Health
Vol. 4 No. 11A (2012) , Article ID: 25009 , 9 pages DOI:10.4236/health.2012.431177
Recent advances in pharmacological therapy of Parkinson’s disease: Levodopa and carbidopa protective effects against DNA oxidative damage
![]()
1Department of Biology, University “Roma TRE”, Rome, Italy; *Corresponding Author: cozzi@uniroma3.it
2Department of Neurosciences, Institute of Neurology, Catholic University, Rome, Italy
3Don Carlo Gnocchi Onlus Foundation, Milan, Italy
4Radiation Biology and Human Health Unit (UT-BIORAD), Research Centre ENEA, Casaccia, Italy
Received 4 October 2012; revised 7 November 2012; accepted 13 November 2012
Keywords: Parkinson’s Disease; Levodopa; Carbidopa; Protective Effects
ABSTRACT
Parkinson’s disease is one of the most common progressive neurodegenerative disorder. It is characterized by the depletion of dopamine in the dopaminergic neurons of the striatum of the brain. Pharmacological treatment involves the administration of a dopamine precursor, levodopa (L-Dopa), which crosses the blood-brain barrier and replaces the loss of dopamine in the brain. One of the main drawbacks of the administration of L-Dopa is its short half-life, due to the presence of enzymes, such as the amino acid decarboxylase (AADC), able to rapidly metabolize L-Dopa. For this reason the intake of L-Dopa takes always place together with an AADC inhibitor such as carbidopa. The assumption of carbidopa increases L-Dopa half-life, but several patients need to increase the dosage of the pharmacological therapy during the progression of the disease. Another area of dispute is represented by the possibility that L-Dopa can exert a toxic effect on the cells, both in peripheral and in central nervous system, increasing the production of ROS following its conversion to dopamine. Past studies reported toxic effects of L-Dopa in vitro and show conflicting data in in vivo experiments. More recent studies have however shown that L-dopa may exert a protective and antioxidant effect on dopaminergic cells, and its combination with carbidopa in pharmacological treatment amplifies antioxidant capability.
1. INTRODUCTION
The first clinical features of Parkinson’s Disease (PD) were described and published by James Parkinson in 1817 [1]. Nowadays PD is the second most common progressive neurodegenerative disorder and its prevalence reaches 1% - 2% in people over the age of 50 [2].
Parkinson’s Disease is characterized by decreased levels of neurotransmitter dopamine (DA) in the striatum of the brain, due to the selective degeneration of the nigro-striatal DA neurons [3] and a progressive loss of DA in the basal ganglia. Pharmacological treatment aims to slow down neurodegeneration replacing the loss of dopamine, but trial with oral dopamine failed because it cannot cross the blood-brain barrier. Following from this, George Cotzias demonstrated that high doses of dopamine’s prodrug, levodopa (L-Dopa) promptly enhanced clinical function in PD patients [4] and corrected the mechanical disorders at the early stage of the disease [2]. To date L-Dopa is the most effective symptomatic agent in the treatment of PD. Since its production more than 30 years ago, L-Dopa therapy has provided marked symptomatic benefits to PD patients. Despite the positive effects, one of the problems of the treatment with L-Dopa alone is its low central nervous system (CNS) bioavailability because of rapid peripheral decarboxylation to dopamine [5,6]. Concomitant administration of a dopa decarboxylase (DDC) inhibitor, such as carbidopa, was later demonstrated to markedly increase L-Dopa CNS bioavailability [7,8]. L-Dopa administration is usually associated with peripheral DDC inhibitors to increase the amount of drug available to cross the blood brain barrier [9], however, the response duration to each dose shortens as the disease progresses [10].
Although L-Dopa remains at present the most powerful drug for the treatment of PD, different areas of controversy exist at least. The main unsolved question is about L-Dopa toxicity due to its oxidative metabolism, which generates reactive oxygen species (ROS). Pharmacological therapy is in the centre of two areas of controversity: whether L-Dopa is toxic and whether L-Dopa directly causes motor complications [11].
The purpose of this review is to present the latest experimental evidences about the effects of L-Dopa and carbidopa treatments.
2. L-DOPA METABOLISM AND OXIDATIVE STRESS
Dopamine production in neurons of the basal ganglia of the brain has an important role in coordinating complex movements. In DA neurons the amino acid tyrosine is converted into L-Dopa by the enzyme tyrosine hydroxylase. L-Dopa is metabolized by the enzyme, aromatic amino acid decarboxylase, to produce dopamine, which is finally sequestered into storage vesicles [12]. Dopamine is metabolized and inactivated in the postsynaptic cleft by the enzymes catechol-O-méthyl transferase (COMT) and mono-amine oxidase (MAO). COMT degrades dopamine by incorporating a methyl group into the catecholamine function. The MAO catalyses the oxidative deamination of the monoamine group (Figure 1). L-Dopa is a natural dopamine precursor that can cross the blood brain barrier to reach the brain and to be converted into dopamine [2], it replenishes the lack of DA in the striatum [13,14], and increases the DA content of the brain [15,16]. Biochemical studies have shown that a portion of administered L-Dopa may undergo decarboxylation to DA [17,18] both in peripheral district and in the striatum of the brain. Oxygen and semiquinone free radicals derived from DA autoxidation are highly reactive
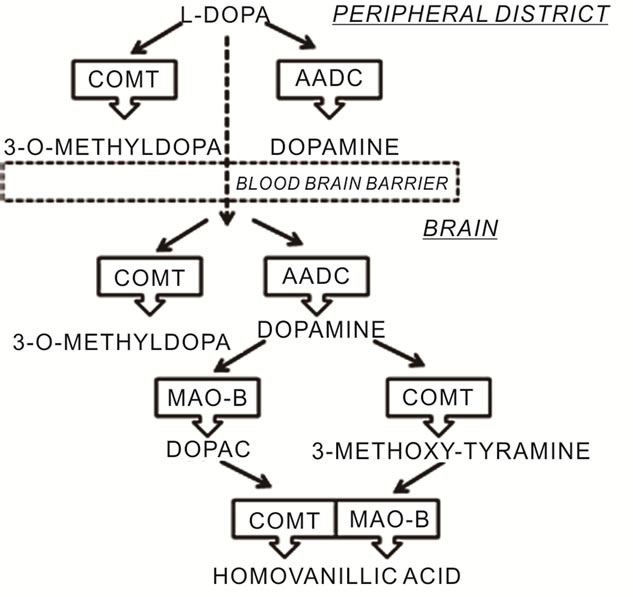
Figure 1. Metabolic pathways of L-Dopa.
