Journal of Cancer Therapy
Vol.4 No.7(2013), Article ID:36128,10 pages DOI:10.4236/jct.2013.47137
The Aryl Hydrocarbon Receptor: A Target for Breast Cancer Therapy
![]()
1Department of Biological Sciences & Center for Cancer Research and Therapeutic Development, Clark Atlanta University, Atlanta, USA; 2Department of Biochemistry & Cancer Biology, Meharry Medical College, Nashville, USA.
Email: *seltom@mmc.edu
Copyright © 2013 Joann B. Powell et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 14th, 2013; revised July 16th, 2013; accepted July 24th, 2013
Keywords: aryl hydrocarbon receptor; therapeutic targeting; breast cancer progression; chemosensitization
ABSTRACT
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that regulates a battery of genes in response to exposure to a broad class of environmental poly aromatic hydrocarbons (PAH). AhR is historically characterized for its role in mediating the toxicity and adaptive responses to these chemicals, however mounting evidence has established a role for it in ligand-independent physiological processes and pathological conditions, including cancer. The AhR is overexpressed and constitutively activated in advanced breast cancer cases and was shown to drive the progression of breast cancer. In this article we will review the current state of knowledge on the possible role of AhR in breast cancer and how it will be exploited in targeting AhR for breast cancer therapy.
1. Introduction
Breast cancer is the second leading cause of cancerrelated death in women in the US [1]. An estimated 40 percent of breast cancer patients relapse and develop metastatic disease and approximately 40,000 women die of breast cancer each year [2]; a mortality rate is largely attributed to systemic metastatic disease [3]. Despite the recently reported decline in death rates the complexity of breast cancer makes treatment of many breast cancer subtypes difficult [4,5]. Therefore, it is imperative to identify and characterize factors associated with breast cancer malignancy, which will have the potential to serve as novel molecular targets for breast cancer therapy. The AhR is one of these emerging factors that have the potential to be targeted for treating certain subtypes of metastatic breast cancer.
The AhR is a member of the basic-helix-loop-helix (bHLH)-Per-ARNT-Sim (PAS) family of transcription factors [6]. It is the only known ligand-activated member of the bHLH-PAS family [7]. It is well characterized for mediating the effects of a large class of polycyclic and poly halogenated aromatic hydrocarbons [8]. However, a growing body of literature suggests a role for AhR protein that diverges from its role as a sensor and adapter for exposure to environmental xenobiotics [9,10]. In particularly, the AhR proved to play a central role in driving the normal mammary gland development, and in an analogous fashion to drive the breast cancer progression. Although this dual double-edge role may make AhR a rather difficult target for breast cancer therapy, the focus will be on the unique aspects of its biology that is more specific to breast cancer invasion and metastasis.
2. Discussion
2.1. Role of AhR in Breast Development
The mammary gland is a complex tissue that undergoes many structural and functional changes during various life stages [11]. The AhR may play a role in breast development in utero, during pregnancy and is also reported to play a critical role in breast cancer development. AhR mRNA and protein is expressed as early as gestational day 10 - 16 in some embryonic mouse tissue [12] . AhR deficient mice exhibit altered development in multiple organs, including the mammary gland. Lack of AhR in the mammary gland results in a 50% decrease in terminal end buds [13] . In utero exposure to the prototypic AhR agonist, 2,3,7.8,-tetrachlorodibenzo-p-dioxin (TCDD), increases the number of terminal end buds and causes sustained defects in mammary gland development and functions in normal mice [14] . TCDD exposed mice also demonstrate reduced epithelial elongation and fewer alveolar buds. Evidence suggests that the alterations to mammary development are permanent in gestationally exposed animals. Mice exposed to TCDD in utero exhibit stunted progression of epithelium through the fat pad, fewer lateral branches and delayed lobule formation that persist past postnatal 68 [15] . However, TCDD exposed mammary glands retain the ability to differentiate in response to estrogen. TCDD exposed tissues express increased levels of estrogen receptor alpha and upon stimulation with estrogen induce mammary gland differentiation. The percentage of lobules I and II in TCDD exposed mammary glands increased significantly following exposure to 17β-estradiol [16] . Pregnant dams exposed to TCDD by gavage also demonstrated severe developmental defects including decreased mammary gland weight and branching. Analysis of hormone levels revealed a significant decrease in prolactin and estrogen on day 17 of pregnancy and at parturition [17]. These phenotypic changes mirror image those of the AhR null mice, underscoring the significance of the lack of the receptor or its activation with subsequent ligand-dependent depletion during those critical time of development.
AhR activation by TCDD during pregnancy has also been reported to delay DMBA-induced tumor formation in adult mice. TCDD exposure resulted in a 4-week delay in tumor formation. Overall tumor incidence was also lower in TCDD exposed group compared to the control group [18]. This is in contrast to alteration of mammary gland differentiation during in utero exposure, which is correlated with increased susceptibility to carcinogenesis. Prenatal TCDD treatment resulted in increased tumor incidence in rats [19]. Varying responses to TCDD exposure at different times during pregnancy have been reported ADDIN EN.CITE ADDIN EN.CITE.DATA [14] . Additional research is needed to determine if these diverse effects are a result of circulating estrogen levels or AhR protein levels.
Transcriptional pattern analysis revealed that AhR and AhR related genes are frequently deregulated in breast cancer. The majority of tumors tested revealed deregulation of AhR related genes [20] . Evaluation of AhR mRNA levels in rat mammary tissue and tumors indicates lower AhR expression in normal mammary epithelial cells in contrast to high AhR levels in DMBA-induced tumors [21] . Together, these findings suggest that AhR mediated mammary tumorigenesis may not require ligand-induced alteration of mammary gland structure and function.
2.2. AhR and Breast Cancer Progression
Elevated levels of AhR expression in human mammary tumors were reported from different laboratories including ours [22,23] . We reported dramatic elevated levels of AhR proteins in human breast carcinoma (HBC) cell lines from advanced malignancy (MDA-MB231, MDAMB468, MDA-MB435s, MT2, NT, MCF7 breast cancer cell lines), while less levels were expressed in HBC derived from early stages of malignancy (T47D, MDAMB-436 cell lines) and in immortalized and primary human mammary epithelial cells. The AhR was also constitutively activated in the advanced malignant cell lines [22]. Our observation on the breast cancer cell lines was later confirmed by others [23] who showed that infinite life-span cell lines had low levels of AhR mRNA compared to immortalized but non-malignant cell lines, which showed a 10-fold increase in AhR mRNA expression. Fully malignant cell lines had an 8-fold increase in AhR expression compared to the normal representative cell lines.
