Journal of Cancer Therapy
Vol. 4 No. 8A (2013) , Article ID: 36233 , 7 pages DOI:10.4236/jct.2013.48A008
Synergistic Anticancer Activity of Topotecan— Cyclin-Dependent Kinase Inhibitor Combinations against Drug-Resistant Small Cell Lung Cancer (SCLC) Cell Lines
![]()
Ludwig Boltzmann Cluster of Translational Oncology, Vienna, Austria.
Email: *gerhard.hamilton@toc.lbg.ac.at
Copyright © 2013 Gerhard Hamilton et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 27th, 2013; revised July 25th, 2013; accepted August 4th, 2013
Keywords: Small Cell Lung Cancer; Chemoresistance; Topotecan; CDK Inhibitor; Olomoucine; Roscovitine
ABSTRACT
Extended-stage small cell lung cancer (SCLC) responds to platinum/vepeside-based first-line chemotherapy but relapses rapidly as drug-resistant tumor. Topotecan (TPT) is the single chemotherapeutic agent approved for second-line treatment of SCLC. However, the response to TPT is short-lived and novel treatment modalities need to be developed. Sequential treatment of cytotoxic drugs and inhibitors of cyclin-dependent kinases (CDKs) showed promising preclinical anticancer activity and, in the present work, combinations of TPT with CDK inhibitors olomoucine, roscovitine and CDK4I are shown to exhibit synergistic cytotoxic activity against SCLC cell lines. Highest activity was found against TPT-resistant NCI-H417 and DMS153 cell lines and moderate chemosensitizing effects against a primary SCLC cell line and sensitive GLC19 cells at levels of CDK inhibitors which exerted low toxicity. A combination of 0.6 µM TPT with 0.6 µM roscovitine, exhibiting no significant cytotoxicity as single agents, reduced viability of the TPT-resistant NCI-H417 line (IC50 > 10 µM) by 50%. In the TPT resistant cell lines olomoucine and roscovitine, targeting CDK1,2,5,7, were highly effective, whereas in the more sensitive cell lines CDK4I, inhibiting mainly CDK4/6, showed activity. In NCI-417 cells, preincubation with roscovitine for one day proved synergistic with TPT. Thus, in good accordance with previous findings, CDK inhibitors are able to convert SCLC cancer cells which are cell-cycle arrested by a blockade of topoisomerase I by TPT to apoptotic cells. Since nowadays several CDK inhibitors are at various phases of clinical testing their combination with TPT seems to constitute a promising approach to improve second-line chemotherapy in SCLC.
1. Introduction
Small cell lung cancer (SCLC) represents approximately 13% of all lung cancer diagnoses and its incidence has reduced over the last 20 years, although the frequency is rising in women due to increased use of tobacco [1-3]. It is a highly malignant neuroendocrine tumor of the lung and treatment of SCLC remains challenging because of its rapid growth, early dissemination and development of drug resistance during the course of the disease [4]. Without treatment, SCLC has the most aggressive clinical course of any type of pulmonary tumor, with median survival from diagnosis of only 2 to 4 months [4]. Compared with other cell types of lung cancer, SCLC has a greater tendency to be widely disseminated by the time of diagnosis but is much more responsive to chemotherapy and radiation therapy first-line. With current chemotherapy regimens survival is prolonged, however, the overall survival at 5 years is only 5% to 10% [4-6]. At the time of diagnosis, approximately 30% of patients with SCLC will have tumor confined to the hemithorax of origin, classified as having limited-stage disease andmedian survival of 16 to 24 months [7]. Patients with extensive-stage disease have a worse prognosis with median survival of 6 to 12 months.
Chemotherapy improves the survival of patients with limited-stage or extensive-stage SCLC, but it is curative in only a minority of patients [4-6]. The standard combination of etoposide and cisplatin chemotherapy with concurrent chest radiation therapy achieves median survivals of 18 to 24 months and 40% to 50% 2-year survival with less than a 3% treatment-related mortality. Carboplatin is an acceptable alternative to cisplatin, with comparable efficacy and a more favorable toxicity profile [8]. Despite high initial response rates, the majority of patients relapse early and exhibit chemoresistance. Regimens that have shown activity as second line treatment include oral etoposide, etoposide/cisplatin, cyclophosphamide/doxorubicin/vincristine (CAV), lomustine/me-thotrexate, paclitaxel, and topotecan (TPT)[4,5]. Pharmacological inhibitors of receptor tyrosine kinases (RTK), including c-Kit, epidermal growth factor receptor (EGFR), insulin-like growth factor-I receptor (IGFR1) and vascular endothelial growth factor receptor (VEGFR), have been investigated as potential anti-tumor agents in SCLC with disappointing results [9].
