Advances in Bioscience and Biotechnology
Vol. 4 No. 12 (2013) , Article ID: 41058 , 10 pages DOI:10.4236/abb.2013.412142
The Cbl family of ubiquitin ligases regulates FcεRI expression and mast cell activation
![]()
1Department of Molecular Medicine, “Sapienza” University of Rome, Rome, Italy
2Lymphocyte Interaction Laboratory, London Research Institute, Cancer Research, London, UK
3Institute Pasteur-Fondazione Cenci Bolognetti, “Sapienza” University of Rome, Rome, Italy
Email: rossella.paolini@uniroma1.it
Copyright © 2013 Rosa Molfetta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 1 November 2013; revised 2 December 2013; accepted 13 December 2013
Keywords: Mast Cells; Basophils; FcεRI; Negative Regulation; Cbl Proteins; Ubiquitination
ABSTRACT
Antigen interaction with specific IgE bound to the high-affinity Fc receptor for IgE, constitutively expressed on the cell-surface of mast cells, generates signals that cause a shift in the resting state equilibrium of phosphorylation and dephosphorylation events that serves to maintain homeostasis. The outcome of this activated state is the release of a wide array of preformed and newly synthesized pro-inflammatory mediators. During the past few years, the existence of a negative feedback loop initiated upon FcεRI engagement has also been envisaged. This negative signal involves the coordinated action of adaptors, phosphatases and ubiquitin ligases that limits the intensity and duration of positive signals, thus modulating mast cell functions. Relevant to this, others and we have demonstrated that Cbl family proteins control the amplitude of FcεRI-generated signals by specific ubiquitin modification of activated receptor subunits and associated protein tyrosine kinases. In this article, we review advances in our understanding of the molecular mechanisms through which Cbl proteins regulate FcεRI expression and signaling.
1. INTRODUCTION
Mast cells have long been recognized, together with basophils, as the critical effector cells in IgE-mediated allergic diseases [1,2].
Mast cells and basophils share the presence of basophilic granules in their cytoplasm and the surface expression of a high affinity receptor for the Fc fragment of IgE (FcεRI).
FcεRI belongs to a family of multisubunit immunoreceptors that lack intrinsic enzymatic activity but transduces intracellular signals through association with cytoplasmic protein tyrosine kinases (PTKs) [3,4]. In rodent and human mast cells and basophils, the intracellular signalling generated upon engagement of receptor-bound IgE with the corresponding allergens is responsible for the release of preformed and newly synthesized mediators including histamine, leukotrienes, IL-4 and IL-13 [4-6].
Besides these positive signals, FcεRI aggregation has been understood to generate negative intracellular signals capable of limiting mast cell functional responses through the action of a variety of multidomain adaptor proteins [7].
Among them, the Cbl family of ubiquitin (Ub) ligases [8-11] has attracted considerable interest due to the finding that it controls the intensity and duration of FcεRImediated signals mainly by specific Ub modification of the activated receptor subunits and associated PTKs [12- 14].
This review is aimed at providing an overview on the mechanisms through which Cbl proteins, acting as Ub ligases, negatively regulate mast cell and basophil functional responses.
1.1. The High Affinity Receptor for IgE: Structure and Function
FcεRI is expressed on mast cells and basophils as a heterotetramer composed by an IgE-binding α subunit, a four transmembrane-spanning β subunit, and two identical disulphide-linked γ subunits. In rodents, all three subunits are needed for surface expression, whereas in humans a trimeric complex lacking the β-chain (FcεRIαγ2) also exists, and is expressed not only on mast cells and basophils but also on antigen presenting cells [4].
Several observations obtained using a rat basophilic leukemia cell line, namely RBL-2H3, have contributed to elucidate the function of the different receptor subunits.
FcεRIα-chain contains two extracellular Ig-like domains involved in IgE binding, a transmembrane region with an aspartic acid residue and a short cytoplasmic tail that lacks signal transduction motifs. The β and γ subunits have no role in ligand binding, but they share a conserved immunoreceptor tyrosine-based activation motif (ITAM) within their long cytoplasmic tails that, upon FcεRI aggregation, is rapidly phosphorylated on tyrosines by the Src family kinase Lyn that binds to FcεRIβ under resting conditions [3,4].
The phosphorylated γ-chains are competent to drive cell activation in the absence of a β-chain ITAM, however FcεRIβ amplifies the intensity of signals from FcεRIβ [15], thus the two subunits act cooperatively in promoting signal transduction.
It has been demonstrated that the full activation of FcεRI requires its migration into lipid rafts, specialized regions of the plasma membrane enriched in cholesterol and glycosphingolipid that form ordered but dynamic structures floating in the less ordered surrounding membrane [16]. Upon FcεRI engagement membrane rafts coalesce into larger and more stable structures where engaged receptors are concentrated [17], and can more easily interact with signalling molecules, such as active Lyn [18,19], thus favouring ITAM phosphorylation.
Phosphorylated ITAMs provides a docking site for the tandem pair of Src homology 2 (SH2) domains of the cytoplasmic kinase Syk, that is, in turn, activated upon tyrosine phosphorylation [3]. The use of Syk specific inhibitors and Syk-negative RBL-2H3 cells has demonstrated an obligatory role for this kinase in FcεRI-mediated signalling [20-23]. For example, bone marrow-derived mast cells (BMMCs) isolated from Syk−/− mice show impaired calcium mobilization, degranulation and cytokine production upon receptor stimulation [21]. In humans, a minority of normal blood donors have basophils that fail to release granules. These “nonreleaser” basophils express a normal level of FcεRI but very low levels of Syk protein [24,25]. Furthermore, variable expression levels of Syk, observed in human basophils from releaser donors, correlate well with the IgE-mediated responsiveness of these cells [25].
Syk activation enables the productive interaction with its many targets including the membrane anchored linker for activation of T cells (LAT) that, once phosphorylated, recruits SH2-containing adaptors such as leukocyte protein of 76 kDa (SLP-76) and Grb2, and enzymes such as phospholipase Cγ (PLCγ). Once in the membrane, PLCγ undergoes activating tyrosine phosphorylation, and hydrolyzes the membrane phosphatidyl inositol 4,5-bisphosphate [PtdIns(4,5)P2] to form the soluble inositol 1,4,5- trisphosphate (IP3) and the membrane bound diacylglycerol (DAG), which are responsible for intracellular calcium mobilization and protein kinase C (PKC) activation, respectively.
The adaptors SLP-76 and Grb2 recruit exchange factors promoting the activation of the small GTPases, Ras, Rac, and Rho. They regulate complex networks of signalling pathways leading to the secretion of preformed and newly synthesized mediators and cytokines.