We further investigated the potential of AhR as a stage specific marker of breast cancer. We examined the expression of AhR by immunohistochemistry in tissue microarrays (TMA) containing 192 specimens of clinically defined three stages of invasive breast cancer: node negative, node-positive and metastatic carcinoma. Statistical analysis showed a highly significant correlation between the AhR expression and the carcinoma case type and the stage of invasive carcinoma (Eltom, et al., unpublished data). These findings provide compelling evidence for AhR as a new predictive clinical marker for metastatic breast cancer and a unique target for the design of novel selective inhibitors for therapeutic intervention of metastatic breast cancer.
To delineate the role of AhR in the breast cancer progression, we employed the knock-in and knock-down approaches. First, we stably expressed high levels of AhR protein in an immortalized normal mammary epithelial cell line. Characterizing these transformed cells reveals that they exhibit several malignant properties. These cells underwent epithelial-mesenchymal-transition, had increased growth rates, abrogated cell cycle control and increased migration and invasive potential [24] (Figure 1). These studies revealed that AhR alone is required and sufficient to induce malignant transformation in mammary epithelial cells.
We also showed that conversely, the stable knock down of inherently high levels of AhR in MDA-MB-231 human metastatic breast cancer cell line has resulted in attenuation of the tumorigenic properties in vitro, including proliferation, anchorage independent growth and migration while it enhanced their apoptosis. Testing the cells with depleted AhR for their ability to form tumors and metastasize in nude mouse xenograft model of breast cancer, we uncovered that lack of AhR has resulted in substantial reduction of the orthotopic xenograft tumor growth and experimental lung metastasis [25] (Figure 2), underscoring the critical role of AhR in driving both the tumor survival and metastasis.
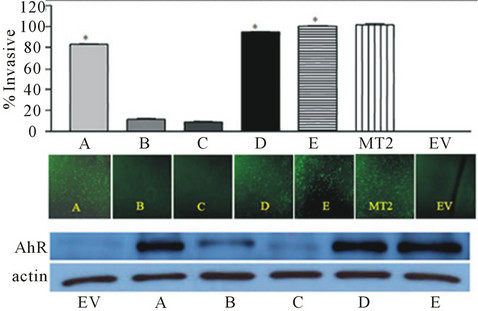
Figure 1. AhR is ectopically over expressed in clones (A-E) of an immortalized normal mammary epithelial cells (EV). Over expression of AhR induced invasion to a degree that correlated to the increase in AhR expression (from Brooks et al., 2011, with permission).
2.3. Constitutive AhR Signaling
Our findings and others clearly affirm the notion of AhR functioning in the progression of breast and other types of cancers, independent of its known ligands. Structurally, AhR contains both nuclear localization signal and nuclear export signal that are required for nuclear-cytoplasmic shuttling of AhR. Nucleocytoplasmic shuttling of AhR is required for inducible expression of CYP1A1 [26] . AhR responsive gene CYP1B1 is expressed in non-small cell lung cancer cells as well as prostate cancer cell lines in the absence of an exogenous ligand. In both cases, CYP1B1 expression was accompanied by increased AhR expression and constitutive activity of the receptor [27,28] (Figure 3). Depletion of AhR protein resulted in subsequent decrease in CYP1B1 expression, confirming that the basal CYP1B1 expression is regulated by constitutive AhR signaling. In addition, pre-malignant and malignant mammary tissues are reported to constitutively express CYP1 B1 mRNA. In these human and rodent mammary tumors, AhR was also over-expressed and constitutively active ADDIN EN.CITE ADDIN EN.CITE.DATA [23] . In addition, transient overexpression of AhR into an AhR null cell line also induced ligand independent transcriptional activity [28,29] .
The studies of the constitutive AhR signaling showed the differential CYP1B1 and not CYP1A1 expression is regulated by constitutively active AhR. The level of CY P1B1 and not CYP1A1 is more closely associated with AhR overexpression and constitutive activity. In the absence of exogenous ligands, AhR overexpression upregulated the expression of CYP1B1 in the early stage of lung adenocarcinoma [30] . However, suspension of wildtype Hepa-1 cells results in nuclear localization and activation of AhR to enhance expression of CYP1A1 [31]. In addition, loss of cell to cell contact experienced at low cell density also induces AhR transcriptional activity. CY P1B1 reporter activity in cells with loss of contact at low density was comparable to the level of activity produced following TCDD exposure. This process, however, was attenuated by depletion of intracellular calcium [32] , pointing to the calcium role in this process.
We reported previously that activation of calcium-dependent calpain was necessary to mediate ligand-induced activation of AhR. Calpain is a member of a family of cytosolic calcium dependent cysteine proteases known to be involved in a number of cellular processes. Regulation of AhR transcriptional activity has been shown to require calcium changes induced by ligand activation. Results indicate that nuclear accumulation of AhR is dependent on calpain activity. Inhibition of calpain activity with a specific inhibitors blocks transcriptional activity of AhR [33] . Furthermore, chemopreventive agent oltipraz which increases intracellular calcium, induces expression of AhR responsive gene, CYP1A1. Data presented demonstrated that oltipraz induces AhR nuclear translocation through activation of calpain [34] (Figure 4). Additional reports confirmed the enhanced elevation of intracellular calcium in response to AhR activation is associated with enhanced activity of the Ca2+/calmodulin (CAM)-dependent protein kinase (CaMK) pathway. In this set of studies, CaMK1α inhibition or knockdown inhibited TCDD induced nuclear translocation of AhR [35] .
Although any of these processes could account for the AhR constitutive activation, an endogenous ligand has long been sought after, and many candidates have been reported. The indole metabolite, indoxyl 3-sulfate (I3S) has been identified as a potent endogenous ligand that activates AhR at nanomolar concentrations in primary human hepatocytes [36] . Kynurenic acid, a tryptophan metabolite of the indoleamine-2,3-dioxygenase pathway has also been identified as an endogenous AhR ligand [37] . Competition binding assays revealed these metabolites as true ligands for AhR but the physiological relevance of their expression and activity in breast cancer needs further investigation. Increased understanding of the role of these potential endogenous ligands in breast cancer cells could provide additional insights on the role of AhR in mediating breast cancer progression.