One possible approach to improve the second-line treatment of SCLC may try to increase the efficacy of the chemotherapeutics in use through modulation of drug resistance of the tumor cells. Topotecan (TPT), a water soluble semisynthetic derivative of camptothecin (CPT), has demonstrated antineoplastic activity in a wide range of cell culture and xenograft systems and is currently approved for second-line therapy in ovarian cancer and SCLC [10-12]. The drug inhibits replication of rapidly dividing cells by disrupting the normal function of the nuclear enzyme topoisomerase I. The mechanism of CPT poisoning of TOP1 rests on inhibition of the religation function of the enzyme resulting in the stabilization of the TOP1-cleavable complex. In the presence of CPTs this enzyme-DNA complex impairs transcription and DNA replication, resulting in fork stalling and the formation of DNA double-strand breaks in proliferating cells [13]. The efficacy of TPT is related to exposure time and the recommended regimen is 1.5 mg/m2 as a 30-minute intravenous infusion, daily for 5 days, repeated every 21 days. In phase II trials of TPT in SCLC the overall response rate in refractory patients (relapse within 90 days after first-line therapy) was low at 2% to 11%, whereas in sensitive patients (relapse after days following firstline therapy) the overall response rate was 14% to 37%. TPT was compared with combined cyclophosphamide/ doxorubicin(adriamycin)/vincristine (CAV) therapy in patients with relapsed, sensitive SCLC and no significant differences were detected for response rates and duration; however, TPT offered superior control of some symptoms. Furthermore, an oral formulation of TPT with similar efficacy to intravenous formulations is available [14, 15]. Noncumulative anaemia, neutropenia and thrombocytopenia are the dose-limiting adverse effects associated with TPT. TPT is currently the only drug licensed in Europe and the US for second-line treatment of SCLC, having been shown in a phase III trial to lead to longer overall survival and better quality of life than with BSC. A phase III trial which compared oral TPT with best supportive care (BSC) in relapsed SCLC patients revealed partial response rate of 7% for the TPT group, with a further 44% gaining disease stabilization, and a median survival time of 25.9 weeks (versus 13.9 weeks for BSC), clearly indicating an urgent need for novel and improved therapeutic options [16,17]. As with most chemotherapeutics, intrinsic and acquired drug resistance represents an obstacle that limits the success of camptothecin (CPT) derivative therapy [13,18]. Resistance to CPT-based drugs might be caused by factors such as poor drug accumulation in the tumor, high rate of drug efflux, mutations in TOP1 leading to failure in CPT docking, or altered signaling triggered by the drugTOP1-DNA complex, as well as expression of DNA repair proteins, and failure to activate cell death pathways.
Inactivation of p16INK4/cyclin-dependent kinase inhibitor 2A (CDKN2A), an inhibitor of cyclin-dependent kinases 4 (CDK4) and 6 (CDK6), may be essential for oncogenesis in non-small cell lung cancer (NSCLC). Two clones of p16INK4-transfected NSCLC cell lines showed more than sevenfold increases in sensitivity to CPT-11 (11,7-ethyl-10-[4-(1-piperidino)-1-piperidino] carbonyloxy-camptothecin), compared with the matching untransformed cells [19]. The topoisomerase I-mediated DNA relaxation activities of ectopic p16INK4-expressing cells were approximately 5 times higher than those of the parent cells, due to an elevated topo I mRNA level and an increase in topo I protein. Therefore, we tested several CDK inhibitors for a possible modulation of TPT sensitivity of SCLC cell lines. These inhibitors recently gained much interest since, for example, Palbociclib (formerly known as PD-0332991), a novel oral selective inhibitor of CDK4/6 that blocks tumor cell progression, has received a “breakthrough therapy designation” from the FDA for the treatment of patients with breast cancer, and similar drugs, like Dinaciclib (SCH727965/CDK1,2, 5,9), Alvocidib (flavopiridol/CDK1,2,4,6) and Seliciclib/ CYC202 (roscovitine/CDK2,5) are at various phases of clinical testing [20]. Our experiments comprised SCLC lines with different sensitivity to TPT and a primary cell line SCLC26A initiated in our institution.
2. Materials and Methods
2.1. Reagents and Cell Lines
Olomoucine and roscovitine were obtained from SigmaAldrich (St. Louis, MO, USA) and the CDK4 inhibitor from Calbiochem (Darmstadt, Germany). Stock solutions of all compounds were prepared in DMSO. All other chemicals were purchased from Sigma-Aldrich, except indicated otherwise. NCI-H417 was obtained from ATCC (Rockville, MD, USA), DMS153 cells from ECACC (Porton Down, Salisbury, UK) and the GLC19 cell line from Dr. Nina Pedersen from the Department of Radiation Biology, The Finsen Centre, National University Hospital, Copenhagen, Denmark and SCLC26A was established from a pleural effusion of an untreated patient with SCLC at our institution. Cells were grown in RPMI-1640 bicarbonate medium (Seromed, Berlin, Germany) supplemented with 10% fetal bovine serum (Seromed), 4 mM glutamine and antibiotics (10× stock formulated to contain ~5000 units penicillin, 5 mg streptomycin and 10 mg neomycin/ml) under tissue culture conditions (37˚C, 5% CO2, 95% humidity) and checked for mycoplasma contamination (Mycoplasma PCR ELISA, Roche Diagnostics, Vienna, Austria). Cells grow as spheroids in suspension and were subcultures two times a week.