and can potentially cause a site-specific oxidative damage [19-22]. The possibility that L-Dopa can induce reactive oxygen species (ROS) is important in PD because the substantia nigra pars compacta is in a state of oxidative stress: post-mortem analysis show increased levels of the pro-oxidant ferrous iron [23-25] and decreased mitochondrial complex I in PD patients [26,27]. Furthermore, there is evidence of oxidative damage to lipids [28], DNA [29] and proteins [30]. During DA metabolism, hydrogen peroxide (H2O2) is generated [31]: H2O2 could undergo autoxidation in the presence of high levels of iron, giving rise to highly toxic hydroxyl radicals [32]. For these reasons oxidative stress, resulting from the imbalance between ROS formation and antioxidant defenses, is thought to play a pivotal role in the pathogenesis of the disease [1,2] Increased markers of lipid peroxidation [24], protein nitration [33], DNA damage [29], decreased mitochondrial complex I activity [27] and lower amount of the reduced form of glutathione (GSH) [34] have all been identified in the SNpc of PD patients.
ROS can damage all biological macromolecules, including DNA, where for example deoxyguanosine is converted to 8-oxo-7,8-dihydro-deoxyguanosine (8-oxodG). This specific base modification is one of the most abundant products of oxidative damage to DNA and induces mutations through G to T transversion [35]. Oxidative DNA damage includes both oxidized purine and pyrimidine bases and structural DNA changes such as single or double DNA strand breakage, results in an elevated genomic damage, expressed as sister chromatid exchanges or chromosomal aberrations [36,37].
Based on these considerations it has been postulated that L-Dopa itself could be toxic to DA neurons [38,39], so that the chronic administration of L-Dopa would exacerbate the production and the accumulation of ROS leading neurons to death. Some studies suggest a toxic effect of L-Dopa in neuronal cells in vitro [40-42] even if the concentrations used in these experiments are higher than those reached in the plasma (10 - 20 µM) after one oral therapy intake [43-45]: in these concentrations L-Dopa is not toxic to cultured dopamine neurons [11] and nor toxicity has been confirmed in healthy rodents [46], nonhuman primates [47] or humans [48]. Moreover, recent clinical trials did not provide evidence for toxic effects of L-Dopa [49]. Indeed there is evidence suggesting that in some circumstances L-Dopa might be protective and have trophic effects [11]. Studies conducted using glia-conditioned media showed that L-Dopa can exert a neurotrophic effect manifested by an increase in cell survival and in growth of neurite [41,50-52]. In addition, exposure to low concentration of L-Dopa can protect dopamine neurons from subsequent exposure to pro-oxidants that would otherwise be toxic [53].
3. OPTIMIZATION OF PHARMACOLOGICAL THERAPY OF PARKINSON’S DISEASE: L-DOPA IN COMBINATION WITH CARBIDOPA
One of the fundamental problems of pharmacological therapy with L-Dopa is its short half-life. L-Dopa is absorbed in the proximal small intestine after oral intake and is transported across the blood-brain barrier by an active amino acid carrier.
L-Dopa is absorbed in the intestine quickly and completely, but bioavailability is less than 1% in the absence of aromatic amino acid decarboxylase (AADC) inhibitors. The half-life of a single standard oral dose of LDopa is approximately 1.5 hours in the presence of an AADC inhibitor and approximately 1 hour without AADC inhibitor [54,55]. Fluctuation in L-Dopa concentration leads to fluctuations in motor performance and to the development of drug-induced involuntary movements: dyskinesias is one of the main problems that occur in the later stages of the disease [56]. Because of these side effects it would be important monitor the levels of L-Dopa in the plasma of patients, in order to determine whether the motor fluctuations are predictable and avoidable through an individual optimization of drug therapy. Despite the use of other medications that increase the stimulation of dopamine receptors in the striatum, the differences in the doses required by patients are obvious and represent the most important factor for the improvement of therapy. Pharmacodynamic factors, such as receptor changes, and pharmacogenetic factors, such as genetic polymorphism in drug-metabolizing enzymes are important to study [56]. The total daily dosage of oral L-Dopa should be individualized, involving two parameters: the amount of each dose and the frequency of dose intakes [10]. The strategy to optimize therapy is to decrease the doses and increase the number of dose intakes [57]. However with the progression of the disease the threshold of L-Dopa concentration for therapeutic effect increases [55] and it is important that each dose is large enough to give an appropriate response [10].
All commercially available L-Dopa formulations now contain an AADC inhibitor: L-Dopa in combination with an inhibitor of AADC is the most effective treatment in the progression of PD [10]. The ratio of carbidopa to L-Dopa was initially 1:10, but was increased to 1:4 [58] because of pharmacokinetic advantages and decreased adverse effects. Time to reach peak plasma concentration may vary between patients and within patients, but it is usually reported to be about 0.5 - 2 hours [54].
Despite dopa-decarboxylase inhibitors were introduced in PD therapy for the first time several decades ago, the time of administration, the duration of the effect of L-Dopa, the dosage of dopa decarboxylase inhibitor request and the real effectiveness of carbidopa have not yet been well defined. [58,59]. Carbidopa is typically administered simultaneously with each dose of L-Dopa. This presumes that the onset of decarboxylase activity inhibition occurs quickly enough to prevent peripheral L-Dopa metabolism and this inhibition persists during the time course of effect of L-Dopa [60,61]. The plasma half-life of carbidopa is approximately 2.5 h and this correlates with the extent of decarboxylase activity inhibition [62-64].
The first marketed product containing a combination of L-Dopa and carbidopa was an immediate-release [3] oral dosage form under the trade name of Sinemet® [2].