In RBL-2H3 cells, a complementary pathway is initiated by another PTK of the Src family, Fyn, and cooperates with the Lyn/Syk pathway in the propagation of FcεRI-mediated signal [26]. Upon receptor engagement, Fyn phosphorylates the molecular adaptor Gab2, favouring membrane recruitment of the p85 regulatory subunit of phosphatidylinositol-3-OH kinase (PI3K). Once activated, PI3K catalyzes the formation of PtdIns(3,4,5)P3 (PIP3) that functions as docking site for pleckstrin homology domain-containing proteins such as Bruton’s tyrosine kinase (Btk) and PLCγ.
1.2. Mechanisms Underlying Negative Regulation of Mast Cell and Basophil Activation
Over the past several years, it has become apparent that mast cell and basophils express surface receptors that counteract FcεRI-mediated activation responses. The common feature of these inhibitory receptors is the presence in their cytoplasmic tail of immunoreceptor tyrosine-based inhibitory motifs (ITIMs) then, once phosphorylated, recruit negative signaling molecules [27,28].
Furthermore, FcεRI signaling itself has been understood to consist of a mixture of positive and negative signals whose integration determines the rate and the extent of functional responses.
In RBL-2H3 cells, the phosphatases SHP-1, SHP-2 and SHIP are tyrosine phosphorylated and activated upon their recruitment to the phosphorylated FcεRIβ, thus promoting dephosphorylation events that contrast FcεRImediated signal propagation [29-31].
A low level of SHIP was detected in “hyper-releaseable” basophils derived from highly allergic donors, and it was found to correlate with a higher sensitivity to stimulation [32], suggesting a role for SHIP as negative regulator of human basophil degranulation as well.
Futhermore, in human mast cells, the absence of another lipid phosphatase, namely PTEN, leads to increased PIP3 levels even in resting conditions. In PTENdeficient cells, the enzymatic activity of c-Jun and p38 MAPKs is increased and is followed by a constitutive secretion of cytokines [33].
Interestingly, signaling molecules other than phosphatases, such as PTKs, adapters and ubiquitin ligases, are also able to generate negative signals, thus contributing to control the intensity and duration of functional mast cell and basophil responses [7].
Among them, Cbl family proteins have emerged as negative regulators of FcεRI-mediated signals.
2. Cbl FAMILY PROTEINS
2.1. Domain Structure and Function
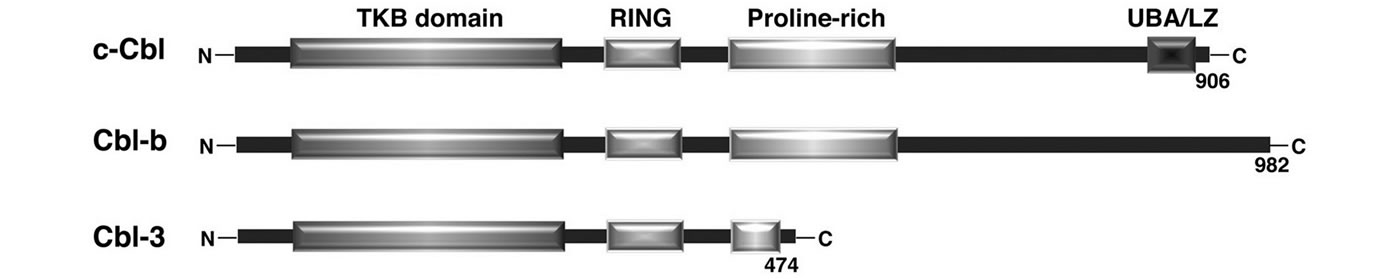
The mammalian Cbl family consists of three proteins encoded by separate genes: c-Cbl, Cbl-b and Cbl-3 [8-11] (Figure 1(a)).
All of them are structurally characterized by the presence of highly conserved regions in their N-terminal: a tyrosine kinase-binding (TKB) domain, a RING finger domain, and a proline-rich domain.
Moreover, c-Cbl and Cbl-b share additional regions in their C-terminal mainly involved in protein-protein interactions [34]: an extensive prolin-rich domain able to interact with several SH3-containing proteins, such as Src family kinases; a region containing several tyrosine residues which are phosphorylated following the stimulation of a diverse array of membrane receptors, and promotes interaction with SH2-domain containing proteins; a region homologous to both Ub-Associated (UBA) domain and to Leucine Zipper (LZ) that mediate Ub binding and intermolecular oligomerization.
Importantly, the highly conserved TKB and RING finger domains define the basic functional unit of c-Cbl and Cbl-b [11,34]. The TKB domain is composed of a four-helix bundle (4H), a calcium-binding EF hand and a modified SH2 domain, and was so named for its ability to bind to phosphotyrosine residues of receptor and nonreceptor PTKs including growth factor receptors, such as EGFR and PDGFR, and cytoplasmic tyrosine kinases of the Syk/ZAP-70 family [35].
The RING Finger domain, interacting with Ub-conjugating enzymes, catalyzes protein ubiquitination [36,37].
Ubiquitination is a post-translational reversible modification whereby Ub, a 76-amino-acid-globular peptide (8 KDa), is covalently attached to lysine residues of acceptor proteins that are then mainly targeted to degradation [38-40]. Ubiquitination is catalyzed by the action of three different enzymes, namely E1, E2 and E3. The Ub-activating enzyme (E1) forms a thiol-ester bond with the carboxy-terminal glycine of Ub in an ATP-dependent process. Activated Ub is, successively, accepted by the Ub-conjugating enzyme (E2) by transthiolation, and finally transferred to the substrate through the action of the Ub protein ligase (E3). Thus, this latter class of enzymes provides specificity to the Ub system being responsible for substrate recognition and Ub ligation to the target protein.
The Cbl family belongs to the E3 Ub ligases characterized by the presence of a RING finger domain [37]: TKB domain, as well as additional domains located in the C-terminal region of both c-Cbl and Cbl-b, determine Cbl substrate specificity serving as a docking site for tyrosine phosphorylated proteins that are then ubiquitinated by the RING-finger associated E2 enzyme (Figure 1(b)).
