— containing proteins, which are required for maintenance of nuclear genome integrity [48] .
A possible problem is a probable defect involving the protein Frataxin, which is a vital protein highly expressed in tissues rich in mitochondria. Frataxin is an inner mitochondrial membrane and mitochondrial matrix protein that is involved in (FeS) cluster and heme biogenesis as well as iron storage [47] . Mitochondrial iron metabolism is extremely complicated and there is much more to learn from future important needed research.
6. Conclusions
The first point to make in this conclusion is that I highly recommend that the readers study the marvelous work of Seyfried [1] [2] especially his book on cancer metabolism published by Wiley. For non-believers it will be difficult to refute. The evidence supports a general hypothesis that cancer is a disease of energy metabolism. Seyfried and I believe that all of the hallmarks of cancer can be linked to impaired mitochondrial function. To maintain viability, cancer cells transition to substrate level phosphorylation uses glucose and glutamine as energy substrates.
The road traveled on this review has convinced me that Warburg was remarkably correct about the role of mitochondrial dysfunction in malignancy. The evidence is accumulating every day, thanks to the work of researchers like Seyfried. We hope to soon contribute more evidence by the work we are doing on mitochondrial organelle transplantation of isolated normal mammary mitochondria into breast cancer cell lines [34] . We are presently designing an in vivo model.
The future discoveries will only make even more remarkable the contributions of Warburg especially in the era he worked. This communication is a humble tribute to his great research and observations. I honor him and now Seyfried for their tremendous contributions. Hopefully, this review will stimulate more debate and work in this area to improve therapy for this dreaded disease. However, I believe that all systemic therapy for cancer should be complemented with support of the host immune system, and an adjunctive metabolic approach. All large cancer centers should have a metabolic unit. The next few years will breed more controversy, heated debate and interesting results; but I believe that cancer as a metabolic disease will win the war.
References
- Seyfried, T.N. and Shelton, L.M. (2010) Cancer as a Metabolic Disease. Nutrition and Metabolism, 7, 1-22. http://dx.doi.org/10.1186/1743-7075-7-7
- Seyfried, T.N. (2012) Cancer as a Metabolic Disease (On the Origin, Management, and Prevention of Cancer). Wiley.
- Elliott, R.L. and Head, J.F. (2005) Host Immunity Ignored in Clinical Oncology: A Medical Opinion. Cancer Biotherapy and Radiopharmaceuticals, 20, 199-121. http://dx.doi.org/10.1089/cbr.2005.20.123
- Warburg, O., Wind, F. and Negleis, E. (1930) On the Metabolism of Tumors in the Body. In: Warburg, O., Ed., The Metabolism of Tumors, Constable, Princeton, 254-270.
- Warburg, O. (1956) On the Origin of Cancer Cells. Science, 123, 309-314. http://dx.doi.org/10.1126/science.123.3191.309
- Hanahan, D. and Weinberg, R.A. (2000) The Hallmarks of Cancer. Cell, 100, 57-70. http://dx.doi.org/10.1016/S0092-8674(00)81683-9
- Seyfried, T.N. and Muckheryee, P. (2005) Targeting Energy Metabolism in Brain Cancer, Review and Hypothesis. Nutrition & Metabolism, 2, 30. http://dx.doi.org/10.1186/1743-7075-2-30
- Semenza, G.L., Cartemor, D., Bede, A., Bhujwolla, Z., Chiles, K., Feldser, O., Laughner, E., Pavi, R., Simons, J., Taghavi, P. and Thong, H. (2001) The Metabolism of Tumors: 70 Years Later. The Tumour Microenvironment: Causes and Consequences of Hypoxia and Acidity: Novartis Foundation Symposium 240, 251-260. http://dx.doi.org/10.1002/0470868716.ch17
- Ristow, M. (2006) Oxidative Metabolism in Cancer Growth. Current Opinion in Clinical Nutrition and Metabolic Care, 9, 339-345. http://dx.doi.org/10.1097/01.mco.0000232892.43921.98
- Gatenby, R.A. and Gillies, F.J. (2004) Why Do Cancers Have High Aerobic Glycolysis. Nature Reviews Cancer, 4, 891-899. http://dx.doi.org/10.1038/nrc1478
- Gogvadze, V., Orrenius, S. and Zhivotorsky, B. (2008) Mitochondria in Cancer Cells: What Is So Special about Them? Trends in Cell Biology, 18, 165-173. http://dx.doi.org/10.1016/j.tcb.2008.01.006
- Elliott, R.L. and Head, J.F. (2012) Cancer: Tumor from Metabolism, Mitochondrial Dysfunction and Tumor Immunog Suppression; “A Tight Partnership—Was Warburg Correct?” Journal of Cancer Therapy, 3, 278-311. http://dx.doi.org/10.4236/jct.2012.34039
- Loeb, L.A. (2001) A Mutator Phenotype in Cancer. Cancer Research, 61, 3230-3239.