4. OXIDATIVE STRESS AND DNA DAMAGE: EVIDENCES TO SUPPORT THE PROTECTIVE EFFECT OF L-DOPA
The presence of oxidative stress, resulting from an imbalance between the formation of ROS and the antioxidant defense system, has been reported in post-mortem tissues of patients with PD: a decrease activity of complex I of the mitochondrial respiratory chain [27] with a decrease in levels of reduced glutathione [34,65, 66] have been observed in the substantia nigra of patients with PD. Lipids [67], proteins [4] and DNA [68,69] can all be damaged by free radicals, and can be used as biomarkers for the quantification of the oxidative stress. The first study that investigated cytogenetic damage in patients with de novo PD using the micronucleus assay was published by Migliore and coworkers (2002) [70]. They performed micronuclei assay, FISH, standard and modified Comet assay on leukocytes of 20 patients with the novo sporadic PD and 16 control subjects showing that spontaneous micronuclei frequencies are higher in PD patients compared with control subjects. FISH analysis showed that the majority of micronuclei is constituted by acentric fragments, suggesting an increased incidence of chromosome breaks in the formation of micronuclei in PD patients. Comet assay showed an increase in the levels of single strand breaks that may well explain the increased centromere-negative micronuclei frequency observed in PD patients (Table 1).
To assess oxidative DNA damage was also performed the modified version of the comet assay. In this technique is used an enzyme (Endo III or FPG) that specifically induces lesions in DNA in correspondence of oxidized bases [71]. In this case Migliore and coworkers have shown that the level of strand breaks due to oxidized purines is higher in PD patients compared to controls.
Since ROS induce chromosomal aberrations with high efficiency, the authors conclude that the observed chromosomal damage in somatic cells in PD patients is due to a high and abnormal oxidative stress in these patients. Furthermore, these data confirmed the hypothesis that
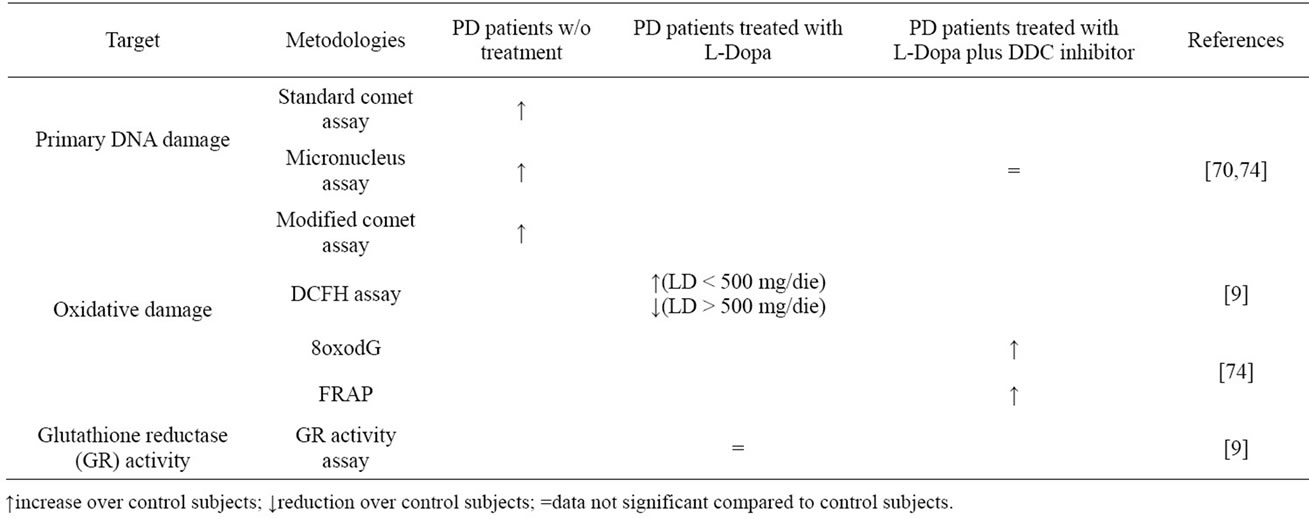
Table 1. Summary of experimental evidences.
oxidative damage to DNA also occurs at the peripheral level and demonstrated for the first time that the oxidative damage to DNA also occurs outside of the central nervous system in PD patients. The presence of DNA oxidative damage outside the CNS supports the hypothesis that a systemic derangement parallels neural abnormalities in PD patients [70].
In addition to these experimental evidences, Prigione et al., in 2006 [9], demonstrated a negative correlation between oxidative stress in peripheral blood mononuclear cells from PD patients and LD dosage. In this study PBMCs from treated patients and healthy subjects were use as dopaminergic non-neural cell model to study the ex-vivo relation between oxidative stress and LD intake by measuring ROS production and glutathione reductase (GR) activity. ROS levels in PBMCs were significantly higher in PD patients than in healthy controls nevertheless the daily dosage of LD showed significant negative correlation with ROS production in PBMCs and significant positive association with GR activity. To clarify whether the production of ROS depends on L-Dopa dosage, patients were divided into two groups: one taking more than 500 mg/day (L-Dopa > 500) and the other taking less than 500 mg/day (L-Dopa < 500). The first group does not present a significant increase in ROS production compared to the controls. Furthermore, LDopa > 500 significantly reduces the production of ROS with respect to L-Dopa < 500.
The detected increase in oxidative stress in PBMCs of PD patients seems to correlate to the intake of L-Dopa, which in fact seems to exert a protective function.
In addition, the group of PD patients treated with additional drugs (for example carbidopa) together with LDopa showed no differences in ROS production compared to patients treated with L-Dopa alone (Table 1). These results were still left open the debate on L-Dopa until 2009, when our research group [72] analyzed the presence of DNA damage in peripheral blood lymphocytes [6] isolated from blood samples of nine PD patients and nine matched controls, during a controlled dosage and washout of L-Dopa. We evaluated the oxidative DNA damage fluctuation after L-Dopa intake through the standard and the Fpg-modified version of Comet assay. L-Dopa intake was suspended the day before the samplings according to the washout procedure. Each patient underwent three consecutive venous blood samplings: the first sampling was performed in early morning (after 15 h of L-Dopa suspension) and the usual L-dopa therapy was administered immediately after. The second and the third samplings were performed 90 and 180 min, respectively, after the therapy, according to the LD halflife.