Another factor essential for the E3 activity of c-Cbl (and in a lesser extent of Cbl-b) is the tyrosine phosphorylation of its linker domain located between the TKB and RING domains. This phosphorylation causes conformational changes in c-Cbl that are required for the
 (a)
(a)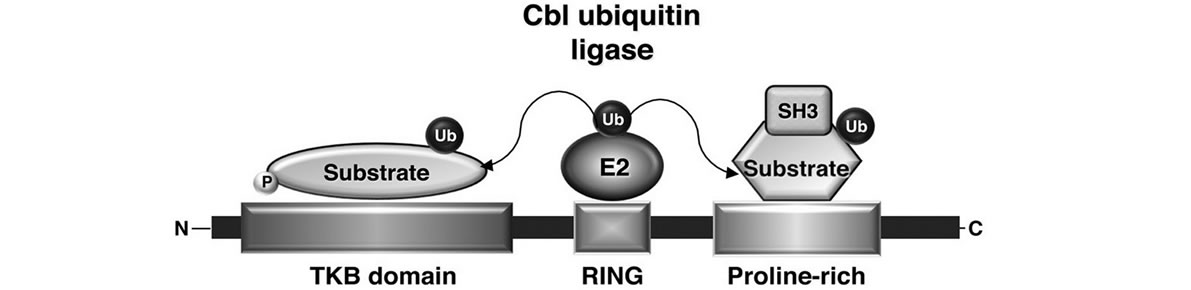 (b)
(b)
Figure 1. (a) Schematic representation of the structural domains of the three mammalian Cbl isoforms c-Cbl, Cbl-b, and Cbl-3; (b) Model of Cbl ubiquitin ligase function. Multiple motifs in Cbl proteins, such as tyrosine-kinase-binding (TKB) domain or the proline-rich domain, serve to recruit the substrates for ubiquitination. The ubiquitin-conjugating enzyme (E2), which interacts with the RING domain of the ubiquitin ligase Cbl, transfers ubiquitin to the target proteins. Modified from Duan L. et al., Immunity, 2004.
release of the RING-finger associated E2, an essential step in Cbl driven ubiquitination [41].
Ubiquitinated substrates are, then, targeted to different degradation pathways depending on the kind of ubiquitination itself. Polyubiquitin chains, whereby Ub acts as a substrate for the attachment of further Ub molecules, is commonly recognized as a signal that targets substrates to degradation by the 26S proteasome [38-40,42]. The attachment of single Ub to one or more lysines of target proteins, namely mono and multiubiquitination respectively, can act as internalization signals driving transport of membrane receptors along the endocytic pathway towards a lysosomal compartment for degradation [43-45].
In vitro, c-Cbl and Cbl-b appear to have equal capacity to act as E3 Ub ligases toward a similar range of substrates. However, unlike c-Cbl, ubiquitination by Cbl-b does not often result in substrate degradation, but rather appears to affect protein localization [34].
2.2. FcεRI Down-Regulation by Cbl-Mediated Ubiquitination
Evidence collected in the past years has strongly supported a crucial role for Cbl family Ub ligases in the down-regulation of immunoreceptors, including FcεRI [12,46,47].
c-Cbl and Cbl-b are both expressed on rodent and mouse mast cells, and upon FcεRI engagement, become tyrosine phosphorylated [48,49] and translocate into lipid rafts [14,50].
c-Cbl is also expressed on cultured human mast cells [51] and basophils [52], where it undergoes antigen-dependent phosphorylation.
The first evidence for a negative role played by c-Cbl in mast cell came from experiments in which c-Cbl overexpression inhibited receptor-mediated serotonin release in RBL-2H3 cells without affecting receptor phosphorylation [53]. The molecular mechanism underlying this inhibition has been elucidated by additional works demonstrating the involvement of c-Cbl in the ubiquitination of FcεRI receptor subunits. An earlier study from our group [54] had demonstrated that FcεRI β and γ subunits were subjected to ubiquitination upon stimulation of RBL-2H3 cells with IgE and multivalent antigen. Subcellular fractionation and confocal microscopy experiments have subsequently demonstrated that c-Cbl colocalizes with FcεRI β and γ subunits into lipid rafts after receptor engagement, suggesting the involvement of Cbl in receptor ubiquitination [50].
Our group has, indeed, identified c-Cbl as the main E3 ligase responsible for the antigen-induced receptor ubiquitination in RBL-2H3 cells [12]. Overexpression of wild type c-Cbl, but not a mutant form deleted in the RING finger domain, strongly increased antigen-induced FcεRI β and γ ubiquitination, providing evidence for a direct role of c-Cbl as Ub ligase.
In line with this finding, we have then demonstrated that FcεRI β and γ subunits are mainly monoubiquitinated by c-Cbl at multiple sites upon antigen stimulation, and provided evidence that this modification controls receptor internalization and sorting along the endocytic compartments through the interaction with adaptor proteins harboring Ub interacting motifs (UIMs), namely epsin, Eps15, Eps15R and Hrs [55,56]. Eps15, eps15R and epsin display similar functions: they control the early steps of the endocytic route coupling ubiquitinated receptors with components of the budding vesicles [57,58]. The key role of Hrs is, instead, the delivery of ubiquitinated proteins to the outer membrane of the late endosomes and the sorting of the cargo into internal vesicles of multivesicular bodies for lysosomal degradation [59].
We demonstrated that Eps15, Eps15R and Epsin play a partial overlapping functions in ubiquitinated FcεRI uptake, whereas Hrs plays a key role in controlling the fate of internalized receptor complexes: Hrs depletion by means of siRNA retains ubiquitinated receptors into early endosomes and partially prevents their sorting into lysosomes [55].
Since FcεRI β and γ subunits and c-Cbl have been reported to translocate into lipid rafts upon antigen stimulation [17,18,50], we have also investigated the implication of a lipid raft environment in regulating Cbl-mediated FcεRI ubiquitination. We have demonstrated that the recruitment of engaged FcεRI subunits into lipid rafts precedes their ubiquitination, and that the integrity of these membrane microdomains is required to allow receptor ubiquitination [55]. We have also shown a strong interdependence between lipid rafts and receptor endocytosis, in line with the finding of Fattakhova and coworkers, who demonstrated that aggregated FcεRI complexes remain associated with lipid rafts upon antigeninduced internalization [60].
All together, our data demonstrate that Cbl-dependent FcεRI ubiquitination initiates into lipid raft and provide evidence that FcεRI may use Ub as an internalization signal. Furthermore, they support a key role for Cbl-mediated receptor ubiquitination in ensuring a proper cargo sorting along the endocytic compartments mainly through the interaction with the Ub-binding adaptor Hrs (see Figure 2).
To understand how Hrs function is regulated upon FcεRI engagement, we first investigated whether Hrs itself undergoes covalent modifications. We found that Hrs is subjected to antigen-dependent tyrosine phosphorylation and monoubiquitination, and we identified Syk as the main kinase regulating both inducible Hrs post-translational modifications in RBL-2H3 cells [61].