2.2. Chemosensitivity Assay
1 × 104 cells in 100 μl medium per well were distributed in 96-well microtiter plates (Greiner, Kremsmuenster, Austria) and the test compound added in another 100 μl. Drugs and solute controls were serially diluted in twofold steps in triplicate. The microtiter plates were incubated under tissue culture conditions for four days and cell viability was measured using a modified MTT (3-(4,5- dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide) assay (EZ4U, Biomedica, Vienna, Austria). Optical density was measured using a microplate reader at 450 nm with an empty well as reference. Values obtained from control wells containing cells and media alone were set to 100% proliferation.
3. Results
NCI-H417 and DMS153 SCLC cell lines were treated with TPT alone and in combination with the CDK inhibitors roscovitine and olomoucine, respectively (Figure 1). Initial concentrations of 10 µM for TPT, 10 µM for roscovitine and 25 µM for olomoucine were diluted in eight twofold dilution steps, the cells incubated for four days in tissue culture and viability assessed by the modified MTT assay. Combinations of TPT and CDK inhibitors comprised full initial concentrations of both drugs. Whereas DMS153 cells show intermediate resistance to TPT with IC50 values of approximately 2.5 µM, NCI-H417 proved to be highly resistant to TPT with IC50 values exceeding 10 µM. Peak plasma concentrations of TPT reaches values up to approximately 0.5 µM. Roscovitine and olomoucine alone had a minor effects on cell viability, not exceeding 10% inhibition at the highest concentrations used. Combinations of the CDK inhibitors with TPT revealed significant synergism for DMS153 and roscovitine (Figure 1(a)) and marked synergism for
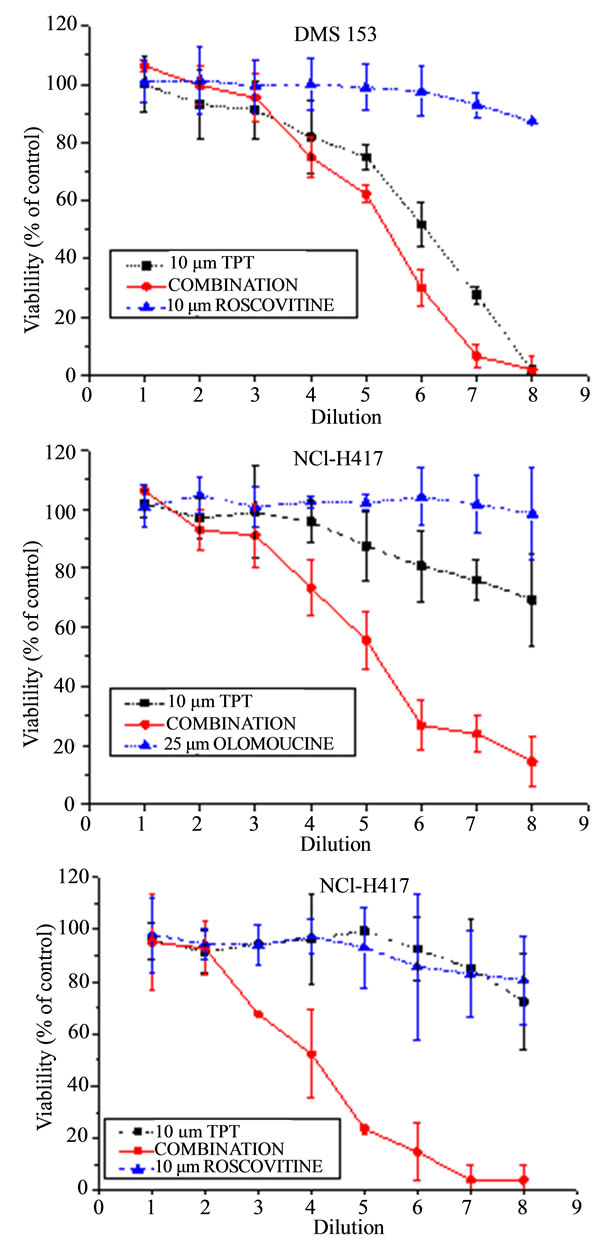
Figure 1. Results from chemosensitivity tests showing doseresponse curves of the combinations of TPT with roscovitine for DMS153 (A) and NCI-H417 (C), respectively, as well as of TPT with olomoucine for NCI-H417 cells (B). Drugs were used at initial concentrations of 10 µM for TPT and roscovitine, and 25 µM for the less active olomoucine, respectively. The combinations contained the full concentration of both compounds and all formulations were diluted twofold in eight steps (mean values ± SD, n = 3).
the NCI-H417 cells cotreated with TPT and olomoucine and roscovitine, respectively (Figures 1(b) and (c)). In fact, the single drugs which reduced viability of these cells to less than 10% - 25%, resulted in complete cell kill when used in combination.