The group of PD patients analyzed after 15 hours washout of therapy showed high levels of DNA strand breaks compared to control subjects. After the intake of L-Dopa a progressive and significant reduction of DNA damage within 3 hours after administration (corresponding to the half-life of L-Dopa) was observed. The values of DNA damage analyzed in control subjects by standard comet assay remained unchanged in the three sampling times. To verify if the DNA damage in leukocytes of these patients was also due to oxidative stress, our research group performed a modified version of the comet assay by using lesion-specific enzymes able to induce strand breaks in the site of oxidized nucleosides. The modified version of comet assay confirmed that only a small portion of the DNA lesions are due to oxidative damage, while the total amount of DNA lesions decreases after the L-Dopa intake.
As regards the control subjects the observed variability likely reflects normal cellular metabolic activities and antioxidant levels in the blood. The concentrations of oxidative stress biomarkers in plasma and urine of healthy subjects follow a circadian rhythm, being very low at night and early in the morning, with an increasing trend toward the evening [73].
In 2010 Oli et al. [74] demonstrated no increased chromosomal damage in L-Dopa-treated patients with PD. In this study 18 PD patients were recruited: patients were treated with L-Dopa and a dopa decarboxilase inhibitor. Life partners of PD patients were also recruited in this study to form a control group. Oli and collaborators analyzed 8-oxodG in lymphocyte DNA and detected significantly elevated levels of oxidative DNA alterations in PD patients compared to the control group. Therefore, FRAP capacity (antioxidant capacity of the plasma) was significantly higher in PD patients than in controls. Analyzing micronucleus frequencies no difference between the groups was found (Table 1).
Results obtained from this study confirmed that the oxidative stress biomarker 8-oxo-dG is elevated in chronically L-Dopa-treated PD patients. A potential explanation of antioxidative capacity of plasma in PD patients and of low micronucleus frequency is that treatment with L-Dopa plus carbidopa in these patients could protect patients from PD-related additional chromosomal damage. This assumption of protective effect is confirmed by correlation between antioxidant FRAP values and daily L-Dopa plus carbidopa dosage. As the increase of cytosolic levels of dopamine induces formation of ROS, semiquinones and quinines [75], it is believed that the increase of 8-oxodG observed in PD patients is caused by the metabolism of dopamine itself, which also determines an increase in of 8-oxodG in PC12 cells [74]. On the basis of these experimental evidences our research group decided to investigate the in vitro effects of various L-Dopa and carbidopa concentrations, alone and in combination, in a human neuroblastoma cell line, SHSY5Y [76]. In particular, we analyzed the effects of L-Dopa treatments by evaluating DNA damage levels and investigating whether this damage was due to oxidative stress. We have analyzed the behavior of L-Dopa in the presence of oxidative stress exogenously induced by hydrogen peroxide and we the effect of carbidopa alone and in combination with L-Dopa, even in the presence of exogenous oxidative stress.
The results obtained from the standard comet assay showed that L-dopa induces a significant increase in DNA damage only at the highest dose used, but we can exclude that this damage is due to oxidative stress because neither the FPG modified-comet assay nor the analysis of intracellular ROS gave significant results. In addition it must be taken into account that the L-Dopa dose showing an increased DNA damage is much higher compared to that present in the cerebrospinal fluid after taking an oral dose of the drug [11].
To evaluate the protective effect of L-Dopa against oxidative stress we performed combined treatments with hydrogen peroxide both short term and 24 hours treatments. We reported a significant protective effect exerted by L-Dopa against H2O2-induced DNA damage at all used doses, particularly evident with the highest ones, showing a close relationship between L-Dopa concentrations and treatment times (Figure 2(a)). We also investigated the effects of carbidopa on neuroblastoma cells: carbidopa per se’ does not induce DNA damage but is effective in reducing the damage caused by reactive oxygen species (Figure 2(b)). Carbidopa in combination with L-Dopa (in a 4:1 ratio) emphasizes the protective effects of L-Dopa (Figure 3), decisively acting both in prolonging L-Dopa half-life as previously shown [2,77], and protecting cells from oxidative intermediate of LDopa metabolism. Previous in vivo studies had demonstrated that co-administration of carbidopa and L-Dopa blocks the generation of hydroxyl radicals due to dopa-
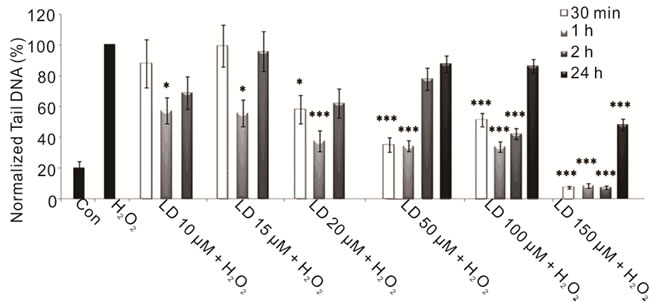 (a)
(a)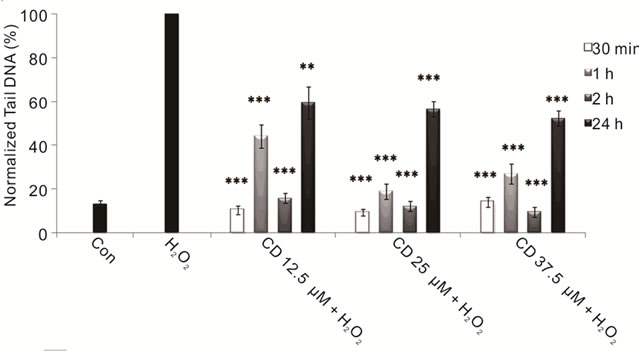 (b)
(b)
Figure 2. Protective effects of LD and CD in SH-SY5Y cells evaluated by alkaline comet assay. (a) Protective effect in SH-SY5Y cells after 30-min-, 1-, 2-, and 24-h treatments with various concentrations of LD in the presence of H2O2 is evaluated in terms of reduction of H2O2-induced DNA damage; (b) The protective effect in SH-SY5Y cells after 30-min-, 1-, 2-, and 24-h treatments with various concentrations of CD in the presence of H2O2 is evaluated in terms of reduction of H2O2- induced DNA damage. Values of Tail DNA are normalized with respect to the percentage of DNA damage induced by H2O2 single treatment (100%) and represent the mean results of three experiments ± SE (Standard Error). H2O2: 100 µM; 30-min treatment. *p < 0.05; **p < 0.01; ***p < 0.001 (at Mann-Whitney U test) treated cells versus H2O2-treated ones [76].
