Advances in Bioscience and Biotechnology
Vol.3 No.7A(2012), Article ID:25130,23 pages DOI:10.4236/abb.2012.327123
Oxidative stress and human health
![]()
Department of Biochemistry and Molecular Biology, Faculty of Biological Sciences, University of Dhaka, Dhaka, Bangladesh
Email: *hossainshekhar@yahoo.com, *shekhardu@hotmail.com
Received 15 August 2012; revised 21 September 2012; accepted 28 October 2012
Keywords: Oxidative Stress; Reactive Oxygen Species; Antioxidant; Disease
ABSTRACT
Redox degenerative reactions of the biological system inevitably produce reactive oxygen species (ROS) and their derivatives. Oxidative stress is the result of an imbalance in pro-oxidant/antioxidant homeostasis that leads to the generation of toxic reactive oxygen species (ROS), such as hydrogen peroxide, organic hydro peroxides, nitric oxide, superoxide and hydroxyl radicals etc. Information are accumulating steadily, supporting the general importance of oxidative damage of tissue and cellular components as a primary or secondary causative factor in many different human diseases and aging processes. Many of the recent landmarks in scientific research have shown that in human beings, oxidative stress has been implicated in the progression of major health problems by inactivating the metabolic enzymes and damaging important cellular components, oxidizing the nucleic acids, leading to cardiovascular diseases, eye disorders, joint disorders, neurological diseases (Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis), atherosclerosis, lung and kidney disorders, liver and pancreatic diseases, cancer, ageing, disease of the reproductive system including the male and female infertility etc. The advent of a growing number of in vitro and in vivo models for evaluating the human disease pathology is aiding scientists in deciphering the detailed mechanisms of the point of intersection of the oxidative stress with other cellular components or events in the growing roadmap leading to different human disorders. The toxic effect of reactive oxygen and nitrogen species in human is balanced by the antioxidant action of non-enzymatic antioxidants, as well as by antioxidant enzymes. Such antioxidant defences are extremely important as they represent the direct removal of free radicals (prooxidants), thus providing maximal protection for biological sites. These systems not only assert with the problem of oxidative damage, but also play a crucial role in wellness, health maintenance, and prevention of chronic and degenerative diseases. In this review we have tried to generate a gross picture on the critical role of ROS in deteriorating human health and the importance of antioxidative defense system in ameliorating the toxicity of ROS.
1. INTRODUCTION
Oxidative stress occurs when the generation of free radicals and active intermediates in a system exceeds the system’s ability to neutralize and eliminate them (Figure 1) [1-3]. The current concept of “oxidative stress” should also include the pathways related to the “nitrosative stress” and, for their implication in cellular and extracellular metabolic events, to the “metabolic stress”. Reactive oxygen intermediate (ROI) and reactive nitrogen intermediate (RNI) are constantly produced under physiological conditions [4,5], is the crucial event in living organisms. At the moment, the concept of oxidative stress confined to ROI such as hydroxyl and superoxide radicals, and hydrogen peroxide and singlet oxygen has been extended onto RNI such as nitric oxide (NO), peroxynitrite and, recently, to S-nitrosothiols [5]. Thus, ROI and RNI react with proteins, carbohydrates and lipids, with consequent alteration both in the intracellular and intercellular homeostasis, leading to possible cell death and regeneration (Figure 2) [6].
To cope with the oxidative stress elicited by aerobic metabolism, animal and human cells have developed a ubiquitous antioxidant defense system, which consists of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) and glutathione reductase together with a number of low molecular-weight antioxidants such as ascorbate, α-tocopherol and glutathione, cysteine, thioredoxin, vitamins, etc. However, this antioxidant defense system may be overwhelmed by various pathological or environmental factors so that a fraction of ROS may escape destruction and form the far more reactive hydroxyl radicals [7,8]. An increase in ROSelicited oxidative damage to DNA and other biomolecules may impair normal functions of tissue cells and lead to human aging and disease [9,10].