- Longauer, C., Kingler, K.W. and Vogelstein, B. (1998) Genetic Instabilities in Human Cancers. Nature, 396, 643-649. http://dx.doi.org/10.1038/25292
- Wokolorczyk, D., Gliniewicz, B., Sikorski, A., Zlowocka, E., Masojc, B., Defniak, T., Matyjasik, J., Miergejewski, M., Medrek, K., Oszutowska, D., Suchy, J., Gronwald, J., Teodrczyk, U., Juzarski, T., Byrski, T., Jakuborwska, A., Gorski, B., Van de Wetering, T., Walczak, S., Nurod, S.A., Lubrinski, J. and Cybulski, C. (2008) A Range of Cancers Is Associated with the rs 6983267 Marker on Chromosome 8. Cancer Research, 68, 9982-9986. http://dx.doi.org/10.1158/0008-5472.CAN-08-1838
- Nowell, P.C. (2002) Tumor Progression: A Brief Historical Perspective. Seminars in Cancer Biology, 12, 261-266. http://dx.doi.org/10.1016/S1044-579X(02)00012-3
- Ortega, A.D., Sanchez-Arago, M., Siner-Sanchez, D., Sanchez-Cenizo, L., Willers, I. and Cuezva, J.M. (2009) Glucose Acidity of Carcinomas. Cancer Letters, 276, 125-135. http://dx.doi.org/10.1016/j.canlet.2008.08.007
- Attenberg, B. and Greulick, K.O. (2004) Genes of Glycolysis Are Ubiquitously Overexpressed in 24 Cancer Classes. Genomies, 84, 1014-1020. http://dx.doi.org/10.1016/j.ygeno.2004.08.010
- Warburg, O. (1969) The Prime Cause of Cancers and Prevention—Part 2. Annual Meeting of Nobelists at Lindau Germany.
- Warburg, O. (1931) The Metabolism of Tumor. Richard R. Smith, New York.
- Moreno, S., Rodriguez, R., Enriguez, S., Soavedro, E., Masin-Hernandez, A. and Gallardo-Perez, J.C. (2009) The Bioenergetics of Cancer: Is Glycolysis the Main ATP Supplier in All Tumor Cells? Biofactors, 35, 209-225. http://dx.doi.org/10.1002/biof.31
- Bonnet, S., Archer, S.L., Allalunis-Turner, J., Hasomy, A., Beculieu, C., Thompson, R., Lee, C.T., Lopasahuk, G.D., Puttagunta, L., Bonnet, S., Harry, G., Hashimoto, K., Porter, C.J., Androcla, M.A., Thebaud, B. and Michelakis, E.D. (2007) A Mitochondria-Rt Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell, 11, 37-51. http://dx.doi.org/10.1016/j.ccr.2006.10.020
- Semenza, G.L. (2007) HIF-1 Mediates the Warburg Effect in Clear Cell Renal Carcinoma. Journal of Bioenergetics and Biomembranes, 39, 231-234. http://dx.doi.org/10.1007/s10863-007-9081-2
- Moreno-Sanchez, R., Rodriguez-Enriquez, S., Morin-Hernandez, A. and Savedra, E. (2007) Energy Metabolism in Tumor Cells. FEBS Journal, 274, 1393-1418. http://dx.doi.org/10.1111/j.1742-4658.2007.05686.x
- Ausinberg, A.C. (1961) The Glycolysis and Respiration of Tumors. Academic Press, New York.