Figure 2. Model depicting how Cbl orchestrates endocytosis of engaged FcεRI complexes. Upon antigen stimulation, the combined action of Syk and Cbl, recruited to phosphorylated FcεRI as a preformed complex, leads to receptor multiubiquitination that may act as an independent signal for receptor endocytosis. Ubiquitinated receptor complexes upon internalization are then sorted along the endocytic pathway throught the sequential action of ubiquitin-adaptor proteins, and finally targeted to lysosomes for degradation.
Moreover, by siRNA knock down of c-Cbl and complementary overexpression studies, we demonstrated that Hrs monoubiquitination is under the control of c-Cbl ligase activity. To address how Hrs modifications might impact on its function as endocytic adapter, we examined the distribution of phosphorylated and ubiquitinated Hrs species between particulate-membrane and cytosolic constituents. We demonstrated that upon FcεRI engagement Hrs is present into both membrane and cytosolic fractions. However, an increase of Hrs phosphorylation was reproducibly observed only in membranes, suggesting that Syk preferentially phosphorylates Hrs located into endosomal sorting site. Consistent with this assumption, a predominant relocalization of Syk in membrane fraction was observed.
Interestingly, we also found that monoubiquitinated forms of Hrs are preferentially confined on cytosolic fractions. The relocation of ubiquitinated Hrs from membrane to cytosolic compartments may be functionally significant. Indeed, Hoeller and collegues previously reported that monoubiquitination of Hrs promotes intramolecular interactions, preventing Hrs ability to bind to ubiquitinated cargo [62]. Thus, the removal of monoubiquitinated Hrs from endosomal membrane could facilitate the clearance of the non-functional adapter and its replacement with non ubiquitinated and sorting-competent Hrs. In conclusion, our findings are compatible with the following scenario depicted in Figure 3.
Upon antigen stimulation ubiquitinated FcεRI complexes are recognized by Hrs that becomes a substrate for Syk and Cbl enzymatic activities. Although we did not address the order in which Hrs phosphorylation and ubiquitination occur, it is likely that Syk-induced Hrs phosphorylation occurs at the endosomal membrane and precedes Hrs ubiquitination.
Monoubiquitinated Hrs is then removed from endosomal sorting sites allowing its replacement with nonubiquitinated Hrs that may need to be tyrosine phosphorylated to interact with other endocytic adapters in order to ensure an efficient transport of ubiquitinated cargos.
In this scenario, Hrs monoubiquitination would serve to relocate Hrs from endosomes to the cytosol, without promoting degradative events.
Although not as extensively studied as in RBL-2H3 cells, evidence indicates that a role for Cbl proteins in FcεRI down-regulation can also be envisaged in mouse mast cells.
In this regard, c-Cbl and Cbl-b knock-out (KO) mice were useful in the highlighting of redundant or distinct
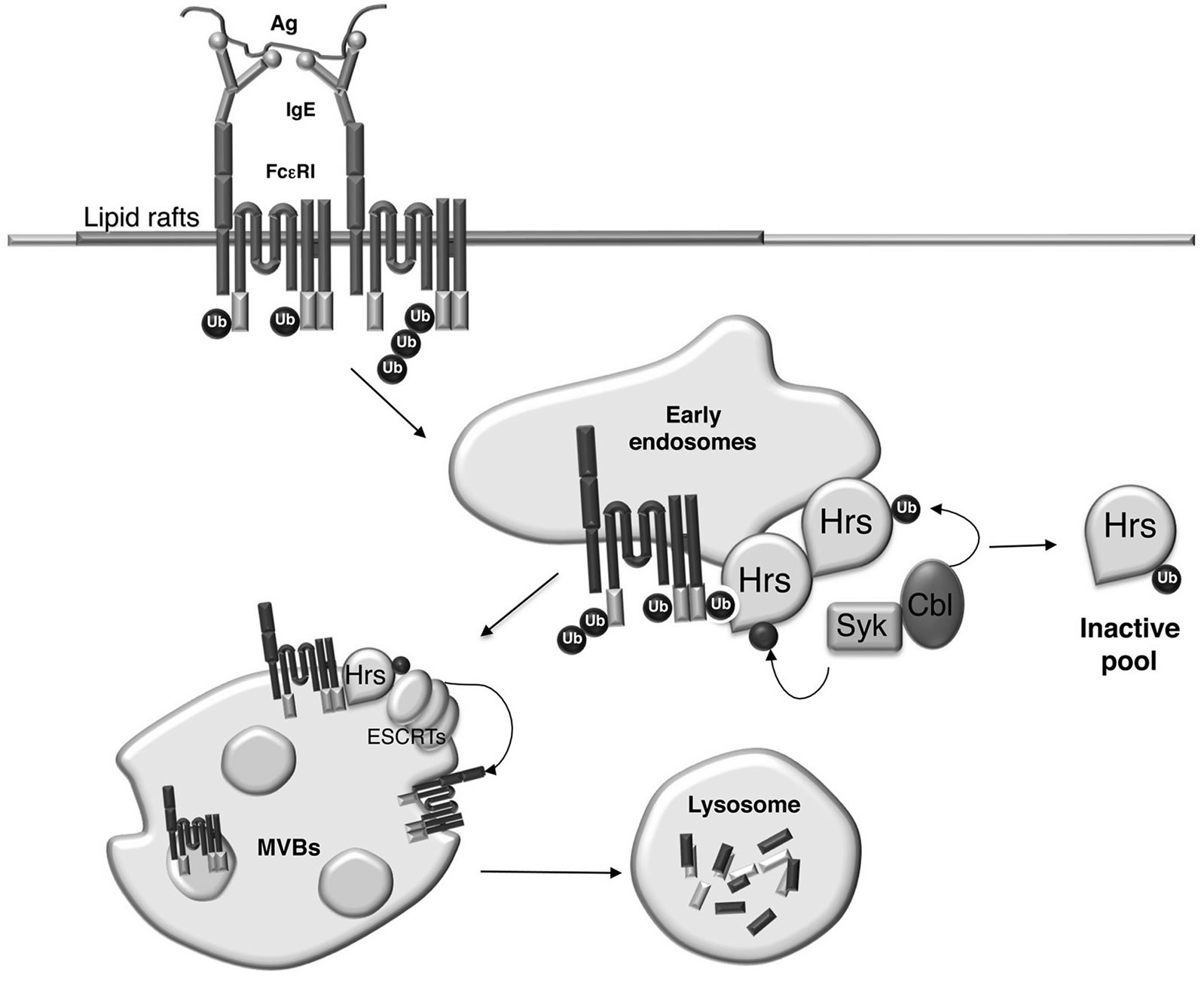
Figure 3. Model depicting how Cbl-mediated monoubiquitination of Hrs affects its subcellular localization. Ubiquitinated FcεRI complexes upon internalization are recognized by Hrs that becomes a substrate for Syk and Cbl enzymatic activities. Monoubiquitinated Hrs is then removed from endosomal sorting sites whereas phosphorylated Hrs interacts with other endocytic adapters of the ESCRT complexes in order to ensure an efficient transport of ubiquitinated FcεRI complexes to the multivesicular bodies (MVBs) and to the lysosomes for degradation. ESCRT, endosomal sorting complex required for transport.