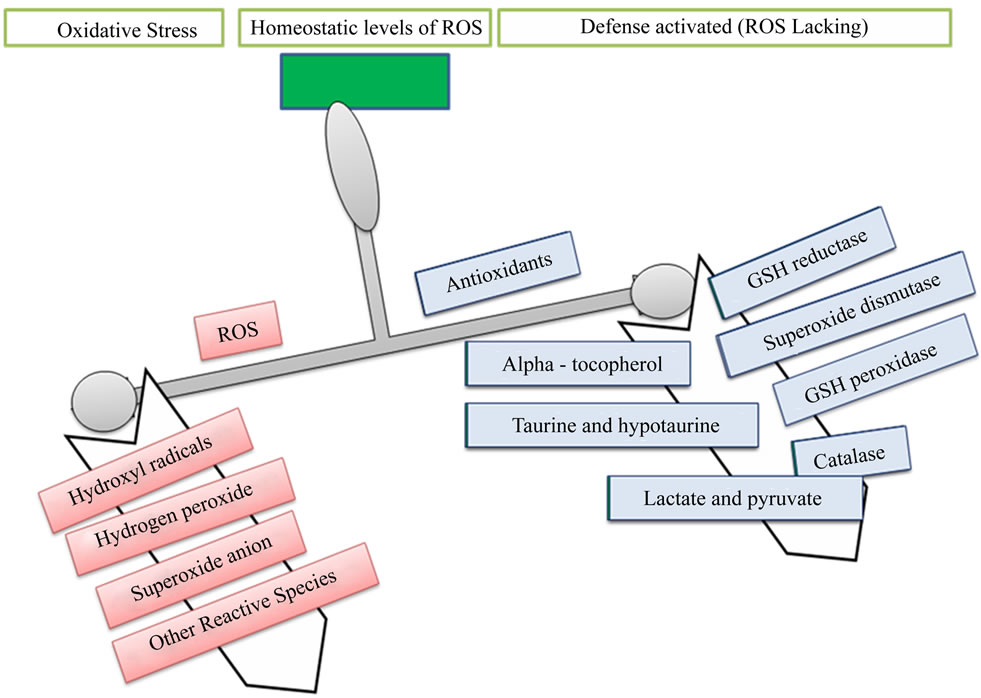
Figure 1. Imbalance between oxidant and antioxidant.

Figure 2. Reactions of ROI and RNI with proteins, carbohydrates and lipids, with consequent alteration both in the intracellular and intercellular homeostasis until possible cell death and regeneration.
2. TYPES OF ROS, SOURCE OF SYNTHESIS AND THE DAMAGE CAUSED BY THE PRODUCTION OF ROS
2.1. Types of Reactive Species
The highly reactive molecules include Reactive oxygen Species (ROS) and Reactive Nitrogen Species (RNS) are listed in Tables 1 and 2. Among these reactive molecules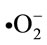 , •NO, ONOO– are the most widely studied species and play important roles in the diabetic cardiovascular complications. Superoxide (
, •NO, ONOO– are the most widely studied species and play important roles in the diabetic cardiovascular complications. Superoxide (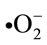 ) is produced by one electron reduction of oxygen by different oxidases including dihydro nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthin oxidases, cycloxigenase as well as by the mitochondrial electron transport chain during the course of normal oxidative phosphorylation which is essential for generating ATP [11].
) is produced by one electron reduction of oxygen by different oxidases including dihydro nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthin oxidases, cycloxigenase as well as by the mitochondrial electron transport chain during the course of normal oxidative phosphorylation which is essential for generating ATP [11].
2.2. Source and Cellular Response of ROS
Oxidants are generated as a result of normal intracellular metabolism in mitochondria and peroxisomes, as well as from a variety of cytosolic enzyme systems. In addition, a number of external agents can trigger ROS production. A sophisticated enzymatic and non-enzymatic antioxidant defense system (Figure 3) including catalase (CAT), superoxide dismutase (SOD) and reduced glutathione(GSH) counteracts and regulates overall ROS levels to maintain physiological homeostasis. Lowering
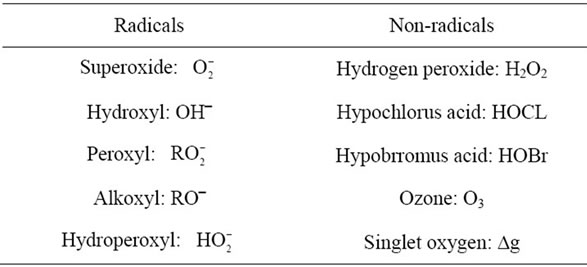
Table 1. Reactive oxygen species (ROS).
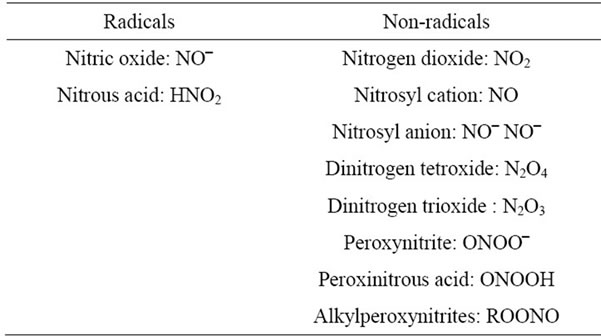
Table 2. Reactive nitrogen species (RNS).