2.4. The Cross Talk between AhR and Estrogen Receptor (ER)
Several studies indicate that constitutive AhR signaling is required for crosstalk with ER. Estrogen-induced activation of BRCA-1 transcription requires unliganded AhR binding to the BRCA-1 promoter. It has also been reported that a physical interaction between the hormoneactivated ER and constitutively active AhR cooperate to induce estrogen regulated genes ADDIN EN.CITE ADDIN EN.CITE.DATA [37] , whereas activation of AhR by TCDD ablates estrogen induced BRCA-1 transcription [38] . In line of these findings, benzo [a] pyrene (B[a]P), an AhR ligand, also inhibited BRCA-1
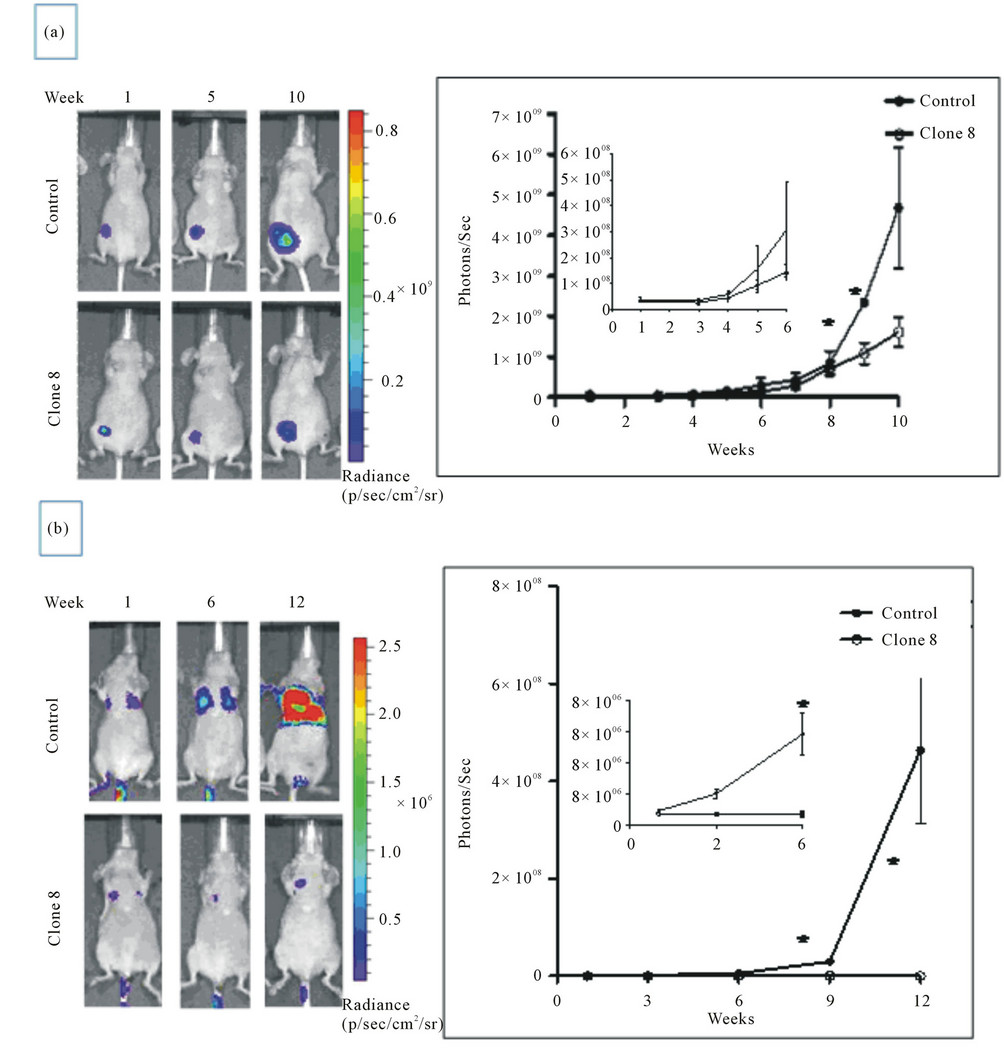
Figure 2. AhR knockdown reduces orthotopic growth of MDA-MB-231 cells in xenograft nude mice model (a) and reduces lung metastas is in nude mice experimental metastasis model (b) (from Goode et al., 2013, with permission).
expression in MCF-7 breast cancer cells and reduced these cell proliferation in a time and dose dependent manner [39] . A constitutively active mutant of AhR that was designed to mimic continuous TCDD activation also inhibited expression of estrogen-dependent cathepsin D and attenuated the estrogen-induced growth of MCF-7 human breast cancer cells [40] . Collectively, these studies indicate that constitutive activation of AhR is estrogenic while ligand activation of AhR appears to have antiestrogenic effects.
Several reports documented the crosstalk between the ER and AhR ligand-induced signaling pathways. Suppression subtractive hybridization studies using MCF-7 cells identified 33 genes that are induced by estrogen and inhibited by AhR agonist [41]. The AhR agonist-induced decline in estrogen-induced gene expression is a result of AhR inhibition of ER binding to ERE and targeting of ER to proteasome degradation [42] . Mutant cell lines deficient
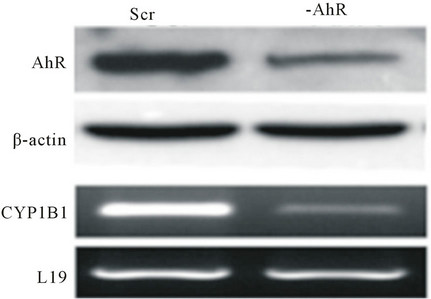
Figure 3. Prostate cancer cells transfected with a scrambled sequence (SCR) have high basal CYP1B1 expression shRNA reduction in AhR protein expression (-AhR) decreases CYP1B1 Mrna levels (from Tran et al., 2013, with permission).
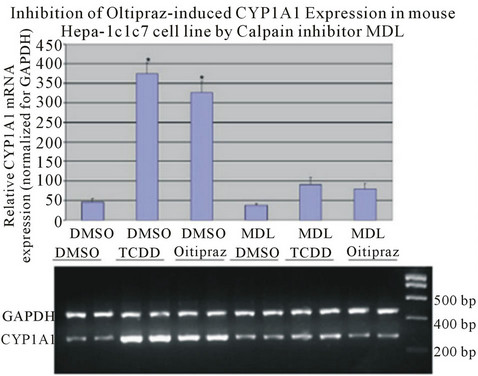
Figure 4. Oltipraz induced CYP1A1 expression to a level comparable to TCDD induction. Oltipraz-induced CYP1A1 expression is inhibited by calpain inhibitor, MDL 28170, in Hepa-1, a murine hepatoma cell line (from Dale & Eltom, 2006, with permission).
in AhR do not retain the ability to inhibit estrogen induced gene expression upon TCDD exposure [43]. Circulating levels of estrogen are significantly decreased by exposure to TCDD in pregnant rats [44] , due to induction of CYP1 B1 and CYP1A1 which metabolizes estrogens [45,46] .