- Gantin, V.R. and Leder, P. (2006) Mitochondriotoxic Compounds for Cancer Therapy. Oncogene, 25, 4787-4797. http://dx.doi.org/10.1038/sj.onc.1209599
- Hervouet, E., Demont, J., Pecina, P., Vojtiskova, A., Houstek, J., Simonnet, H. and Godinot, C. (2005) A New Role for the Von Hippel-Landau Tumor Suppressor Protein: Stimulation of Mitochondrial Oxidative Phosphorylation Complex Biogenesis. Carcinogenesis, 26, 531-539. http://dx.doi.org/10.1093/carcin/bgi001
- Weinhouse, S. (1956) On Respiratory Impairment in Cancer Cells. Science, 124, 267-269. http://dx.doi.org/10.1126/science.124.3215.267
- Weinhouse, S. (1976) The Warburg Hypothesis Fifty Years Later. Zeitschrift für Krebsforschung und Klinische Onkologie, 87, 115-126. http://dx.doi.org/10.1007/BF00284370
- Kim, J.W. and Dang, C.V. (2006) Cancer’s Molecular Sweet Tooth and the Warburg Effect. Cancer Research, 66, 8927-8930. http://dx.doi.org/10.1158/0008-5472.CAN-06-1501
- Hsu, P.P. and Sabatini, D.M. (2008) Cancer Cell Metabolism: Warburg and Beyond. Cell, 134, 703-707. http://dx.doi.org/10.1016/j.cell.2008.08.021
- Shaw, R.J. (2006) Glucose Metabolism and Cancer. Current Opinion in Cell Biology, 18, 598-608. http://dx.doi.org/10.1016/j.ceb.2006.10.005
- Jones, R.G. and Thompson, C.B. (2009) Tumor Suppressors and Cell Metabolism: A Recipe for Cancer Growth. Genes & Development, 23, 537-548. http://dx.doi.org/10.1101/gad.1756509
- Elliott, R.L., Jiang, X.P. and Head, J.F. (2012) Mitochondrial Organelle Transplantation: Intraduction of Normal Epithelial Mitochondria into Human Cancer Cells Inhibits Proliferation and Increases Drug Sensitivity. Breast Cancer Research and Treatment, 136, 347-354. http://dx.doi.org/10.1007/s10549-012-2283-2
- Sebastian, C., Zwaans, B., Silberman, D.M., Gymsek, M., Gosen, A., Zhong, L., Ram, O., Tudlove, J., Guimaralo, A.R., Toiber, D., Cosentino, C., Greenson, J.K., Macdonald, A.I., MCGlynse, L., Maxwell, F., Edwards, J., Giacosa, S., Guccione, E., Weissleder, R., Bernstein, B.E., Regeu, A., Shiels, P.G., Lombard, D.B. and Mostoslavsky, R. (2012) The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell, 151, 1185-1199. http://dx.doi.org/10.1016/j.cell.2012.10.047
- Hershkov, C. (1994) Control of Disease by Selective Iron Depletion: A Novel Therapeutic Strategy Utilizing Iron Chelators. Balliese’s Clinical Hematology, 7, 965-1000. http://dx.doi.org/10.1016/S0950-3536(05)80133-7
- Buss, J.L., Greene, B.T., Turner, J., Torti, F.M. and Torti, S.V. (2004) Iron Chelaters in Cancer Chemotherapy. Current Topics in Medicinal Chemistry, 4, 1623-1635. http://dx.doi.org/10.2174/1568026043387269
- Andrews, N.C. (2000) Disorders of Iron Metabolism. The New England Journal of Medicine, 342, 1293-1294. http://dx.doi.org/10.1056/NEJM200004273421716
- Commack, R., Wrigglesworth, J.M. and Baum, H. (1990) Iron-Dependent Enzymes in Mammalian Systems in Iron Transport and Storage. In: Ponka, P., Schulman, H.M. and Wodworth, R.C., Eds., Iron Transport and Storage, CRC Press, Boca Baton, 17-40.