In order to further investigate the interaction of TPT with CDK inhibitors, the experiments were extended to include the GLC19 SCLC cell line obtained after chemoradiotherapy, including cyclophosphamide/adriamycin/ vincristine (CAV), and the freshly isolated, primary SCLC26A cell line, at first passages in tissue culture. Furthermore, CDK4I targeted primarily to CDK4/6, in contrast to roscovitine and olomoucine, was tested (Table 1). GLC19 is TPT-sensitive, with an IC50 value of 17 nM TPT and the SCLC26A cell line has a higher IC50 value of 125 ± 43 nM TPT (results not shown). Figure 2 shows the reduction of viable SCLC cells in MTT assays resulting from combing TPT with the respective CDK4 inhibitors in the range of IC50 concentrations for TPT, except for NCI-H417, where the IC50 values of the drug combination had to be used, since this value was not reched for TPT (Figure 2). The results demonstrate, that sensitization to TPT by CDK inhibitors is highest for the drug-resistant cell lines NCI-H417 and DMS153. For NCI-H417 cells, roscovitine is the most efficient resistance
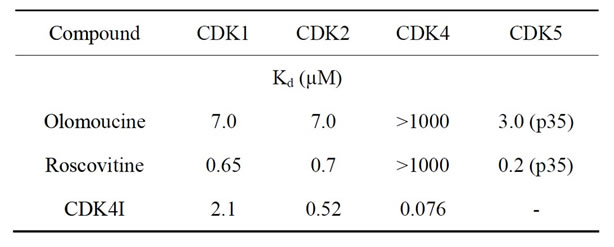
Table 1. Specificity of selected CDK inhibitors.
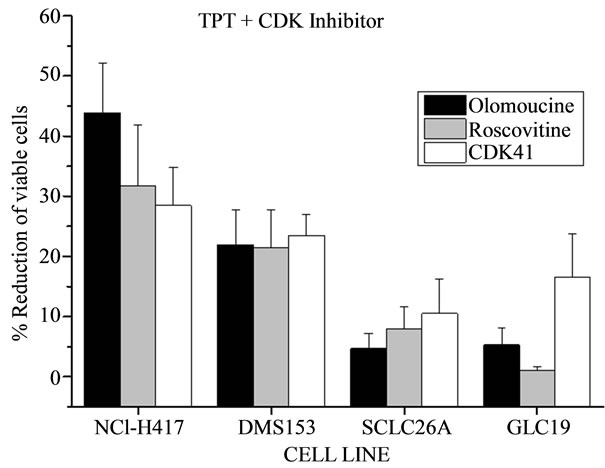
Figure 2. Overview of the reductions in the number of viable SCLC cancer cells due to the combined cytotoxicity of TPT in combination with the respective CDK inhibitors in MTT assays. Except for NCI-H417, where the IC50 values of the drug combination had to be used for calculation since this value was not reached for TPT, the enhanced cytotoxicity against the other three cell lines was quantified for the IC50 values of TPT in the dose-response curves (mean values ± SD, n = 3).
modulator, followed by olomoucine, which had to be applied at higher concentration (×2.5) and CDK4I, whereas in the more TPT-sensitive cell lines SCLC26A and GLC19 the chemosensitizing effect is much smaller and the CDK4I inhibitor proved to more effective compared to roscovitine and olomoucine (Figure 2).
For NCI-H417 cells the cytotoxic activity of TPT was measured following preincubation of the cells with 5 µM CDK4I, 10 µM roscovitine or 25 µM olomoucine, respectively for 24 hrs. Pretreatment of the cells with CDK4I and olomoucine lowered TPT-sensitivity (approximately 4- and 2-fold, respectively), whereas roscovitine preincubation still resulted in increased chemosensitivity (approximately 3fold; results not shown).