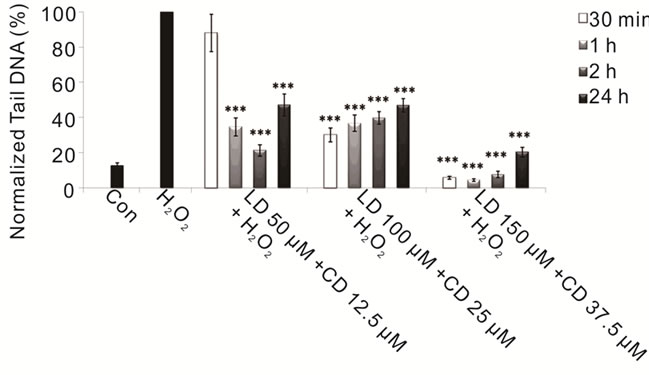
Figure 3. Protective effects of combined treatment LD/CD in a 4:1 ratio in SH-SY5Y cells evaluated by alkaline comet assay. The protective effect in SH-SY5Y cells after 30-min-, 1-, 2-, and 24-h treatments with various concentrations of LD/CD in the presence of H2O2 is evaluated in terms of reduction of H2O2-induced DNA damage. Values of Tail DNA are normalized with respect to the percentage of DNA damage induced by H2O2 single treatment (100%) and represent the mean results of three experiments ± SE (Standard Error). H2O2: 100 µM; 30- min treatment. ***p < 0.001 (at Mann-Whitney U test) treated cells versus H2O2-treated ones [76].
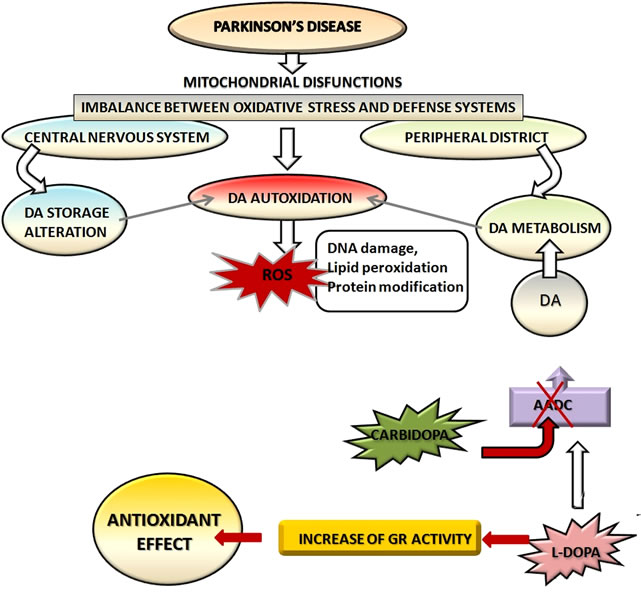
Figure 4. Metabolic pathways of L-Dopa.
mine autoxidation [22]. Therefore, from our results we can deduce that carbidopa can act by inhibiting the formation of intermediates of dopamine metabolism.
5. CONCLUSIONS
Parkinson’s disease is one of the most common agerelated diseases, so the improvement of pharmacological therapy is of great interest.
Since L-Dopa has begun to be used to relieve the symptoms of neurodegeneration [4] it showed conflicting results about its neurotoxicity [40,41] and its ability to increase oxidative stress inside the cell [28-30,42]. The most recent experimental evidences have shown that Parkinson’s disease causes an imbalance between oxidative stress and defense systems in the cell, both at the level of the central nervous system and in the peripheral districts [25-30] (Figure 4).
This imbalance causes DNA damage in PD patients [70] and increases levels of cellular ROS [9,74]. Apart from baseline damage present in PD patients, L-Dopa does not seem to have an adverse effect, but rather a protective effect both on lymphocytes [72] and on human dopaminergic cell line [76].
The concomitant use of drugs that inhibit the peripheral metabolism of L-dopa can increase its half-life and bioavailability: in particular carbidopa amplifies the antioxidant effects of L-Dopa (Figure 4) and has beneficial effects in both the peripheral districts [74] and human dopaminergic cells [76]. In the assessment of the effects of the pharmacological therapy is also important to consider the physio-pathological and genetic variables among patients. In this regard it’s fundamental to investigate effects of the substances used in combination with L-Dopa in order to restrict the side effects and to improve drug therapy basing on the individual characteristics of each patient.
![]()
![]()
REFERENCES
- Parkinson, J. (1817) An essay on the shaking palsy. SherWood, Neely and Jones, London.