roles of the two adapter proteins. In Cbl-b-deficient BMMCs FcεRI-mediated phosphorylation, Ca2+ mobilization, histamine release, and induction of pro-inflammatory cytokines were increased compared to wild type and c-Cbl KO mice [49,63]. Moreover, loss of Cbl-b, but not c-Cbl, retards FcεRI internalization in BMMCs [63], but Cbl-b ligase activity is dispensable for receptor down-modulation [64].
All together, the data obtained in RBL-2H3 cells and in BMMCs support the requirement of Cbl protein for the negative regulation of FcεRI-induced signal propagation. However, while in RBL-2H3 cells c-Cbl plays a major role in receptor down-regulation mainly through its ligase activity, in mouse mast cells the main function of Cbl-b is to work as an adapter protein.
The role of Cbl proteins in regulating FcεRI surface expression in human mast cells and basophils has not yet been investigated.
2.3. Cbl Ligase Activity Regulates Non-Receptor PTK Expression and Function
Cbl proteins induce ubiquitination of a number of nonreceptor PTKs, including Syk and several members of Src family kinases, and in most case ubiquitination of activated PTKs correlates with downregulation of their protein level and kinase activity [12,52,65-68].
Several lines of evidence have demonstrated that human basophils show a wide variability in the rate of FcεRI-mediated degranulation and, among them, a small percentage of basophils completely fail to release histamine. Their unresponsiveness has been linked to lower levels of Lyn and to the absence of Syk despite a normal level of their mRNA, suggesting the action of a posttranslational mechanism responsible for kinase degradation [24,25]. Although differences in Cbl protein levels between releaser and nonreleaser basophils have not been observed, it is likely that alterations in Cbl ligase activity contribute to Syk instability in the nonreleaser phenotype. In this regard, the involvement of the Ubproteasome pathway has been reported [69], and may contribute to the regulation of Syk levels in unstimulated basophils isolated from nonreleaser donors. Relevant to this, we have more recently demonstrated that Cbl ligase activity is influenced by the protein abundance of specific positive and negative regulators in unstimulated RBL-2H3 cells. Indeed, we found that overexpression of a specific positive regulator of Cbl ligase activity promotes an antigen-independent Syk ubiquitination and degradation with consequent functional defects [70].
In RBL-2H3 we have also demonstrated that, following FcεRI stimulation, c-Cbl is responsible for Syk ubiquitination, and that Syk activity controls its own ubiquitination [12].
Syk and c-Cbl have been previously reported to be constitutively associated in RBL-2H3 cells [48]. Thus, it is conceivable that Syk/Cbl interaction allows the enzymes to become reciprocal substrates: Syk phosphorylates and activates Cbl that, in turn, ubiquitinates activated Syk, as depicted in Figure 2. In line with this model, we have also demonstrated that ubiquitination preferentially affects the phosphorylated and active forms of Syk [12]. Moreover, proteasome inhibition induced a persistence of activated kinase forms, supporting the involvement of Cbl-dependent ubiquitination in the downregulation of the active pool of Syk.
A similar role for Cbl in targeting Syk ubiquitination and degradation has also been envisaged in stimulated human basophils. FcεRI engagement, achieved by antigen or anti-IgE antibodies, promotes a progressive loss of Syk protein that correlates with Syk ubiquitination and is sensitive to proteasome inhibitors [52]. Moreover, an antigen-dependent interaction between Syk and Cbl was observed, and the level of protein association correlates with that of Cbl tyrosine phosphorylation.
More recent lines of evidence support this finding showing that also IgE-independent stimuli are able to induce c-Cbl phophorylation, and are responsible for Syk degradation and for the induction of nonreleasing phenotype [71].
Following FcεRI engagement, Cbl proteins can also ubiquitinate Lyn and Fyn in RBL-2H3 cells [13,14]. Both c-Cbl and Cbl-b act as Ub ligases targeting the active forms of Lyn, further supporting our previous finding that ubiquitination preferentially affects active forms of the kinases responsible for signal propagation. The same study [14] provided also evidence for the requirement of a lipid raft environment in Cbl-mediated ubiquitination events: targeting Cbl-b into lipid raft enhances Lyn ubiquitination. Moreover, overexpression of a mutant form of Cbl-b that constitutively localizes in lipid rafts reduces antigen-mediated degranulation and cytokine production by negatively regulating both Lyn-SykLAT and Fyn-Gab2-mediated signalling pathways. At molecular level, the membrane-targeted overexpression of Cbl-b inhibited receptor phosphorylation and kinase activity, and dramatically downregulated the protein amount of Gab2 by promoting its ubiquitination.
Thus, overexpression studies in RBL-2H3 cells have shown that c-Cbl and Cbl-b play similar negative regulatory roles in FcεRI-mediated signaling. However, additional studies are needed to demonstrate a formal role of Cbl proteins in promoting PTK ubiquitination and degradation in human basophils.
3. CONCLUSIONS
We have provided on overview of the mechanisms through which Cbl proteins control the intensity and duration of FcεRI-generated signals.
Cbl-dependent receptor down-modulation occurs mainly by means of Cbl ligase activity that promotes FcεRI β and γ multiubiquitination providing signals for receptor internalization and sorting into endocytic compartments, a process required for receptor degradation.
Remarkably, Cbl proteins also negatively regulate the expression level of PTKs, including Lyn and Syk, by the selective ubiquitination and degradation of the activated kinase forms.
In conclusion, Cbl ligase activities, by regulating the half-life of activated receptor complexes and cytoplasmic PTKs, may contribute to the down-regulation of FcεRImediated signaling leading to basophil and mast cell effector functions.