4. Discussion
In the present work we employed several CDK inhibitors at concentrations that exhibited low toxicity in order to investigate the putative modulation of TPT cytotoxicity against several SCLC cell lines. CDK4I is a cell-permeable, unsymmetrical indolocarbazole compound that displays antiproliferative properties through reversible and ATP-competitive inhibition of CDK4. This inhibitor blocks tumor cells growth (IC50 < 3.0 µM in HCT-116 and NCI-H460 cells) by impairing retinoblastoma suspectibility protein (Rb) phosphorylation and inducing G1 cell cycle arrest. Olomoucine is a purine derivative which inhibits CDK1,2,5,7 and induces G1 arrest, and roscovitine/Seliciclib has the same specificity as olomoucine, but shows 10fold improved efficacy in stabilization of p53 as well as induction of cell cycle arrest and apoptosis [21]. Our results demonstrating the chemosensitizing effects of CDK inhibitors on the TPT-sensitivity of SCLC cell lines corroborate the findings obtained in NSCLC upon manipulation of the CDKN2A inhibitor of CDKs [19]. The highest synergistic effect of the CDK inhibitors was detected for the SCLC cell lines with high resistance to TPT, namely NCI-H417 and DMS153, although a significant effect was still visible in the freshly isolated SCLC26A cells and the GLC19 cell line, which acquired resistance to CAV chemotherapy/irradiation in vivo. Whereas CDK4I directed to CDK4/6 showed good potency against the partly TPT-sensitive SCLC26A and GLC19 cell lines, olomoucine and roscovitine, targeting CDK1,2,5,7, were more effective against the TPT-resistant cell lines NCI-H417 and DMS153. Thus, inhibition of CDK4/6 works for SCLC cells sensitive to TPT, in contrast to resistant cells which respond best to impairment of the other CDKs.
The mechanisms underlying the synergism of TPT and the CDK inhibitors are not clear. Inhibition of cancer cell proliferation and induction of apoptosis are important mechanisms for a chemotherapeutic agent [22]. During proliferation, progression through the cell cycle is first regulated at G1 by cyclin D1 and CDK4/6 which are expressed continuously throughout the early G1 phase before cells pass through the restriction point and contribute to activating genes for entry into S phase. Activation of the p53 tumor suppressor gene leads to G1 phase arrest and helps to maintain genetic stability by preventing entry into S phase. If DNA repair is compromised cells commit to exit G1 phase and proceed to undergo apoptotic cell death [23]. p16ink4 has been implicated as a tumor suppressor that is lost from a variety of human tumors and human cell lines [24]. The tumor suppressor p16ink4 specifically binds and inhibits CDK4/6, preventing activation of the suppressor protein RB by phosphorylation. This tumor suppressor circuit is eliminated in SCLC. Only 11% of 55 SCLC samples had absent p16INK4 protein, and all of these belonged to the rare subset of SCLC with wildtype RB expression. Conversely of 48 SCLC samples with absent or mutant RB, all showed detectable levels of p16INK4 protein [25,26]. Furthermore, the p53 status of the SCLC26A has not been checked, but the three other SCLC cell lines, NCIH417, DMS153 and GLC19 feature mutated p53 [27,28]. Since the DNA-damage checkpoint depends on p53 activation, the status of p53 might critically influence the response to camptothecins. For the p53-mutated colon cancer cell line HT29 gain of sensitivity to CPT-11/irinotecan was increased by either restoration of wild-type p53 function or by sequential treatment with the roscovitine/CYC-202 CDK inhibitor [29]. In a similar fashion, roscovitine resulted rather an induction of cell death instead of cell cycle arrest Lovo colorectal carcinoma cells and in hepatocellular carcinoma cell lines, roscovitine pretreatment considerably enhanced the carboplatinand quercetin-induced cell death via down regulation of AKT, procaspases and Bcl-2 [30,31].
Mutations in the tumor-suppressor gene p53 allow the growing tumor with multiple genetic alterations to evade cell-cycle arrest and apoptosis [32]. Agents damaging DNA may initiate postdamage responses by activating cell-cycle checkpoints and these events might also influence treatment sensitivity and apoptotic potential [33]. Leukemia U-937 cells rapidly underwent characteristic morphological changes, caspase activation, and DNA fragmentation typical of apoptosis on treatment with the DNA topoisomerase I inhibitor CPT. Accumulation of these cells at G1 was not associated with major changes in expression level or kinase activities of CDK2, CDK4, and CDK6 after CPT treatment [34]. These results indicate that the G1 arrest of U937 cells does not correlate with a classical driven cell cycle checkpoint but with the known effect of CPT in mediating inhibition of DNA replication and RNA transcription after stabilization of topoisomerase I-linked DNA strand breaks. Treatment of the colon cancer cells HCT116 with SN-38, the active metabolite of CPT11/irinotecan, resulted in G2 cell-cycle arrest without cell death; however, subsequent exposure to flavopiridol induced apoptosis [35]. The broadband CDK inhibitor flavopiridol is not only cytostatic, but also cytotoxic to a variety of human tumor cell lines with equal activity against an active cycling and noncycling NSCLC cell line [36]. Flavopiridol enhanced paclitaxelinduced apoptosis only when administered after cytotoxic drug treatment, most likely through activation of caspases, specifically caspase 3 [37]. In A549 NSCLC cells in vitro, paclitaxel, cytarabine, TPT, doxorubicin, and etoposidein combinations of flavopiridol exhibited higher synergy when the agents were administered before flavopiridol rather than concomitant with or following flavopiridol [38]. Similarly, overruling of the CPT-induced S phase checkpoint by the protein kinase C inhibitor staurosporine enhanced the cytotoxicity of CPT, particularly in p53-mutated tumors [39].