ROS levels below the homeostatic set point may interrupt the physiological role of oxidants in cellular proliferation and host defense. Similarly, increased ROS may also be detrimental and lead to cell death or to acceleration in ageing and age-related diseases. Traditionally, the impairment caused by increased ROS is thought to result from random damage to proteins, lipids and DNA. In addition to these effects, a rise in ROS levels may also constitute a stress signal that activates specific redoxsensitive signaling pathways. Once activated, these diverse signaling pathways may have either damaging or potentially protective functions (Table 3) [12].
The primary ROS produced in the course of oxygen metabolism is superoxide, which is a highly reactive, cytotoxic ROS. Superoxide is converted to a far less reactive product, hydrogen peroxide (H2O2), by a family of metalloenzymes known as superoxide dismutase (SOD) [13]. The ubiquitous superoxide dismutase’s (SODs) catalyze the disproportionation of superoxide to molecular oxygen and peroxide and thus are critical for protecting the cell against the toxic products of aerobic respiretion.
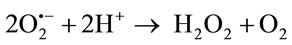
GSH is by far the most important antioxidant in most mammalian cells. This ubiquitous tripeptide, γ-Glu-CysGly, performs many cellular functions. In particular, the thiol containing moiety is a potent reducing agent [14]. GSH has the important function of destroying reactive oxygen intermediates and free radicals that are constantly formed in metabolism [15].

Intracellular GSH is converted to GSSG by seleniumcontaining GSH peroxidase, which catalyzes the reduction of H2O2 in the presence of GSH and GSH peroxidase is coupled with oxidation of glucose-6-phosphate and of 6-phosphogluconate, which provides NADPH for reduction of GSSG by GSSG reductase. This is a major pathway of H2O2 metabolism in many cells. It is thus important for the protection of membrane lipids against oxidation. Hydrogen peroxide (H2O2) can also be neutralized to molecular oxygen (O2) and water (H2O) by one of the most important antioxidant enzyme Catalase. It also catalyses the oxidation of various hydrogen donors in the presence of relatively lower concentrations of hydrogen peroxide. Lipids when react with free radicals, they undergo peroxidation to form lipid peroxides. Lipid peroxides decompose to form numerous products including malondialdehyde [16]. The reaction course is largely influenced by the presence of radical trapping
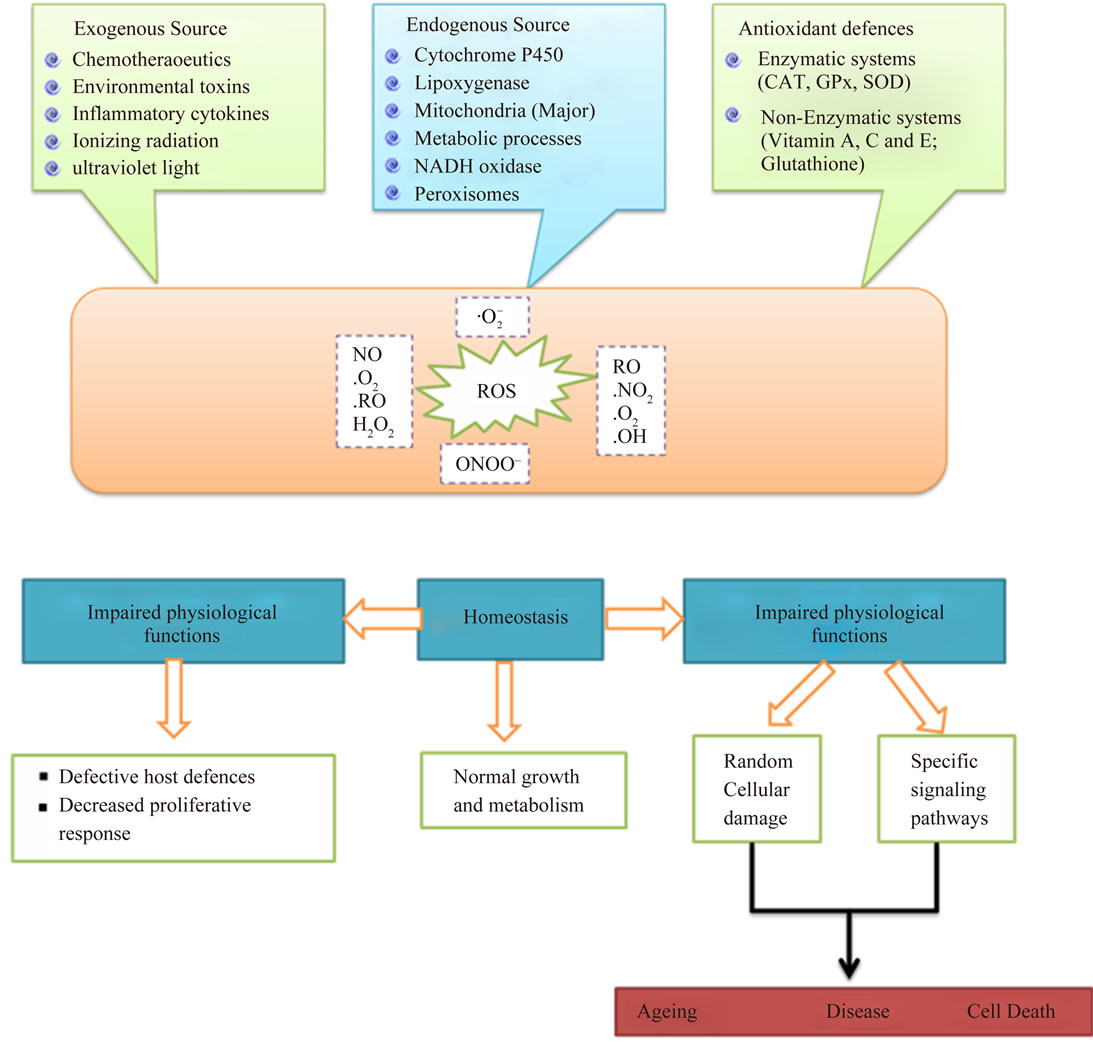
Figure 3. The source and cellular responses to reactive oxygen species (ROS).