- Goole, J., Amighi K. (2009) Levodopa delivery systems for the treatment of Parkinson’s disease: An overview. International Journal of Pharmaceutics, 380, 1-15. doi:10.1016/j.ijpharm.2009.07.026
- Nagatsua, T., Sawadab, M. (2009) L-dopa therapy for Parkinson’s disease: Past, present, and future. Parkinsonism & Related Disorders, 15, S3-S8. doi:10.1016/S1353-8020(09)70004-5
- Cotzias, C.G., Van Woert, M.H., Schiffer, L.M. (1967) Aromatico amino acids and modification of Parkinsonism. The New England Journal of Medicine, 276, 374-379. doi:10.1056/NEJM196702162760703
- Nutt, J.G., Woodward, W.R., Hammerstad, J.P., Carter, J.H. and Anderson, J.L. (1984) The “on-off” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. The New England Journal of Medicine, 310, 483-488. doi:10.1056/NEJM198402233100802
- Olanow, C.W. (2004) The scientific basis for the current treatment of Parkinson’s disease. Annual Reviews of Medicine, 55, 41-60. doi:10.1146/annurev.med.55.091902.104422
- Boomsma, F., Meerwaldt, J.D., Intveld, A.J.M., Hovestadt, A. and Schalekamp, M. (1989) Treatment of idiopathic Parkinsonism with L-Dopa in the absence and presence of decarboxylase inhibitors—Effects on plasma-levels of L-Dopa, Dopa decarboxylase, catecholamines and 3-Omethyl-dopa. Journal of Neurology, 236, 223-230. doi:10.1007/BF00314504
- Hauser, R.A., Panisset, M., Abbruzzese, G., Mancione, L., Dronamraju, N., Kakarieka, A., et al. (2009) Doubleblind trial of levodopa/carbidopa/entacapone versus levodopa/carbidopa in early Parkinson’s disease. Movement Disorders, 24, 541-550. doi:10.1002/mds.22343
- Prigione, A., Begni, B., Galbussera, A., Beretta, S., Brighina, L., Garofalo, R., et al. (2006) Oxidative stress in peripheral blood mononuclear cells from patients with Parkinson’s disease: Negative correlation with levodopa dosage. Neurobiology of Disease, 23, 36-43. doi:10.1016/j.nbd.2006.01.013
- Nyholm, D. (2006) Pharmacokinetic optimisation in the treatment of Parkinson’s disease: An update. Clinical Pharmacokinetics, 45, 109-136. doi:10.2165/00003088-200645020-00001
- Olanow, C.W., Agid, Y., Mizuno, Y., Albanese, A., Bonucelli, U., Damier, P., et al. (2004) Levodopa in the treatment of Parkinson’s disease: Current controversies. Movement Disorders, 19, 997-1005. doi:10.1002/mds.20243
- Lawlor, P.A. and During, M.J. (2004) Gene therapy for Parkinson’s disease. Expert Reviews in Molecular Medicine, 6, 1-18. doi:10.1017/S146239940400746X
- Rinne, U.K., Sonninen, V. and Hyyppa, M. (1971) Effect of L-dopa on brain monoamines and their metabolites in Parkinson's disease. Life Sciences, 10, 549-557. doi:10.1016/0024-3205(71)90040-3
- Snyder, G.L. and Zigmond, M.J. (1990) The effects of L-dopa on in vitro dopamine release from striatum. Brain Research, 508, 181-187. doi:10.1016/0006-8993(90)90394-Q
- Lloyd, K.G., Davidson, L. and Hornykiewicz, O. (1975) The neurochemistry of Parkinson’s disease: Effect of L-dopa therapy. Journal of Pharmacology and Experimental Therapeutics, 195, 453-464.
- Schoenfeld, R.I. and Uretsky, N.J. (1973) Enhancement by 6-hydroxydopamine of the effects of DOPA upon the motor activity of rats. Journal of Pharmacology and Experimental Therapeutics, 186, 616-624.
- Arai, R., Karasawa, N., Geffard, M. and Nagatsu, I. (1995) L-dopa is converted to dopamine in serotonergic fibers of the striatum of the rat—A double-labeling immunofluorescence study. Neuroscience Letters, 195, 195-198. doi:10.1016/0304-3940(95)11817-G
- Everett, G.M. and Borcherding, J.W. (1970) L-DOPA: Effect on concentrations of dopamine, norepinephrine, and serotonin in brains of mice. Science, 168, 849-850.
- Fornstedt, B., Brun, A., Rosengren, E. and Carlsson, A. (1989) The apparent autoxidation rate of catechols in dopamine-rich regions of human brains increases with the degree of depigmentation of substantia nigra. Journal of Neural Transmission Parkinson’s Disease and Dementia Section, 1, 279-295. doi:10.1007/BF02263482
- Fornstedt, B., Pileblad, E. and Carlsson, A. (1990) In vivo autoxidation of dopamine in guinea-pig striatum increases with age. Journal of Neurochemistry, 55, 655-659. doi:10.1111/j.1471-4159.1990.tb04183.x
- Halliwell, B. (1989) Oxidants and the central nervous system: Some fundamental questions. Is oxidant damage relevant to Parkinson's disease, Alzheimer’s disease, traumatic injury or stroke? Acta Neurologica Scandinavica, Supplementum, 126, 23-33. doi:10.1111/j.1600-0404.1989.tb01779.x
- Obata, T. and Yamanaka, Y. (1996) Protective effect of carbidopa on hydroxyl radical generation in the rat striatum by dopamine. Neuroscience Letters, 27, 13-16.
- Dexter, D.T., Wells, F.R., Agid, F., Agid, Y., Lees, A.J., Jenner, P., et al. (1987) Increased nigral iron content in postmortem Parkinsonian brain. Lancet, 2, 1219-1220. doi:10.1016/S0140-6736(87)91361-4
- Dexter, D.T., Carter, C.J., Wells, F.R., Javoyagid, F., Agid, Y., Lees, A., et al. (1989) Basal lipid-peroxidation in substantia nigra is increased in parkinsons-disease. Journal of Neurochemistry, 52, 381-389. doi:10.1111/j.1471-4159.1989.tb09133.x
- Sofic, E., Paulus, W., Jellinger, K., Riederer, P. and Youdim, M.B.H. (1991) Selective increase of iron in substantia-nigra zona compacta of Parkinsonian brains. Journal of Neurochemistry, 56, 978-982. doi:10.1111/j.1471-4159.1991.tb02017.x
- Mizuno, Y., Ohta, S., Tanaka, M., Takamiya, S., Suzuki, K., Sato, T., et al. (1989) Deficiencies in complex-i subunits of the respiratory-chain in Parkinson’s-disease. Biochemical and Biophysical Research Communications, 163, 1450-1455. doi:10.1016/0006-291X(89)91141-8
- Schapira, A.H.V., Cooper, J.M., Dexter, D., Clark, J.B., Jenner, P. and Marsden, C.D. (1990) Mitochondrial complex I deficiency in Parkinson’s-disease. Journal of Neurochemistry, 54, 823-827. doi:10.1111/j.1471-4159.1990.tb02325.x
- Dexter, D.T., Holley, A.E., Flitter, W.D., Slater, T.F., Wells, F.R., Daniel, S.E., et al. (1994) Increased levels of lipid hydroperoxides in the parkinsonian substantia-nigra —an hplc and esr study. Movement Disorders, 9, 92-97. doi:10.1002/mds.870090115
- Alam, Z.I., Jenner, A., Daniel, S.E., Lees, A.J., Cairns, N., Marsden, C.D., et al. (1997) Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. Journal of Neurochemistry, 69, 1196-1203. doi:10.1046/j.1471-4159.1997.69031196.x
- Yoritaka, A., Hattori, N., Uchida, K., Tanaka, M., Stadtman, E.R. and Mizuno, Y. (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proceedings of National Academy of Sciences USA, 93, 2696-2701. doi:10.1073/pnas.93.7.2696
- Graham, D.G. (1984) Catecholamine toxicity: A proposal for the molecular pathogenesis of manganese neurotoxicity and Parkinson’s disease. Neurotoxicology, 5, 83-95.