The results regarding flavopiridol discussed above were published almost a decade ago and since then the further development of this CDK inhibitor was detained by disappointing clinical results. Although in phase I clinical trials concentrations of flavopiridol needed for CDK inhibition were achieved safely, the drug proved to be ineffective in phase II trials against a large range of different tumor entities [40]. Dose-limiting toxicities became evident, especially in combinations with other chemotherapeutics. Recently, with better understanding of the tumor biology and the availability of several other CDK inhibitors these agents are studied increasingly alone or in drug combinations in susceptible tumor entities and patient subpopulations [20]. In conclusion, CDK inhibitors seem to constitute potent chemosensitizers of TPT in SCLC cell lines. The genetic background of these tumor cells, lacking a functional cyclin D1-p16INK4- CDK4/6-RB tumor suppressor pathway in conjunction with mutated p53 seem to provide an ideal background for the anticancer activity of TPT-CDK inhibitor combinations. In contrast to published results, here the agents need not to be applied in the specific sequence of TPT and CDK inhibitor subsequently, but synergize in concomitant administration [38].
5. Acknowledgements
This work was funded by the “Medical Scientific Fund of the Mayor of the City of Vienna” project number 11016.
REFERENCES
- D. R. Youlden, S. M. Cramb and P. D. Baade, “The International Epidemiology of Lung Cancer: Geographical Distribution and Secular Trends,” Journal of Thoracic Oncology, Vol. 3, No. 8, 2008, pp. 819-831. doi:10.1097/JTO.0b013e31818020eb
- S. G. Spiro and G. A. Silvestri, “One Hundred Years of Lung Cancer,” American Journal of Respiratory Critical Care Medicine, Vol. 172, No. 5, 2005, pp. 523-529. doi:10.1164/rccm.200504-531OE
- A. Califano, Z. Abidin, R. Peck, C. Faivre-Finn and P. R. Lorigan, “Management of Small Cell Lung Cancer: Recent Developments for Optimal Care,” Drugs, Vol. 72, No. 4, 2012, pp. 471-490. doi:10.2165/11597640-000000000-00000
- R. Govindan, N. Page, D. Morgensztern, et al., “Changing Epidemiology of Small-Cell Lung Cancer in the United States over the Last 30 Years: Analysis of the Surveillance, Epidemiologic, and End Results Database,” Journal of Clinical Oncology, Vol. 24, No. 28, 2006, pp. 4539-4544. doi:10.1200/JCO.2005.04.4859
- W. N. William and B. S. Glisson, “Novel Strategies for the Treatment of Small-Cell Lung Carcinoma,” Nature Reviews Clinical Oncology, Vol. 8, No. 10, 2011, pp. 611-619. doi:10.1038/nrclinonc.2011.90
- Y. Agra, M. Pelayo, M. Sacristan, et al., “Chemotherapy versus Best Supportive Care for Extensive Small Cell Lung Cancer,” Cochrane Database Systematic Review, Vol. 4, 2003, Article ID: CD001990.
- M. Takada, M. Fukuoka, M. Kawahara, et al., “Phase III Study of Concurrent versus Sequential Thoracic Radiotherapy in Combination with Cisplatin and Etoposide for Limited-Stage Small-Cell Lung Cancer,” Journal of Clinical Oncology, Vol. 20, No. 14, 2002, pp. 3054-3060. doi:10.1200/JCO.2002.12.071
- A. Rossi, M. Di Maio, P. Chiodini, et al., “Carboplatinor Cisplatin-Based Chemotherapy in First-Line Treatment of Small-Cell Lung Cancer: The COCIS Meta-Analysis of Individual Patient Data,” Journal of Clinical Oncology, Vol. 30, No. 14, 2012, pp. 1692-1698. doi:10.1200/JCO.2011.40.4905
- B. Fischer and A. Arcaro, “Current Status of Clinical Trials for Small Cell Lung Cancer,” Review of Recent Clinical Trials, Vol. 3, No. 1, 2008, pp. 40-61. doi:10.2174/157488708783330503
- D. Ormrod and C. M. Spencer, “Topotecan: A Review of Its Efficacy in Small Cell Lung Cancer,” Drugs, Vol. 58, No. 3, pp. 533-551. doi:10.2165/00003495-199958030-00020
- J. L. Hurwitz, F. McCoy, P. Scullin and D. A. Fennell, “New Advances in the Second-Line Treatment of Small Cell Lung Cancer,” Oncologist, Vol. 14, No. 10, 2009, pp. 986-994. doi:10.1634/theoncologist.2009-0026
- A. Schmittel, “Second-Line Therapy for Small-Cell Lung Cancer,” Expert Review in Anticancer Therapy, Vol. 11, No. 4, 2011, pp. 631-637. doi:10.1586/era.11.7
- M. T. Tomicic and K. B. aina, “Topoisomerase Degradation, DSB Repair, p53 and IAPs in Cancer Cell Resistance to Camptothecin-Like Topoisomerase I Inhibitors,” Biochimica et Biophysica Acta, Vol. 1835, No. 1, 2013, pp. 11-27.