2.5. AhR Antagonists/Agonists Protect against Tumor Progression
Antagonists for the AhR appear to have protective effects against carcinogen-mediated tumor initiation. Epigallocatechin-3 gallate (EGCG), a green tea polyphenol, reversed epithelial to mesenchymal transition (EMT) in DMBA treated mammary tumor cells. EGCG also reduced levels of c-Rel and the protein kinase CK2 [47] . Considering AhR is a known inducer of c-Rel and EGCG reduced expression of this target gene by AhR ligand DMBA, EGCG is suggested to inhibit EMT through direct inhibition of AhR signaling.
Genistein, a soy phytoestrogen, reduced the overall rate of DMBA induced mammary tumors. The treated rats appeared with larger mammary glands and increased terminal ducts as a result of increased proliferation [48]. Furthermore, genistein was able to inhibit estrogenic effects and induction of cell proliferation by ginsenoside Rg1 in MCF-7 cells [49] . A separate screening for potential AhR antagonist revealed significant suppression of dioxin induced AhR activation by genistein [50] . The studies were performed using Hepa-1c1c7 cells stably transfected with a secreated alkaline phosphatase (SEAP) gene under the control of the XRE/DRE consensus sequences. The established clones secreated SEAP following stimulation with TCDD in a dose dependent manner. Inhibition of SEAP secretion by genistein, therefore, confirms competitive inhibition of TCDD binding to XREs.
Resveratrol is another natural chemopreventive agent that was identified as an antagonist of the AhR [51]. Stilbene derivatives of resveratrol have been developed with high affinity for the AhR [52] . These are of particular interest due to reports that pterostilbene, a natural stilbene isolated from blueberries, has anticancer properties. Pterostilbene inhibited tumor associated macrophage (TAM) induced-migration and invasion of MCF-7 and MDAMB-231 breast cancer cells. When the breast cancer cells were co-cultured with TAMs, they expressed increased levels of HIF-1α and NF-κB. Pterostilbene inhibited the TAM induced increase in NF-κB. Pterostilbene also suppressed tumor formation in mice inoculated with TAM co-cultured MDA-MB-231 cells [53]. Considering the established role for AhR in modulating NF-κB signaling, it can be surmised that this is an AhR dependent effect.
Ginkgo biloba extracts are known to have estrogenic activity [54] . A CYP1A1 dependent EROD assay revealed increased activity following incubation of MCF-7 cells with ginkgo biloba. CYP1A1 mRNA levels were also elevated following exposure to ginkgo biloba, indicating activation of the AhR [55], and confirming that ginkgo biloba is a natural agonist for the AhR.
2.6. AhR as a Transcription Regulator
AhR represents an attractive druggable target due to its ability to regulate a wide range of physiological processes. As a transcription factor, AhR affects a number of genes besides those involved in xenobiotic metabolism. Evidence shows that the four and a half LIM domain 2 (FHL2), a transcriptional coactivator, interacts directly with AhR in MCF7 and PC3 cells to enhance AhR transcriptional activity. However, FHL2 suppressed AhR activity in T47D and LNCaP cells. It’s worth noting that previous reports have shown constitutive expression of CYP1B1 in MCF-7 and PC3 cells while ligand activation is required in T47D and LNCaP cells. These observations suggest that overexpression of FHL2 is required for ligand-independent signaling of AhR to enhance transcription [56]. AhR can also interact with Sp1 proteins to regulate basal expression of some genes; it appears that both the GC-rich motif that bind Sp1 protein and adjacent dioxin responsive elements (DRE) are required for maximal basal expression of Sp1 regulated genes [57] .
Altered mammary gland development resulting from in utero exposure to AhR ligands is the result of a direct effect on mammary epithelial cells that includes alterations of cell cycle regulator, cyclin D1. Cyclin D1 levels were also decreased in mammary epithelial cells isolated from mice exposed to TCDD [58]. AhR null cells have decreased expression of Cdc2 and Plk, two kinases important for G2/M cell cycle transition [59] . Mouse hepatoma cells deficient in AhR showed decrease proliferation resulting from a prolonged G1 phase [60] . Multiple A549 clones overexpressing AhR have increased proliferation rates proportional to the amounts of AhR [61]. Increases in TGF-β in AhR null cells appear to be the primary factor that causes low proliferation, thus AhR depletion in fibroblast resulted in increased TGF-β gene expression accompanied by decreased proliferation [62] . These studies collectively reveal estrogen-independent mechanisms by which AhR may regulate cell cycle progression.
AhR regulates expression of the zinc finger transcription factor slug which is critical for the induction of epithelial-mesenchymal transition (EMT). AhR directly binds to XREs in the promoter region to enhance transcription of slug [63] . Activation of AhR also represses T-cadherin expression which contains a XRE in the 5’ untranslated region [64] . Therefore, AhR may affect cell adhesion and migration through enhancement of slug and T-cadherin expression. Depletion of AhR expression in mouse embryonic fibroblast results in cytoskeleton alterations due to downregulation of Vav3 expression. Vav3 is a guanosine diphosphate/guanosine triphosphate exchange factor for Rho/Rac GTPases and provides additional insight into AhR’s regulation of cell shape, adhesion and migration [65] . As with slug and T-cadherin, AhR regulates Vav3 signaling by direct binding to the promoter region.
Matrix metalloproteinases (MMP) are key players in cancer invasion and metastasis and provide a possible mechanism by which AhR may modulate the invasive potential of cancer cells. Activation of AhR induces transcription of MMP-9 in advanced prostate cancer cells [66]. TCDD induces expression of MMP-1 through two AP-1 elements in the promoter of the MMP-1 gene [67] . Physical interaction between the RelA subunit of NF-κB and AhR increases transcription of the c-myc oncogene [68] . Modulation of c-myc expression could allow AhR to regulate neoplastic transformation.
3. Closing Summary Statement
There is a growing practice of molecularly targeted therapies in oncology for the treatment of malignancy. We now have remarkable success with agents that enable disease specific treatment with reduced normal tissue toxicity. However, as more and more molecularly targeted agents enter clinical evaluation, problems of more clinical remissions are arising, limiting the utility of a targeted agent within certain patient population. Therefore, identification of novel therapeutic targets is essential to combat breast cancers, especially those lacking estrogen receptor/progesterone receptor and ErbB2 receptor (triple negative breast cancer). Interestingly, many of the triple negative breast cancers cells have increased expression of AhR protein [22].
As increased amount of AhR protein contributes to tumor cell aggressiveness and survival, depletion of AhR reduced orthotopic xenograft tumor growth and metastasis in vivo; making it an ideal candidate for targeted therapy. More relevantly, depletion of AhR in metastatic breast cancer cell line potentiates the efficacy of chemotherapeutic agents and ionizing radiation, increasing the percentage of cells undergoing apoptosis in response to both treatment modalities [25]. As challenges to targeted therapies include acquired and primary resistance, targeting AhR could be a possible way to circumvent the emergence of targeted therapy resistance and cancer recurrence.