- Slivka, A. and Cohen, G. (1985) Hydroxyl radical attack on dopamine. The Journal of Biological Chemistry, 260, 15466-15472.
- Good, P.F., Hsu, A., Werner, P., Perl, D.P. and Olanow, C.W. (1998) Protein nitration in Parkinson’s disease. Journal of the Neuropathology & Experimental Neurology, 57, 338-342. doi:10.1097/00005072-199804000-00006
- Sofic, E., Lange, K.W., Jellinger, K. and Riederer, P. (1992) Reduced and oxidized glutathione in the substantianigra of patients with Parkinson’s-disease. Neuroscience Letters, 142, 128-130.
- Cheng, K.C., Cahill, D.S., Kasai, H., Nishimura, S. and Loeb, L.A. (1992) 8-hydroxyguanine, an abundant form of oxidative DNA damage, causes g -> t and a -> c substitutions. The Journal of Biological Chemistry, 267, 166- 172.
- Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature, 362, 709-715.
- Hastings, T.G. and Zigmond, M.J. (1994) Identification of catechol-protein conjugates in neostriatal slices incubated with h-3 dopamine—Impact of ascorbic-acid and glutathione. Journal of Neurochemistry, 3, 1126-1132. doi:10.1046/j.1471-4159.1994.63031126.x
- Andersen, J.K. (2004) Iron dysregulation and Parkinson’s disease. Journal of Alzheimer’s Disease, 6, S47-S52.
- Smith, T.S., Parker, W.D. and Bennett, J.P. (1994) L-dopa increases nigral production of hydroxyl radicals in-vivo— potential l-dopa toxicity. Neuroreport, 5, 1009-1011.
- Basma, A.N., Morris, E.J., Nicklas, W.J. and Geller, H.M. (1995) L-dopa cytotoxicity to pc12 cells in culture is via its autoxidation. Journal of Neurochemistry, 64, 825-832. doi:10.1046/j.1471-4159.1995.64020825.x
- Mytilineou, C., Han, S.K. and Cohen, G. (1993) Toxic and protective effects of L-dopa on mesencephalic cellcultures. Journal of Neurochemistry, 61, 1470-1478. doi:10.1111/j.1471-4159.1993.tb13642.x
- Mytilineou, C., Walker, R.H., Jnobaptiste, R. and Olanow, C.W. (2003) Levodopa is toxic to dopamine neurons in an in vitro but not an in vivo model of oxidative stress. Journal of the Pharmacology and Experimental Therapeutics, 304, 792-800. doi:10.1124/jpet.102.042267
- Muenter, M.D. and Tyce, G.M. (1971) L-dopa therapy of Parkinson’s disease: Plasma L-dopa concentration, therapeutic response, and side effects. Mayo Clinic Proceedings, 46, 231-239.
- Olanow, C.W., Gauger, L.L. and Cedarbaum, J.M. (1991) Temporal relationships between plasma and cerebrospinal-fluid pharmacokinetics of levodopa and clinical effect in Parkinson’s-disease. Annals of Neurology, 29, 556-559. doi:10.1002/ana.410290516
- Benetello, P., Furlanut, M., Zara, G., Baraldo, M. and Hassan, E. (1993) Plasma-levels of levodopa and its main metabolites in parkinsonian-patients after conventional and controlled-release levodopa-carbidopa associations. European Neurology, 33, 69-73. doi:10.1159/000116905
- Perry, T.L., Yong, V.W., Ito, M., Foulks, J.G., Wall, R.A., Godin, D.V., et al. (1984) Nigrostriatal dopaminergic neu-rons remain undamaged in rats given high doses of L-DOPA and carbidopa chronically. Journal of Neurochemistry, 43, 990-993. doi:10.1111/j.1471-4159.1984.tb12834.x
- Zeng, B.Y., Pearce, R.K.B., MacKenzie, G.M. and Jenner, P. (2001) Chronic high dose L-dopa treatment does not alter the levels of dopamine D-1, D-2 or D-3 receptor in the striatum of normal monkeys: An autoradiographic study. Journal of Neural Transmission, 108, 925-941. doi:10.1007/s007020170013
- Quinn, N., Parkes, D., Janota, I. and Marsden, C.D. (1986) Preservation of the substantia nigra and locus coeruleus in a patient receiving levodopa (2 kg) plus decarboxylase inhibitor over a four-year period. Movement Disorders, 1, 65-68. doi:10.1002/mds.870010109
- Fahn, S., Shoulson, I., Kieburtz, K., Rudolph, A., Lang, A., Olanow, C.W., et al. (2004) Levodopa and the progression of Parkinson's disease. The New England Journal of Medicine, 351, 2498-2508. doi:10.1056/NEJMoa033447
- Hastings, T.G. and Zigmond, M.J. (1994) Identification of catechol-protein conjugates in neostriatal slices incubated with h-3 dopamine—Impact of ascorbic-acid and glutathione. Journal of Neurochemistry, 63, 1126-1132. doi:10.1046/j.1471-4159.1994.63031126.x
- Mena, M.A., Davila, V., Bogaluvsky, J. and Sulzer, D. (1998) A synergistic neurotrophic response to I-dihydroxyphenylalanine and nerve growth factor. Molecular Pharmacology, 54, 678-686.
- Mena, M.A., Davila, V. and Sulzer, D. (1997) Neurotrophic effects of L-DOPA in postnatal midbrain dopamine neuron cortical astrocyte cocultures. Journal of Neurochemistry, 69, 1398-408. doi:10.1046/j.1471-4159.1997.69041398.x
- Han, S.K., Mytilineou, C. and Cohen, G. (1996) L-DOPA up-regulates glutathione and protects mesencephalic cultures against oxidative stress. Journal of Neurochemistry, 66, 501-510. doi:10.1046/j.1471-4159.1996.66020501.x
- Cedarbaum, J.M. (1987) Clinical pharmacokinetics of antiparkinsonian drugs. Clinical Pharmacokinetics, 13, 141- 178. doi:10.2165/00003088-198713030-00002
- Bredberg, E., Tedroff, J., Aquilonius, S.M., et al. (1990) Pharmacokinetics and effects of levodopa in advanced Parkinson’s disease. European Journal of Clinical Pharmacology, 39, 385-389. doi:10.1007/BF00315415
- Contin, M., Riva, R., Albani, F., et al. (1996) Pharmacokinetic optimisation in the treatment of Parkinson's disease. Clinical Pharmacokinetics, 30, 463-481. doi:10.2165/00003088-199630060-00004
- Calne, D.B., Claveria, L.E. and Allen, J.G. (1974) Plasma levodopa and the “on-off” effect. Advances in Neurology, 5, 341-344.