- Thelander, L., Grislund, A. and Thelander, M. (1983) Continued Presence of Oxygen and Iron Required for Mammalian Ribonucleotide Reductase: Possible Regulation Mechanism. Biochemical and Biophysical Research Communications, 110, 859-865. http://dx.doi.org/10.1016/0006-291X(83)91040-9
- Thelander, M., Giaslund, A. and Thelander, L. (1985) Subunit M2 of Mammalian Ribonucleotide Reductase. Journal of Biological Chemistry, 260, 2737-2741.
- Trowbridge, I.S., Newman, A., Domingo, D.L. and Salvage, C. (1984) Transferrin Receptors: Structure and Formation. Biochemical Pharmacology, 925-932.
- Klausner, R.D., Ashwell, G., Van Renswoude, J., Harford, J.B. and Bridges, K.R. (1983) Binding of Apotransferrin to K562 Cells: Elplanation of the Transferrin Cycle. Proceedings of the National Academy of Sciences of the United States of America, 80, 2263-2266. http://dx.doi.org/10.1073/pnas.80.8.2263
- Richardson, D.R. and Ponka, P. (1997) The Molecular Mechanisms of the Metabolism and Transport of Iron in Normal and Neoplastic Cells. Biochemica et Biophysica Acta, 1331, 1-40. http://dx.doi.org/10.1016/S0304-4157(96)00014-7
- Elliott, R.L., Elliott, M.C., Wang, F. and Head, J.F. (1993) Breast Carcinoma and the Role of Iron Metabolism: A Cytochemical, Tissue Culture and Ultrastructural Study. Annals of the New York Academy of Sciences, 698, 159-166. http://dx.doi.org/10.1111/j.1749-6632.1993.tb17204.x
- Elliott, R.L., Stzernholm, R. and Elliott, M.C. (1988) Preliminary Evaluation of Platinumun Transferrin (MPTC-63) a Potential Non-Toxic Treatment for Breast Cancer. Cancer Detection and Prevention, 12, 469-480.
- Richardson, D.R., Lane, D.J., Becker, F.M., Huang, M.L., Whitnall, M., Susyo Rahmonto, Y., Sheftel, A.D. and Panka, P. (2010) Mitochondrial Iron Trafficking and the Integration of Iron Metablolis between the Mitochondrial and Cytosol. Proceedings of the National Academy of the Sciences of the United States of America, 107, 10775-10782. http://dx.doi.org/10.1073/pnas.0912925107
- Veatch, J.R., McMurray, M.A., Nelson, Z.W. and Gottschling, D.E. (2009) Mitochondrial Dysfunction Leads to Nuclear Genome Instability via an Iron-Sulfer Cluster Defect. Cell, 137, 1247-1258. http://dx.doi.org/10.1016/j.cell.2009.04.014
Abbreviations
HIF-1a: Hypoxia inducible factor 1-alpha Glut III: Glucose transporter III
(PDK)1: pyruvate dehydrogenase kinase 1 LDH-A: lactate dehydrogenase FeS: iron sulfur cluster TF: Transferrin TFR1: Transferrin receptor 1 TF-TFR1: Transferrin-transferrin receptor 1 complex DMT1: divalent metal transporter 1