- S. Agelaki, E. Kontopodis, A. Kotsakis, et al., “A Phase I Clinical Trial of Weekly Oral Topotecan for Relapsed Small Cell Lung Cancer,” Cancer Chemotherapy and Pharmacology, 2013, in press. doi:10.1007/s00280-013-2167-0
- R. Riemsma, J. P. Simons, Z. Bashir, C. L. Gooch and J. Kleijnen, “Systematic Review of Topotecan (Hycamtin) in Relapsed Small Cell Lung Cancer,” BMC Cancer, Vol. 10, 2010, p. 436.
- M. E. O’Brien, T. E.Ciuleanu, H. Tsekov, et al., “Phase III Trial Comparing Supportive Care Alone with Supportive Care with Oral Topotecan in Patients with Relapsed Small-Cell Lung Cancer,” Journal of Clinical Oncology, Vol. 24, No. 34, 2006, pp. 5441-5447. doi:10.1200/JCO.2006.06.5821
- D. Hartwell, J. Jones, E. Loveman, P. Harris, A. Clegg and A. Bird, “Topotecan for Relapsed Small Cell Lung Cancer: A Systematic Review and Economic Evaluation,” Cancer Treatment Reviews, Vol. 37, No. 3, 2011, pp. 242-249. doi:10.1016/j.ctrv.2010.07.005
- Z. A. Rasheed and E. H. Rubin, “Mechanisms of Resistance to Topoisomerase I-Targeting Drugs,” Oncogene, Vol. 22, No. 47, 2003, pp. 7296-7304.
- K. Fukuoka, K. Nishio, H. Fukumoto, et al., “p16INK4 Expression Is Associated with the Increased Sensitivity of Human Non-Small Cell Lung Cancer Cells to DNA Topoisomerase I Inhibitors,” Japanese Journal of Cancer Research, Vol. 88, No. 10, 1997, pp. 1009-1016. doi:10.1111/j.1349-7006.1997.tb00322.x
- L. Esposito, P. Indovina, F. Magnotti, D. Conti and A. Giorda, “Anticancer Therapeutic Strategies Based on CDK Inhibitors,” Current Pharmaceutical Design, 2013, in press. doi:10.2174/13816128113199990377
- B. Schutte, L. Nieland, M. van Engeland, M. E. Henfling, L. Meijer and F. C. Ramaekers, “The Effect of the Cyclin-Dependent Kinase Inhibitor Olomoucine on Cell Cycle Kinetics,” Experimental Cell Research, Vol. 236, No. 1, 1997, pp. 4-15. doi:10.1006/excr.1997.3700
- D. Hanahan and R. A. Weinberg, “Hallmarks of Cancer: The Next Generation,” Cell, Vol. 144, No. 5, 2011, pp. 646-674. doi:10.1016/j.cell.2011.02.013
- K. X. Shu, B. Li and L. X. Wu, “The p53 Network: p53 and Its Downstream Genes,” Colloids and Surfaces B: Biointerfaces, Vol. 55, No. 1, 2007, pp. 10-18. doi:10.1016/j.colsurfb.2006.11.003
- R. H. Medema, R. E. Herrera, F. Lam and R. A. Weinberg, “Growth Suppression by p16ink4 Requires Functional Retinoblastoma Protein,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, No. 14, 1995, pp. 6289-6293. doi:10.1073/pnas.92.14.6289
- G. A. Otterson, R. A. Kratzke, A. Coxon, Y. W. Kim and F. J. Kaye, “Absence of p16INK4 Protein Is Restricted to the Subset of Lung Cancer Lines that Retains Wildtype RB,” Oncogene, Vol. 9, No. 11, 1994, pp. 3375-3378.