REFERENCES
- Galli, S.J. (2000) Mast cells and basophils. Current Opinion in Hematology, 7, 32-39. http://dx.doi.org/10.1097/00062752-200001000-00007
- Prussin, C. and Metcalfe, D.D. (2003) IgE, mast cells, basophils, and eosinophils. Journal of Allergy and Clinical Immunology, 111, 486-494. http://dx.doi.org/10.1067/mai.2003.120
- Siraganian, R.P. (2003) Mast cell signal transduction from the high-affinity IgE receptor. Current Opinion Immunology, 15, 639-646. http://dx.doi.org/10.1016/j.coi.2003.09.010
- Kraft, S. and Kinet, J.P. (2007) New developments in FcεRI regulation, function and inhibition. Nature Reviews Immunology, 7, 365-378. http://dx.doi.org/10.1038/nri2072
- Falcone, F.H., Haas, H. and Gibbs, B.F. (2000) The human basophil: A new appreciation of its role in immune responses. Blood, 96, 4028-4038.
- MacGlashan Jr., D.W. (2008) IgE receptor and signal transduction in mast cells and basophils. Current Opinion Immunology, 20, 717-723. http://dx.doi.org/10.1016/j.coi.2008.08.004
- Molfetta, R., Peruzzi, G., Santoni, A. and Paolini, R. (2007) Negative signals from FcεRI engagement attenuate mast cell functions. Archivum Immunologiae et Therapiae Experimentalis, 55, 219-229. http://dx.doi.org/10.1007/s00005-007-0028-4
- Thien, C.B. and Langdon, W.Y. (2001). Cbl: Many adaptations to regulate protein tyrosine kinases. Nature Reviews in Molecular Cell Biology, 2, 294-307. http://dx.doi.org/10.1038/35067100
- Rao, N., Dodge, I. and Band, H. (2002) The Cbl family of ubiquitin ligases: Critical negative regulators of tyrosine kinase signaling in the immune system. Journal of Leukocyte Biology, 71, 753-763.
- Dikic, I., Szymkiewicz, I. and Soubeyran, P. (2003) Cbl signaling networks in the regulation of cell function. Cellular and Molecular Life Science, 60, 1805-1827. http://dx.doi.org/10.1007/s00018-003-3029-4
- Swaminathan, G. and Tsygankov, A.Y. (2006) The Cbl family proteins: Ring leaders in regulation of cell signaling. Journal of Cellular Physiology, 209, 21-43. http://dx.doi.org/10.1002/jcp.20694
- Paolini, R., Molfetta, R., Beitz, L.O., Zhang, J., Scharenberg, A.M., Piccoli, M., Frati, L., Siraganian, R. and Santoni, A. (2002) Activation of Syk tyrosine kinase is required for c-Cbl-mediated ubiquitination of FcεRI and Syk in RBL cells. Journal of Biological Chemistry, 277, 36940-36947. http://dx.doi.org/10.1074/jbc.M204948200
- Kyo, S., Sada, K., Qu, X., Maeno, K., Shahjahan Miah, S.M., Kawauchi-Kamata, K. and Yamamura, H. (2003) Negative regulation of Lyn protein-tyrosine kinase by cCbl ubiquitin-protein ligase in FcεRI-mediated mast cell activation. Genes Cells, 8, 825-836. http://dx.doi.org/10.1046/j.1365-2443.2003.00679.x
- Qu, X., Sada, K., Kyo, S., Maeno, K., Miah, S.M. and Yamamura, H. (2004) Negative regulation of FcεRI-mediated mast cell activation by the ubiquitin-protein ligase Cbl-b. Blood, 103, 1779-1786. http://dx.doi.org/10.1182/blood-2003-07-2260
- Lin, S., Cicala, C., Scharenberg, A.M. and Kinet J.P. (1996) The FcεRI subunit functions as an amplifier of FcεRImediated cell activation signals. Cell, 85, 985-995. http://dx.doi.org/10.1016/S0092-8674(00)81300-8
- Simons, K. and Toomre, D. (2000) Lipid rafts and signal transduction. Nature Reviews in Molecular Cell Biology, 1, 31-39. http://dx.doi.org/10.1038/35036052
- Field, K.A., Holowka, D. and Baird, B. (1997) Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. Journal of Biological Chemistry, 272, 4276-4280. http://dx.doi.org/10.1074/jbc.272.7.4276
- Sheets, E.D., Holowka, D. and Baird, B. (1999) Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcεRI and their association with detergent-resistant membranes. Journal of Cell Biology, 145, 877- 887. http://dx.doi.org/10.1083/jcb.145.4.877
- Young, R.M., Holowka, D. and Baird, B. (2003) A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. Journal of Biological Chemistry, 278, 20746-20752. http://dx.doi.org/10.1074/jbc.M211402200
- Oliver, J.M., Burg, D.L., Wilson, B.S., McLaughlin, J.L. and Geahlen, R.L. (1994) Inhibition of mast cell FcεR1- mediated signalling and effector function by the Syk-selective inhibitor, piceatannol. Journal of Biological Chemistry, 269, 29697-29703.
- Costello, P.S., Turner, M., Walters, A.E., Cunningham, C.N., Bauer, P.H., Downward, J. and Tybulewicz, V.L. (1996) Critical role for the tyrosine kinase Syk in signaling through the high affinity IgE receptor of mast cells. Oncogene, 13, 2595-2605.
- Zhang, J., Berenstein, E.H., Evans, R.L. and Siraganian, R.P. (1996) Transfection of Syk protein tyrosine kinase reconstitutes high affinity IgE receptor-mediated degranulation in a Syk-negative variant of rat basophilic leukemia RBL-2H3 cells. Journal of Experimental Medicine, 184, 71-79. http://dx.doi.org/10.1084/jem.184.1.71
- Moriya, K., Rivera, J., Odom, S., Sakuma, Y., Muramato, K., Yoshiuchi, T., Miyamoto, M. and Yamada, K. (1997) ER-27319, an acridone-related compound, inhibits release of antigen-induced allergic mediators from mast cells by selective inhibition of Fcepsilon receptor I-mediated activation of Syk. Proceeding of the National Academy of Sciences USA, 94, 12539-12544. http://dx.doi.org/10.1073/pnas.94.23.12539
- Kepley, C.L., Youssef, L., Andrews, R.P., Wilson, B.S. and Oliver, J.M. (1999) Syk deficiency in nonreleaser basophils. Journal of Allergy and Clinical Immunology, 104, 279-284. http://dx.doi.org/10.1016/S0091-6749(99)70367-2
- Lavens-Phillips, S.E. and MacGlashan Jr., D.W. (2000) The tyrosine kinases p53/56lyn and p72syk are differentially expressed at the protein level but not at the messenger RNA level in nonreleasing human basophils. American Journal of Respiratory Cell and Molecular Biology, 23, 566-571. http://dx.doi.org/10.1165/ajrcmb.23.4.4123
- Parravicini, V., Gadina, M., Kovarova, M., Odom, S., Gonzalez-Espinosa, C., Furumoto, Y., Saitoh, S., Samelson, L.E., O’Shea, J.J. and Rivera, J. (2002) Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nature Immunology, 3, 741- 748.