The development of selective AhR modulator (SAhRM) could prove beneficial in preventing breast cancer progression and/or metastasis. Such SAhRM include 3,3’- Diindolylmethane (DIM), which was shown to inhibit cell proliferation by inducing apoptosis and delaying cell cycle progression [69] . Not only can the AhR be targeted independently, it can be targeted in combination with other cancer treatments, such as chemotherapy or radiation therapy. As better understanding of AhR activity in breast cancer has shed light on some aspects of AhR signaling, identifying downstream targets will constitute even better option for targeted therapy, given the essential role of AhR in maintenance physiological functions. In regards to the role of AhR in breast cancer, large clinical studies as well as further investigations into the molecular mechanism of AhR function are essential. Early detection and increasing the list of therapeutic targets remains essential as the fight against breast cancer continues.
4. Acknowledgements
We acknowledge our research funding from NIH grant: G12 RR003062 (J.B.P.); NIH training grants: R25GM 059994, T32HL007735 (G.D.G.), NIH grants: G12RR 003032 and SC1CA1553326 (S.E.E.).
REFERENCES
- C. DeSantis, R. Siegel, P. Bandi and A. Jemal, “Breast Cancer Statistics, 2011,” CA: A Cancer Journal for Clinicians, Vol. 61, No. 6, 2011, pp. 409-418. doi:10.3322/caac.20134
- R. Alteri, P. Bandi, L. Brinton, C. Casares, V. Cokkinides, T. Gansler, et al., “Breast Cancer Facts & Figures 2011- 2012,” American Cancer Society, Inc., Atlanta, 2011.
- B. D. Robinson, G. L. Sica, Y. F. Liu, T. E. Rohan, F. B. Gertler, J. S. Condeelis, et al., “Tumor Microenvironment of Metastasis in Human Breast Carcinoma: A Potential Prognostic Marker Linked to Hematogenous Dissemination,” Clinical Cancer Research, Vol. 15, No. 7, 2009, pp. 2433-2441. doi:10.1158/1078-0432.CCR-08-2179
- M. V. Blagosklonny, “Why Therapeutic Response May Not Prolong the Life of a Cancer Patient: Selection for Oncogenic Resistance,” Cell Cycle, Vol. 4, No. 12, 2005, pp. 1693-1698. doi:10.4161/cc.4.12.2259
- M. Parton, M. Dowsett and I. Smith, “Studies of Apoptosis in Breast Cancer,” British Medical Journal, Vol. 322, No. 7301, 2001, pp. 1528-1532. doi:10.1136/bmj.322.7301.1528
- K. M. Burbach, A. Poland and C. A. Bradfield, “Cloning of the Ah-Receptor cDNA Reveals a Distinctive LigandActivated Transcription Factor,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 89, No. 17, 1992, pp. 8185-8189. doi:10.1073/pnas.89.17.8185
- J. C. Rowlands and J. A. Gustafsson, “Aryl Hydrocarbon Receptor-Mediated Signal Transduction,” Critical Reviews in Toxicology, Vol. 27, No. 2, 1997, pp. 109-134. doi:10.3109/10408449709021615
- A. Poland and J. C. Knutson, “2,3,7,8-Tetra Chlorodibenzo-P-Dioxin and Related Halogenated Aromatic Hydrocarbons: Examination of the Mechanism of Toxicity,” Annual Review of Pharmacology and Toxicology, Vol. 22, 1982, pp. 517-554. doi:10.1146/annurev.pa.22.040182.002505
- R. Barouki, X. Coumoul and P. M. Fernandez-Salguero, “The Aryl Hydrocarbon Receptor, More than a Xenobiotic-Interacting Protein,” FEBS Letters, Vol. 581, No. 19, 2007, pp. 3608-3615. doi:10.1016/j.febslet.2007.03.046
- D. H. Sherr, “Another Important Biological Function for the Aryl Hydrocarbon Receptor,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 31, No. 6, 2011, pp. 1247-1248. doi:10.1161/ATVBAHA.111.227553
- M. M. Richert, K. L. Schwertfeger, J. W. Ryder and S. M. Anderson, “An Atlas of Mouse Mammary Gland Development,” Journal of Mammary Gland Biology and Neoplasia, Vol. 5, No. 2, 2000, pp. 227-241. doi:10.1023/A:1026499523505
- B. D. Abbott, L. S. Birnbaum and G. H. Perdew, “Developmental Expression of Two Members of a New Class of Transcription Factors: I. Expression of Aryl Hydrocarbon Receptor in the C57BL/6N Mouse Embryo,” Developmental Dynamics, Vol. 204, No. 2, 1995, pp. 133-143. doi:10.1002/aja.1002040204
- L. J. Hushka, J. S. Williams and W. F. Greenlee, “Characterization of 2,3,7,8-Tetrachlorodibenzofuran-Dependent Suppression and AH Receptor Pathway Gene Expression in the Developing Mouse Mammary Gland,” Toxicology and Applied Pharmacology, Vol. 152, No. 1, 1998, pp. 200-210. doi:10.1006/taap.1998.8508
- B. J. Lew, L. L. Collins, M. A. O’Reilly and B. P. Lawrence, “Activation of the Aryl Hydrocarbon Receptor during Different Critical Windows in Pregnancy Alters Mammary Epithelial Cell Proliferation and Differentiation,” Toxicological Sciences, Vol. 111, No. 1, 2009, pp. 151-162. doi:10.1093/toxsci/kfp125
- S. E. Fenton, J. T. Hamm, L. S. Birnbaum and G. L. Youngblood, “Persistent Abnormalities in the Rat Mammary Gland Following Gestational and Lactational Exposure to 2,3,7,8-Tetrachlorodibenzo-P-Dioxin (TCDD),” Toxicological Sciences, Vol. 67, No. 1, 2002, pp. 63-74. doi:10.1093/toxsci/67.1.63
- B. C. Lewis, S. Hudgins, A. Lewis, K. Schorr, R. Sommer, R. E. Peterson, et al., “In Utero and Lactational Treatment with 2,3,7,8-Tetrachlorodibenzo-P-Dioxin Impairs Mammary Gland Differentiation but Does Not Block the Response to Exogenous Estrogen in the Postpubertal Female Rat,” Toxicological Sciences, Vol. 62, No. 1, 2001, pp. 46-53. doi:10.1093/toxsci/62.1.46
- B. A. Vorderstrasse, S. E. Fenton, A. A. Bohn, J. A. Cundiff and B. P. Lawrence, “A Novel Effect of Dioxin: Exposure during Pregnancy Severely Impairs Mammary Gland Differentiation,” Toxicological Sciences, Vol. 78, No. 2, 2004, pp. 248-257. doi:10.1093/toxsci/kfh062
- T. Wang, H. M. Gavin, V. M. Arlt, B. P. Lawrence, S. E. Fenton, D. Medina, et al., “Aryl Hydrocarbon Receptor Activation during Pregnancy, and in Adult Nulliparous Mice, Delays the Subsequent Development of DMBAInduced Mammary Tumors,” International Journal of Cancer, Vol. 128, No. 7, 2011, pp. 1509-1523. doi:10.1002/ijc.25493
- N. M. Brown, P. A. Manzolillo, J. X. Zhang, J. Wang and C. A. Lamartiniere, “Prenatal TCDD and Predisposition to Mammary Cancer in the Rat,” Carcinogenesis, Vol. 19, No. 9, 1998, pp. 1623-1629. doi:10.1093/carcin/19.9.1623
- I. A. Dialyna, D. A. Arvanitis and D. A. Spandidos, “Genetic Polymorphisms and Transcriptional Pattern Analysis of CYP1A1, AhR, GSTM1, GSTP1 and GSTT1 Genes in Breast Cancer,” International Journal of Molecular Medicine, Vol. 8, No. 1, 2001, pp. 79-87.