- Kaakkola, S., Mannisto, P.T., Nissinen, E., et al. (1985) The effect of an increased ratio of carbidopa to levodopa on the pharmacokinetics of levodopa. Acta Neurologica Scandinavica, 72, 385-391. doi:10.1111/j.1600-0404.1985.tb00888.x
- Hadjiconstantinou, M. and Neff, N.H. (2008) Enhancing aromatic L-amino acid decarboxylase activity: Implications for L-DOPA treatment in Parkinson’s disease. CNS Neuroscience & Therapeutics, 14, 340-351.
- [61] Jonkers, N., Sarre, S., Ebinger, G. and Michotte, Y. (2001) Benserazide decreases central AADC activity, extracellular dopamine levels and levodopa decarboxylation in striatum of the rat. Journal of Neural Transmission, 108, 559- 570. doi:10.1007/s007020170056
- [62] Pinder, R.M., Brogden, R.N., Sawyer, P.R., Speight, T.M. and Avery, G.S. (1976) Levodopa and decarboxylase inhibitors: A review of their clinical pharmacology and use in the treatment of Parkinsonism. Drugs, 11, 329-377. doi:10.2165/00003495-197611050-00001
- [63] Da Prada, M., Kettler, R., Zurcher, G., Schaffner, R. and Haefely, W.E. (1987) Inhibition of decarboxylase and levels of dopa and 3-O-methyldopa: A comparative study of benserazide versus carbidopa in rodents and of Madopar standard versus Madopar HBS in volunteers. European Neurology, 27, 9-20. doi:10.1159/000116170
- [64] Korten, J.J., Keyser, A., Joosten, E.M. and Gabreels, F.J. (1975) Madopar versus sinemet. A clinical study on their effectiveness. European Neurology, 13, 65-71. doi:10.1159/000114663
- [65] Lieberman, A., Goodgold, A., Jonas, S. and Leibowitz, M. (1975) Comparison of dopa decarboxylase inhibitor (carbidopa) combined with levodopa and levodopa alone in Parkinson’s disease. Neurology, 25, 911-916. doi:10.1212/WNL.25.10.911
- [66] Perry, T.L., Yong, V.W., Ito, M., Foulks, J.G., Wall, R.A., Godin, D.V., et al. (1984) Nigrostriatal dopaminergic neurons remain undamaged in rats given high doses of L-DOPA and carbidopa chronically. Journal of Neurochemistry, 43, 990-993. doi:10.1111/j.1471-4159.1984.tb12834.x
- [67] Sian, J., Dexter, D.T., Lees, A.J., Daniel, S., Agid, Y., Javoyagid, F., et al. (1994) Alterations in glutathione levels in parkinsons-disease and other neurodegenerative disorders affecting basal ganglia. Annals of Neurology, 36, 348-355. doi:10.1002/ana.410360305
- [68] Facchinetti, F., Dawson, V.L. and Dawson, T.M. (1998) Free radicals as mediators of neuronal injury. Cellular and Molecular Neurobiology, 18, 667-682. doi:10.1023/A:1020221919154
- [69] Dizdaroglu, M. (1992) Oxidative damage to DNA in mammalian chromatin. Mutation Research, 275, 331-342. doi:10.1016/0921-8734(92)90036-O
- [70] Aust, A.E. and Eveleigh, J.F. (1999) Mechanisms of DNA oxidation. Proceedings of the Society for Experimental Biology and Medicine, 222, 246-252. doi:10.1046/j.1525-1373.1999.d01-141.x
- [71] Migliore, L., Petrozzi, L., Lucetti, C., Gambaccini, G., Bernardini, S., Scarpato, R., et al. (2002) Oxidative damage and cytogenetic analysis in leukocytes of Parkinson’s disease patients. Neurology, 58, 1809-1815. doi:10.1212/WNL.58.12.1809
- [72] Collins, A.R., Duthie, S.J. and Dobson, V.L. (1993) Direct enzymatic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis, 14, 1733-1735. doi:10.1093/carcin/14.9.1733
- [73] Cornetta, T., Palma, S., Aprile, I., Padua, L., Tonali, P., Testa, A., et al. (2009) Levodopa therapy reduces DNA damage in peripheral blood cells of patients with Parkinson’s disease. Cell Biology and Toxicology, 25, 321-330. doi:10.1007/s10565-008-9086-6
- [74] Kanabrocki, E.L., Murray, D., Hermida, R.C., Scott, G.S., Bremner, W.F., Ryan, M.D., et al. (2002) Circadian variation in oxidative stress markers in healthy and type II diabetic men. Chronobiology International, 19, 423-439. doi:10.1081/CBI-120002914
- [75] Oli, R.G., Fazeli, G., Kuhn, W., Walitza, S., Gerlach, M. and Stopper, H. (2010) No increased chromosomal damage in L-DOPA-treated patients with Parkinson’s disease: A pilot study. Journal of Neural Transmission, 117, 737-746. doi:10.1007/s00702-010-0401-z
- [76] Kostrzewa, R.M., Kostrzewa, J.P. and Brus, R. (2002) Neuroprotective and neurotoxic roles of levodopa (LDOPA) in neurodegenerative disorders relating to Parkinson’s disease. Amino Acids, 23, 57-63. doi:10.1007/s00726-001-0110-x
- [77] Colamartino, M., Padua, L., Meneghini, C., Leone, S., Cornetta, T., Testa, A. and Cozzi, R. (2012) Protective effects of L-Dopa and carbidopa combined treatments on human catecholaminergic cells. DNA and Cell Biology, 31, 1572-1579. doi:10.1089/dna.2011.1546
- [78] Seeberger, L.C. and Hauser, R.A. (2007) Optimizing bio-availability in the treatment of Parkinson’s disease. Neuropharmacology, 53, 791-800. doi:10.1016/j.neuropharm.2007.08.019