- Daëron, M., Jaeger, S., Du Pasquier, L. and Vivier, E. (2008) Immunoreceptor tyrosine-based inhibition motifs: A quest in the past and future. Immunological Reviews, 224, 11-43. http://dx.doi.org/10.1111/j.1600-065X.2008.00666.x
- Kepley, C.L., Cambier, J.C., Morel, P.A., Lujan, D., Ortega, E., Wilson, B.S. and Oliver, J.M. (2000) Negative regulation of FcεRI signaling by FcγRII costimulation in human blood basophils. Journal of Allergy and Clinical Immunology, 106, 337-348. http://dx.doi.org/10.1067/mai.2000.107931
- Kimura, T., Zhang, J., Sagawa, K., Sakaguchi, K., Appella, E. and Siraganian, R.P. (1997) Syk-independent tyrosine phosphorylation and association of the protein tyrosine phosphatases SHP-1 and SHP-2 with the high affinity IgE receptor. Journal of Immunology, 159, 4426- 4434.
- Kimura, T., Sakamoto, H., Appella, E. and Siraganian, R.P. (1997) The negative signaling molecule SH2 domain-containing inositol-polyphosphate 5-phosphatase (SHIP) binds to the tyrosine-phosphorylated beta subunit of the high affinity IgE receptor. Journal of Biological Chemistry, 272, 13991-13996. http://dx.doi.org/10.1074/jbc.272.21.13991
- Leung, W.H. and Bolland S. (2007) The inositol 5’-phosphatase SHIP-2 negatively regulates IgE-induced mast cell degranulation and cytokine production. Journal of Immunology, 179, 95-102.
- Vonakis, B.M., Gibbons Jr., S., Sora, R., Langdon, J.M. and MacDonald, S.M. (2001) Src homology 2 domaincontaining inositol 5’ phosphatase is negatively associated with histamine release to human recombinant histamine-releasing factor in human basophils. Journal of Allergy and Clinical Immunology, 108, 822-831. http://dx.doi.org/10.1067/mai.2001.119159
- Furumoto, Y., Brooks, S., Olivera, A., Takagi, Y., Miyagishi, M., Taira, K., Casellas, R., Beaven, M.A., Gilfillan, A.M. and Rivera, J. (2006) Cutting Edge: Lentiviral short hairpin RNA silencing of PTEN in human mast cells reveals constitutive signals that promote cytokine secretion and cell survival. Journal of Immunology, 176, 5167- 5171.
- Thien, C.B. and Langdon, W.Y. (2005) c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochemical Journal, 391, 153-166. http://dx.doi.org/10.1042/BJ20050892
- Meng, W., Sawasdikosol, S., Burakoff, S.J. and Eck, M.J. (1999) Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature, 398, 84-90. http://dx.doi.org/10.1038/18050
- Zheng, N., Wang, P., Jeffrey, P.D. and Pavletich, N.P. (2000) Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell, 102, 533- 539. http://dx.doi.org/10.1016/S0092-8674(00)00057-X
- Joazeiro, C.A. and Weissman, A.M. (2000) RING finger proteins: Mediators of ubiquitin ligase activity. Cell, 102, 549-552. http://dx.doi.org/10.1016/S0092-8674(00)00077-5
- Ciechanover, A. (1998) The ubiquitin-proteasome pathway: On protein death and cell life. EMBO Journal, 17, 7151-7160. http://dx.doi.org/10.1093/emboj/17.24.7151
- Laney, J.D. and Hochstrasser, M. (1999) Substrate targeting in the ubiquitin system. Cell, 97, 427-430. http://dx.doi.org/10.1016/S0092-8674(00)80752-7
- Weissman, A.M. (2001) Themes and variations on ubiquitylation. Nature Reviews. Molecular Cell Biology, 2, 169-178. http://dx.doi.org/10.1038/35056563
- Kassenbrock, C.K. and Anderson, S.M. (2004) Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. Journal of Biological Chemistry, 279, 28017-28027. http://dx.doi.org/10.1074/jbc.M404114200
- Thrower, J.S., Hoffman, L., Rechesteiner, M. and Pickart, C.M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO Journal, 19, 94-102. http://dx.doi.org/10.1093/emboj/19.1.94
- Hicke, L. and Dunn, R. (2003) Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annual Review of Cell and Developmental Biology, 19, 141-172. http://dx.doi.org/10.1146/annurev.cellbio.19.110701.154617
- Haglund, K., Sigismund, S., Polo, S., Szymkiewicz, I., Di Fiore, P.P. and Dikic, I. (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nature Cell Biology, 5, 461-466. http://dx.doi.org/10.1038/ncb983
- Mosesson, Y., Shtiegman, K., Katz, M., Zwang, Y., Vereb, G., Szollosi, J. and Yarden, Y. (2003) Endocytosis of receptor tyrosine kinases is driven by mono-, not poly-ubiquitylation. Journal of Biological Science, 278, 31323- 31326.
- Katkere, B., Rosa, S. and Drake, J. R. (2012) The Sykbinding ubiquitin ligase c-Cbl mediates signaling-dependent B cell receptor ubiquitination and B cell receptormediated antigen processing and presentation. Journal of Biological Chemistry, 287, 16636-16644. http://dx.doi.org/10.1074/jbc.M112.357640
- Wang, H.Y., Altman, Y., Fang, D., Dai, Y., Shao, Y. and Liu, Y.C. (2001) Cbl promotes ubiquitination of the T cell receptor through an adaptor function of Zap-70. Journal of Biological Chemistry, 276, 26004-26011. http://dx.doi.org/10.1074/jbc.M010738200
- Ota, Y., Beitz, L.O., Scharenberg, A.M., Donovan, J.A., Kinet, J.P. and Samelson, L.E. (1996) Characterization of Cbl tyrosine phosphorylation and a Cbl-Syk complex in RBL-2H3 cells. Journal of Experimental Medicine, 184, 1713-1723. http://dx.doi.org/10.1084/jem.184.5.1713
- Zhang, J., Chiang, Y.J., Hodes, R.J. and Siraganian, R.P. (2004) Inactivation of c-Cbl or Cbl-b differentially affects signaling from the high affinity IgE receptor. Journal of Immunology, 173, 1811-1818.