- A. F. Trombino, R. I. Near, R. A. Matulka, S. Yang, L. J. Hafer, P. A. Toselli, et al., “Expression of the Aryl Hydrocarbon Receptor/Transcription Factor (AhR) and AhRRegulated CYP1 Gene Transcripts in a Rat Model of Mammary Tumorigenesis,” Breast Cancer Research and Treatment, Vol. 63, No. 2, 2000, pp. 117-131. doi:10.1023/A:1006443104670
- S. E. Eltom, A. Gasmelseed and D. Saoudi-Guentri, “The Aryl Hydrocarbon Receptor Is Over-Expressed and Constitutively Activated in Advanced Breast Carcinoma,” Proceedings of the American Association for Cancer Research, 2006, p. 47. doi:10.1002/jcb.21630
- X. Yang, S. Solomon, L. R. Fraser, A. F. Trombino, D. Liu, G. E. Sonenshein, et al., “Constitutive Regulation of CYP1B1 by the Aryl Hydrocarbon Receptor (AhR) in Pre-Malignant and Malignant Mammary Tissue,” Journal of Cellular Biochemistry, Vol. 104, No. 2, 2008, pp. 402- 417. doi:10.2174/156800911795655967
- J. Brooks and S. E. Eltom, “Malignant Transformation of Mammary Epithelial Cells by Ectopic Overexpression of the Aryl Hydrocarbon Receptor,” Current Cancer Drug Targets, Vol. 11, No. 5, 2011, pp. 654-669.
- G. Goode, B. R. Ballard, H. C. Manning, M. L. Freeman, Y. Kang and S. E. Eltom, “Knockdown of Aberrantly Upregulated Aryl Hydrocarbon Receptor Reduces Tumor Growth and Metastasis of MDA-MB-231 Human Breast Cancer Cell Line,” International Journal of Cancer, 2013. doi:10.1002/ijc.28297
- T. Ikuta, T. Tachibana, J. Watanabe, M. Yoshida, Y. Yoneda and K. Kawajiri, “Nucleocytoplasmic Shuttling of the Aryl Hydrocarbon Receptor,” Journal of Biochemistry, Vol. 127, No. 3, 2000, pp. 503-509. doi:10.1093/oxfordjournals.jbchem.a022633
- P. Lin, H. Chang, W. L. Ho, M. H. Wu and J. M. Su, “Association of Aryl Hydrocarbon Receptor and Cytochrome P4501B1 Expressions in Human Non-Small Cell Lung Cancers,” Lung Cancer, Vol. 42, No. 3, 2003, pp. 255-261. doi:10.1016/S0169-5002(03)00359-3
- C. Tran, O. Richmond, L. Aaron and J. B. Powell, “Inhibition of Constitutive Aryl Hydrocarbon Receptor (AhR) Signaling Attenuates Androgen Independent Signaling and Growth in (C4-2) Prostate Cancer Cells,” Biochemical Pharmacology, Vol. 85, No. 6, 2013, pp. 753-762. doi:10.1016/j.bcp.2012.12.010
- C. Y. Chang and A. Puga, “Constitutive Activation of the Aromatic Hydrocarbon Receptor,” Molecular and Cellular Biology, Vol. 18, No. 1, 1998, pp. 525-535.
- J. T. Chang, H. Chang, P. H. Chen, S. L. Lin and P. Lin, “Requirement of Aryl Hydrocarbon Receptor Overexpression for CYP1B1 Up-Regulation and Cell Growth in Human Lung Adenocarcinomas,” Clinical Cancer Research, Vol. 13, No. 1, 2007, pp. 38-45. doi:10.1158/1078-0432.CCR-06-1166
- C. M. Sadek and B. L. Allen-Hoffmann, “SuspensionMediated Induction of Hepa 1c1c7 Cyp1a-1 Expression Is Dependent on the Ah Receptor Signal Transduction Pathway,” Journal of Biological Chemistry, Vol. 269, No. 50, 1994, pp. 31505-31509.