- H. Wikman and E. Kettunen, “Regulation of the G1/S Phase of the Cell Cycle and Alterations in the RB Pathway in Human Lung Cancer,” Expert Reviews in Anticancer Therapy, Vol. 6, No. 4, 2006, pp. 515-530. doi:10.1586/14737140.6.4.515
- A. Okamoto, S. P. Hussain, K. Hagiwara, et al., “Mutations in the p16INK4/MTS1/CDKN2, p15INK4B/MTS2, and p18 Genes in Primary and Metastatic Lung Cancer,” Cancer Research, Vol. 55, No. 7, 1995, pp. 1448-1451.
- R. Zandi, G. Selivanova and C. L. Christensen, “PRIMA- 1Met/APR-246 Induces Apoptosis and Tumor Growth Delay in Small Cell Lung Cancer Expressing Mutant p53,” Clinical Cancer Research, Vol. 17, No. 9, 2011, pp. 2830-2841. doi:10.1158/1078-0432.CCR-10-3168
- M. Abal, R. Bras-Goncalves, J. G. Judde, et al., “Enhanced Sensitivity to Irinotecan by Cdk1 Inhibition in the p53-Deficient HT29 Human Colon Cancer Cell Line,” Oncogene, Vol. 23, No. 9, 2004, pp. 1737-1744. doi:10.1038/sj.onc.1207299
- S. J. McClue, D. Blake, R. Clarke, et al., “In Vitro and in Vivo Antitumor Properties of the Cyclin Dependent Kinase Inhibitor CYC202 (R-Roscovitine),” International Journal of Cancer, Vol. 102, No. 5, 2002, pp. 463-468. doi:10.1002/ijc.10738
- A. Sharma and M. K. Bhat, “Enhancement of Carboplatinand Quercetin-Induced Cell Death by Roscovitine Is Akt Dependent and p53 Independent in Hepatoma Cells,” Integrative Cancer Therapy, Vol. 10, No. 4, 2011, pp. 4-14. doi:10.1177/1534735411423922
- J. W. Arends, “Molecular Interactions in the Vogelstein Model of Colorectal Carcinoma,” Journal of Pathology, Vol. 190, No. 4, 2000, pp. 412-416. doi:10.1002/(SICI)1096-9896(200003)190:4<412::AID-PATH533>3.0.CO;2-P
- R. W. Johnstone, A. A. Ruefli and S. W. Lowe, “Apoptosis: A Link between Cancer Genetics and Chemotherapy,” Cell, Vol. 108, No. 2, 2002, pp. 153-164. doi:10.1016/S0092-8674(02)00625-6
- A. T. Sané and R. Bertrand, “Caspase Inhibition in Camptothecin-Treated U-937 Cells Is Coupled with a Shift from Apoptosis to Transient G1 Arrest Followed by Necrotic Cell Death,” Cancer Research, Vol. 59, No. 15, 1999, pp. 3565-3569.
- M. Motwani, C. Jung, F. M. Sirotnak, et al., “Augmentation of Apoptosis and Tumor Regression by Flavopiridol in the Presence of CPT-11 in Hct116 Colon Cancer Monolayers and Xenografts,” Clinical Cancer Research, Vol. 7, No. 12, 2001, pp. 4209-4219.
- K. C. Bible and S. M. Kaufmann, “Flavopiridol: A Cytotoxic Flavone That Induces Cell Death in Noncycling A549 Human Lung Carcinoma Cells,” Cancer Research, Vol. 56, No. 21, 1996, pp. 4856-4861.
- M. Motwani, T. M. Delohery and G. K. Schwartz, “Sequential Dependent Enhancement of Caspase Activation and Apoptosis by Flavopiridol on Paclitaxel-Treated Human Gastric and Breast Cancer Cells,” Clinical Cancer Research, Vol. 5, No. 7, 1999, pp. 1876-1883.
- K. C. Bible and S. H. Kaufmann, “Cytotoxic Synergy between Flavopiridol (NSC 649890, L86-8275) and Various Antineoplastic Agents: The Importance of Sequence of Administration,” Cancer Research, Vol. 57, No. 16, 1997, pp. 3375-3380.
- R.-G. Shao, C.-X. Cao, T. Shimizu, P. M. O’Connor, K. W. Kohn and Y. Pommier, “Abrogation of an S-Phase Checkpoint and Potentiation of Camptothecin Cytotoxicity by 7-Hydroxystaurosporine (UCN-O1) in Human Cancer Cell Lines, Possibly Influenced by p53 Function,” Cancer Research, Vol. 57, No. 18, 1997, pp. 4029-4035.
- M. V. Blagosklonny, “Flavopiridol, an Inhibitor of Transcription: Implications, Problems and Solutions,” Cell Cycle, Vol. 3, No. 12, 2004, pp. 1537-1542. doi:10.4161/cc.3.12.1278
NOTES
*Corresponding author.