- Lafont, F. and Simons, K. (2001) Raft-partititioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proceeding of the National Academy of Sciences of the United States of America, 98, 3180-3184. http://dx.doi.org/10.1073/pnas.051003498
- Suzuki, H., Takei, M., Yanagida, M., Nakahata, T., Kawakami, T. and Fukamachi, H. (1997) Early and late events in Fc epsilon RI signal transduction in human cultured mast cells. Journal of Immunology, 159, 5881-5888.
- Macglashan, D. and Miura, K. (2004) Loss of syk kinase during IgE-mediated stimulation of human basophils. Journal of Allergy and Clinical Immunology, 114, 1317-1324. http://dx.doi.org/10.1016/j.jaci.2004.08.037
- Ota, Y. and Samelson, L.E. (1997) The product of the proto-oncogene c-cbl: A negative regulator of the Syk tyrosine kinase. Science, 276, 418-420. http://dx.doi.org/10.1126/science.276.5311.418
- Paolini, R. and Kinet, J.P. (1993) Cell surface control of the multiubiquitination and deubiquitination of high-affinity immunoglobulin E receptors. EMBO Journal, 12, 779-786.
- Molfetta, R., Gasparrini, F., Peruzzi, G., Vian, L., Piccoli, M., Frati, L., Santoni, A. and Paolini, R. (2009) Lipid raft-dependent FcεRI ubiquitination regulates receptor endocytosis through the action of Ubiquitin Binding Adaptors. PLoS ONE, 4, Article ID: e5604. http://dx.doi.org/10.1371/journal.pone.0005604
- Molfetta, R., Gasparrini, F., Santoni, A. and Paolini. R. (2010) Ubiquitination and endocytosis of the high affinity receptor for IgE. Molecular Immunology, 47, 2427-2434. http://dx.doi.org/10.1016/j.molimm.2010.06.003
- Carbone, R., Fré, S., Iannolo, G., Belleudi, F., Mancini, P., Pelicci, P.G., Torrisi, M.R. and Di Fiore, P.P. (1997) Eps15 and eps15R are essential components of the endocytic pathway. Cancer Research, 57, 5498-5504.
- Chen, H., Fre, S., Slepnev, V.I., Capua, M.R., Takei, K., Butler, M.H., Di Fiore, P.P. and De Camilli, P. (1998) Epsin is an EH-domain-binding protein implicated in clathrinmediated endocytosis. Nature, 394, 793-797. http://dx.doi.org/10.1038/28660
- Raiborg, C., Bache, K.G., Gillooly, D.J., Madshus, I.H., Stang, E. and Stenmark, H. (2002) Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nature Cell Biology, 4, 394-398. http://dx.doi.org/10.1038/ncb791
- Fattakhova, G., Masilamani, M., Borrego, F., Gilfillan, A.M., Metcalfe, D.D. and Coligan, J.E. (2006) The highaffinity immunoglobulin-E receptor (FcεRI) is endocytosed by an AP-2/clathrin-independent, dynamin-dependent mechanism. Traffic, 7, 673-685. http://dx.doi.org/10.1111/j.1600-0854.2006.00423.x
- Gasparrini, F., Molfetta, R., Quatrini, L., Frati, L., Santoni, A. and Paolini, R. (2012) Syk-dependent regulation of Hrs phosphryaltion and ubiquitination upon FcεRI engagement: impact on Hrs membrane/cytosol localization. European Journal of Immunology, 42, 2744-2753. http://dx.doi.org/10.1002/eji.201142278
- Hoeller, D., Crosetto, N., Blagoev, B., Raiborg, C., Tikkanen, R., Wagner, S., Kowanetz, K., Breitling, R., Mann, M., Stenmark, H. and Dikic, I. (2006) Regulation of ubiquitin-binding proteins by monoubiquitination. Nature Cell Biology, 8, 163-169. http://dx.doi.org/10.1038/ncb1354
- Gustin, S.E., Thien, C.B. and Langdon, W.Y. (2006) Cbl-b is a negative regulator of inflammatory cytokines produced by IgE-activated mast cells. Journal of Immunology, 177, 5980-5989.
- Oksvold, M.P., Dagger, S.A., Thien, C.B. and Langdon, W.Y. (2008) The Cbl-b RING finger domain has a limited role in regulating inflammatory cytokine production by IgE-activated mast cells. Molecular Immunology, 45, 925-936. http://dx.doi.org/10.1016/j.molimm.2007.08.002
- Andoniou, C.E., Lill, N.L., Thien, C.B., Lupher Jr., M.L., Ota, S., Bowtell, D.D.L., Scaife, R.M., Langdon, W.Y. and Band, H. (2000) The Cbl proto-oncogene product negatively regulates the Src-family tyrosine kinase Fyn by enhancing its degradation. Molecular Cell Biology, 20, 851-867. http://dx.doi.org/10.1128/MCB.20.3.851-867.2000
- Rao, N., Ghosh, A.K., Ota, S., Zhou, P.C., Reddi, A.L., Hakezi, K., Druker, B.K., Wu, J. and Band, H. (2001). The non-receptor tyrosine kinase Syk is a target of Cblmediated ubiquitylation upon B-cell receptor stimulation. EMBO Journal, 20, 7085-7095. http://dx.doi.org/10.1093/emboj/20.24.7085
- Rao, N., Miyake, S., Reddi, A.L., Douillard, P., Ghosh, A.K., Dodge, I.L., Zhou, P., Fernandes, N.D. and Band, H. (2002) Negative regulation of Lck by Cbl ubiquitin ligase. Proceeding of the National Academy of Sciences USA, 99, 3794-3799.
- Sohn, H.W., Gu, H. and Pierce, S.K. (2003) Cbl-b negatively regulates B cell antigen receptor signaling in mature B cells through ubiquitination of the tyrosine kinase Syk. Journal of Experimental Medicine, 197, 1511-1524. http://dx.doi.org/10.1084/jem.20021686
- Youssef, L.A., Wilson, B.S. and Oliver, J.M. (2002) Proteasome-dependent regulation of Syk tyrosine kinase levels in human basophils. Journal of Allergy and Clinical Immunology, 110, 366-373. http://dx.doi.org/10.1067/mai.2002.127562
- Peruzzi, G., Molfetta, R., Gasparrini, F., Vian, L., Morrone, S., Piccoli, M., Frati, L., Santoni, A. and Paolini, R. (2007) The adaptor molecule CIN85 regulates Syk tyrosine kinase level by activating the ubiquitin-proteasome degradation pathway. Journal of Immunology, 179, 2089- 2096.
- MacGlashan Jr., D.W., Ishmael, S., MacDonald, S.M., Langdon, J.M., Arm, J.P. and Slogane, D.E. (2008) Induced loss of Syk in human basophils by non-IgE-dependent stimuli. Journal of Immunology, 180, 4208-4217.