- Y. C. Cho, W. Zheng and C. R. Jefcoate, “Disruption of Cell-Cell Contact Maximally but Transiently Activates AhR-Mediated Transcription in 10T1/2 Fibroblasts,” Toxicology and Applied Pharmacology, Vol. 199, No. 3, 2004, pp. 220-238. doi:10.1016/j.taap.2003.12.025
- Y. R. Dale and S. E. Eltom, “Calpain Mediates the Dioxin-Induced Activation and Down-Regulation of the Aryl Hydrocarbon Receptor,” Molecular Pharmacology, Vol. 70, No. 5, 2006, pp. 1481-1487. doi:10.1124/mol.106.027474
- Y. Dale and S. E. Eltom, “The Induction of CYP1A1 by Oltipraz Is Mediated through Calcium-Dependent-Calpain,” Toxicology Letters, Vol. 166, No. 2, 2006, pp. 150- 159. doi:10.1016/j.toxlet.2006.06.645
- P. Monteiro, D. Gilot, E. Le Ferrec, C. Rauch, D. Lagadic-Gossmann and O. Fardel, “Dioxin-Mediated Up-Regulation of Aryl Hydrocarbon Receptor Target Genes Is Dependent on the Calcium/Calmodulin/CaMKIalpha Pathway,” Molecular Pharmacology, Vol. 73, No. 3, 2008, pp. 769-777. doi:10.1124/mol.107.043125
- J. C. Schroeder, B. C. Dinatale, I. A. Murray, C. A. Flaveny, Q. Liu, E. M. Laurenzana, et al., “The Uremic Toxin 3-Indoxyl Sulfate Is a Potent Endogenous Agonist for the Human Aryl Hydrocarbon Receptor,” Biochemistry, Vol. 49, No. 2, 2010, pp. 393-400. doi:10.1021/bi901786x
- B. C. DiNatale, I. A. Murray, J. C. Schroeder, C. A. Flaveny, T. S. Lahoti, E. M. Laurenzana, et al., “Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand that Synergistically Induces Interleukin-6 in the Presence of Inflammatory Signaling,” Toxicological Sciences, Vol. 115, No. 1, 2010, pp. 89-97. doi:10.1093/toxsci/kfq024
- J. K. Hockings, P. A. Thorne, M. Q. Kemp, S. S. Morgan, O. Selmin and D. F. Romagnolo, “The Ligand Status of the Aromatic Hydrocarbon Receptor Modulates Transcriptional Activation of BRCA-1 Promoter by Estrogen,” Cancer Research, Vol. 66, No. 4, 2006, pp. 2224-2232. doi:10.1158/0008-5472.CAN-05-1619
- B. D. Jeffy, E. U. Schultz, O. Selmin, J. M. Gudas, G. T. Bowden and D. Romagnolo, “Inhibition of BRCA-1 Expression by Benzo[a]pyrene and Its Diol Epoxide,” Molecular Carcinogenesis, Vol. 26, No. 2, 1999, pp. 100- 118. doi:10.1002/(SICI)1098-2744(199910)26:2<100::AID-MC5>3.0.CO;2-1
- C. Kohle, I. Hassepass, B. S. Bock-Hennig, K. Walter Bock, L. Poellinger and J. McGuire, “Conditional Expression of a Constitutively Active Aryl Hydrocarbon Receptor in MCF-7 Human Breast Cancer Cells,” Archives of Biochemistry and Biophysics, Vol. 402, No. 2, 2002, pp. 172-179. doi:10.1016/S0003-9861(02)00076-0
- I. Chen, T. Hsieh, T. Thomas, S. Safe, I. Chen, T. Hsieh, et al., “Identification of Estrogen-Induced Genes Downregulated by AhR Agonists in MCF-7 Breast Cancer Cells Using Suppression Subtractive Hybridization,” Gene, Vol. 262, No. 1-2, 2001, pp. 207-214. doi:10.1016/S0378-1119(00)00530-8
- M. Wormke, M. Stoner, B. Saville, K. Walker, M. Abdelrahim, R. Burghardt, et al., “The Aryl Hydrocarbon Receptor Mediates Degradation of Estrogen Receptor Alpha through Activation of Proteasomes,” Molecular Cellular Biology, Vol. 23, No. 6, 2003, pp. 1843-1855. doi:10.1128/MCB.23.6.1843-1855.2003
- I. Kharat and F. Saatcioglu, “Antiestrogenic Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Are Mediated by Direct Transcriptional Interference with the Liganded Estrogen Receptor. Cross-Talk between Aryl Hydrocarbonand Estrogen-Mediated Signaling,” The Journal of Biological Chemistry, Vol. 271, No. 18, 1996, pp. 10533- 10537. doi:10.1074/jbc.271.18.10533
- C. L. Chaffin, R. E. Peterson and R. J. Hutz, “In Utero and Lactational Exposure of Female Holtzman Rats to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: Modulation of the Estrogen Signal,” Biology of Reproduction, Vol. 55, No. 1, 1996, pp. 62-67. doi:10.1095/biolreprod55.1.62
- D. C. Spink, D. W. Lincoln 2nd, H. W. Dickerman and J. F. Gierthy, “2,3,7,8-Tetra Chlorodibenzo-p-Dioxin Causes an Extensive Alteration of 17 Beta-Estradiol Metabolism in MCF-7 Breast Tumor Cells,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 87, No. 17, 1990, pp. 6917-6921. doi:10.1073/pnas.87.17.6917
- D. C. Spink, H. P. Eugster, D. W. Lincoln 2nd, J. D. Schuetz, E. G. Schuetz, J. A. Johnson, et al., “17 Beta-Estradiol Hydroxylation Catalyzed by Human Cytochrome P450 1A1: A Comparison of the Activities Induced by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in MCF-7 Cells with Those from Heterologous Expression of the cDNA,” Archives of Biochemistry and Biophysics, Vol. 293, No. 2, 1992, pp. 342-348. doi:10.1016/0003-9861(92)90404-K
- K. Belguise, S. Guo, S. Yang, A. E. Rogers, D. C. Seldin, D. H. Sherr, et al., “Green Tea Polyphenols Reverse Cooperation between c-Rel and CK2 that Induces the Aryl Hydrocarbon Receptor, Slug, and an Invasive Phenotype,” Cancer Research, Vol. 67, No. 24, 2007, pp. 11742-11750. doi:10.1158/0008-5472.CAN-07-2730
- C. A. Lamartiniere, J. B. Moore, N. M. Brown, R. Thompson, M. J. Hardin and S. Barnes, “Genistein Suppresses Mammary Cancer in Rats,” Carcinogenesis, Vol. 16, No. 11, 1995, pp. 2833-2840. doi:10.1093/carcin/16.11.2833
- W. S. Lau, W. F. Chen, R. Y. Chan, D. A. Guo and M. S. Wong, “Mitogen-Activated Protein Kinase (MAPK) Pathway Mediates the Oestrogen-Like Activities of Ginsenoside Rg1 in Human Breast Cancer (MCF-7) Cells,” British Journal of Pharmacology, Vol. 156, No. 7, 2009, pp. 1136-1146. doi:10.1111/j.1476-5381.2009.00123.x
- A. Kasai, N. Hiramatsu, K. Hayakawa, J. Yao and M. Kitamura, “Blockade of the Dioxin Pathway by Herbal Medicine Formula Bupleuri Minor: Identification of Active Entities for Suppression of AhR Activation,” Biological and Pharmaceutical Bulletin, Vol. 31, No. 5, 2008, pp. 838-846. doi:10.1248/bpb.31.838
- R. F. Casper, M. Quesne, I. M. Rogers, T. Shirota, A. Jolivet, E. Milgrom, et al., “Resveratrol Has Antagonist Activity on the Aryl Hydrocarbon Receptor: Implications for Prevention of Dioxin Toxicity,” Molecular Pharmacology, Vol. 56, No. 4, 1999, pp. 784-790.
- P. de Medina, R. Casper, J. F. Savouret and M. Poirot, “Synthesis and Biological Properties of New Stilbene Derivatives of Resveratrol as New Selective Aryl Hydrocarbon Modulators,” Journal of Medicinal Chemistry, Vol. 48, No. 1, 2005, pp. 287-291. doi:10.1021/jm0498194
- K. K. Mak, A. T. Wu, W. H. Lee, T. C. Chang, J. F. Chiou, L. S. Wang, et al., “Pterostilbene, a Bioactive Component of Blueberries, Suppresses the Generation of Breast Cancer Stem Cells within Tumor Microenvironment and Metastasis via Modulating NF-kappaB/microRNA 448 Circuit,” Molecular Nutrition & Food Research, Vol. 57, No. 7, 2013, pp. 1123-1134. doi:10.1002/mnfr.201200549
- S. M. Oh and K. H. Chung, “Estrogenic Activities of Ginkgo biloba Extracts,” Life Sciences, Vol. 74, No. 11, 2004, pp. 1325-1335. doi:10.1016/j.lfs.2003.06.045
- S. M. Oh and K. H. Chung, “Antiestrogenic Activities of Ginkgo biloba Extracts,” The Journal of Steroid Biochemistry and Molecular Biology, Vol. 100, No. 4-5, 2006, pp. 167-176. doi:10.1016/j.jsbmb.2006.04.007
- A. Kollara and T. J. Brown, “Modulation of Aryl Hydrocarbon Receptor Activity by Four and a Half LIM Domain 2,” The International Journal of Biochemistry & Cell Biology, Vol. 41, No. 5, 2009, pp. 1182-1188. doi:10.1016/j.biocel.2008.10.019
- F. Wang, W. Wang and S. Safe, “Regulation of Constitutive Gene Expression through Interactions of Sp1 Protein with the Nuclear Aryl Hydrocarbon Receptor Complex,” Biochemistry, Vol. 38, No. 35, 1999, pp. 11490-11500. doi:10.1021/bi982578f
- B. J. Lew, R. Manickam and B. P. Lawrence, “Activation of the Aryl Hydrocarbon Receptor during Pregnancy in the Mouse Alters Mammary Development through Direct Effects on Stromal and Epithelial Tissues,” Biology of Reproduction, Vol. 84, No. 6, 2011, pp. 1094-1102. doi:10.1095/biolreprod.110.087544
- G. Elizondo, P. Fernandez-Salguero, M. S. Sheikh, G. Y. Kim, A. J. Fornace, K. S. Lee, et al., “Altered Cell Cycle Control at the G(2)/M Phases in Aryl Hydrocarbon Receptor-Null Embryo Fibroblast,” Molecular Pharmacology, Vol. 57, No. 5, 2000, pp. 1056-1063.
- [61] Q. Ma and J. P. Whitlock Jr., “The Aromatic Hydrocarbon Receptor Modulates the Hepa 1c1c7 Cell Cycle and Differentiated State Independently of Dioxin,” Molecular and Cellular Biology, Vol. 16, No. 5, 1996, pp. 2144- 2150.
- [62] S. Shimba, K. Komiyama, I. Moro and M. Tezuka, “Overexpression of the Aryl Hydrocarbon Receptor (AhR) Accelerates the Cell Proliferation of A549 Cells,” The Journal of Biochemistry, Vol. 132, No. 5, 2002, pp. 795- 802. doi:10.1093/oxfordjournals.jbchem.a003289
- [63] X. Chang, Y. Fan, S. Karyala, S. Schwemberger, C. R. Tomlinson, M. A. Sartor, et al., “Ligand-Independent Regulation of Transforming Growth Factor Beta1 Expression and Cell Cycle Progression by the Aryl Hydrocarbon Receptor,” Molecular and Cellular Biology, Vol. 27, No. 17, 2007, pp. 6127-6139. doi:10.1128/MCB.00323-07
- [64] T. Ikuta and K. Kawajiri, “Zinc Finger Transcription Factor Slug Is a Novel Target Gene of Aryl Hydrocarbon Receptor,” Experimental Cell Research, Vol. 312, No. 18, 2006, pp. 3585-3594. doi:10.1016/j.yexcr.2006.08.002
- [65] T. Niermann, S. Schmutz, P. Erne and T. Resink, “Aryl Hydrocarbon Receptor Ligands Repress T-Cadherin Expression in Vascular Smooth Muscle Cells,” Biochemical and Biophysical Research Communications, Vol. 300, No. 4, 2003, pp. 943-949. doi:10.1016/S0006-291X(02)02970-4
- [66] J. M. Carvajal-Gonzalez, S. Mulero-Navarro, A. C. Roman, V. Sauzeau, J. M. Merino, X. R. Bustelo, et al., “The Dioxin Receptor Regulates the Constitutive Expression of the Vav3 Proto-Oncogene and Modulates Cell Shape and Adhesion,” Molecular Biology of Cell, Vol. 20, No. 6, 2009, pp. 1715-1727. doi:10.1091/mbc.E08-05-0451
- [67] M. Haque, J. Francis and I. Sehgal, “Aryl Hydrocarbon Exposure Induces Expression of MMP-9 in Human Prostate Cancer Cell Lines,” Cancer Letters, Vol. 225, No. 1, 2005, pp. 159-166. doi:10.1016/j.canlet.2004.11.043
- [68] K. A. Murphy, C. M. Villano, R. Dorn and L. A. White, “Interaction between the Aryl Hydrocarbon Receptor and Retinoic Acid Pathways Increases Matrix Metalloproteinase-1 Expression in Keratinocytes,” The Journal of Biological Chemistry, Vol. 279, No. 24, 2004, pp. 25284- 25293. doi:10.1074/jbc.M402168200
- [69] D. W. Kim, L. Gazourian, S. A. Quadri, R. RomieuMourez, D. H. Sherr and G. E. Sonenshein, “The RelA NF-kappaB Subunit and the Aryl Hydrocarbon Receptor (AhR) Cooperate to Transactivate the c-myc Promoter in Mammary Cells,” Oncogene, Vol. 19, No. 48, 2000, pp. 5498-5506. doi:10.1038/sj.onc.1203945
- [70] X. F. Yin, J. Chen, W. Mao, Y. H. Wang and M. H. Chen, “A Selective Aryl Hydrocarbon Receptor Modulator 3,3’-Diindolylmethane Inhibits Gastric Cancer Cell Growth,” Journal of Experimental & Clinical Cancer Research, Vol. 31, 2012, p. 46. doi:10.1186/1756-9966-31-46
NOTES
*Corresponding author.