Table 3. Types of ROS, source of synthesis and the damage caused by the production of ROS.
species, particularly oxygen [17]. The most common way to measure lipid peroxides is to estimate malondialdehyde (MDA) content. The amount of malondialdehyde is then determined colorometrically after reaction with thiobarbituric acid [18].
superoxide dismutase (SOD), reduced glutathione (GSH) and catalase (CAT) whose activities contribute to eliminate superoxide, hydrogen peroxide and hydroxyl radicals. Humans have evolved with antioxidant systems to protect against free radicals. These systems include some antioxidants produced in the body (endogenous) and others obtained from diet (exogenous). The first includes a) enzymatic defenses, such as glutathione peroxidase, catalase, and super oxide dismutase, which metabolize superoxide, hydrogen peroxide, and lipid peroxides, thus preventing most of the formation of the toxic OH• and b) nonenzymatic defenses, such as glutathion, histidine-peptides, the iron binding proteins transferrin and ferritin, dihydrolipoic acid, melatonin, urate, and plasma protein thiols [19,20].
3. SOME OF THE HUMAN DISORDERS CLINICALLY LINKED TO OXIDATIVE STRESS
Oxidative stress has been implicated in several diseases including cancer, atherosclerosis, malaria, chronic fatigue syndrome, rheumatoid arthritis and neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease (Figure 4) [21,22]. Indirect evidence via monitoring biomarkers such as reactive oxygen species, and reactive nitrogen species production, antioxidant defense indicates oxidative damage may be involved in the pathogenesis of these diseases [23,24] Oxidative stress also contributes to tissue injury following irradiation and hyperoxia, as well as in diabetes and is likely to be involved in age-related development of cancer. Infection by Helicobacter pylori which increases the production of reactive oxygen and nitrogen species in human stomach is also thought to be important in the development of gastric cancer [25].
ROS also have been reported to damage cellular components in cartilage by impairing the chondrocyte responses to growth factors and migrating to the sites of cartilage injury leading to osteoarthritis [26]. There is increasing evidence suggesting the role of ROS in damageing the islets of pancreas [27]. Increased ROS in human heart is associated with arotic valve stenosis [28]. Excess ROS in human kidney leads to urolithiasis [29]. It was also observed that hyperglycemia triggers the generation of ROS in both mesangial and tubular cells of human kidney, making structural and functional changes in glomeruli causing diabetic nephropathy [30]. Increased ROS production in high altitudes may also be involved in acute mountain sickness (AMS), high altitude pulmonary edema (HAPE) and high altitude cerebral edema (HACE) [31-33].
3.1. Oxidative Stress and Neurodegenerative Diseases
Neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Amyotrophic lateral sclerosis (ALS), are defined by the progressive loss of specific neuronal cell populations and are associated with protein aggregates. A common feature of these diseases is oxidative damage of neurons, which might be responsible for the dysfunction or death of neuronal cells that contributes to disease pathogenesis.
3.1.1. Alzheimer’s Disease (AD)
4-hydroxy-2,3-nonenal (HNE), acrolein, malondialdehyde (MDA) and F2-isoprostanes are important break down products of lipid peroxidation. Elevated HNE levels have been observed in Alzheimer’s disease (AD) [34,35]. Acrolein, thiobarbituric acid-reactive substances (TBARs, the most prevalent substrate of which is malondialdehyde) and F2-isoprostanes are all increased
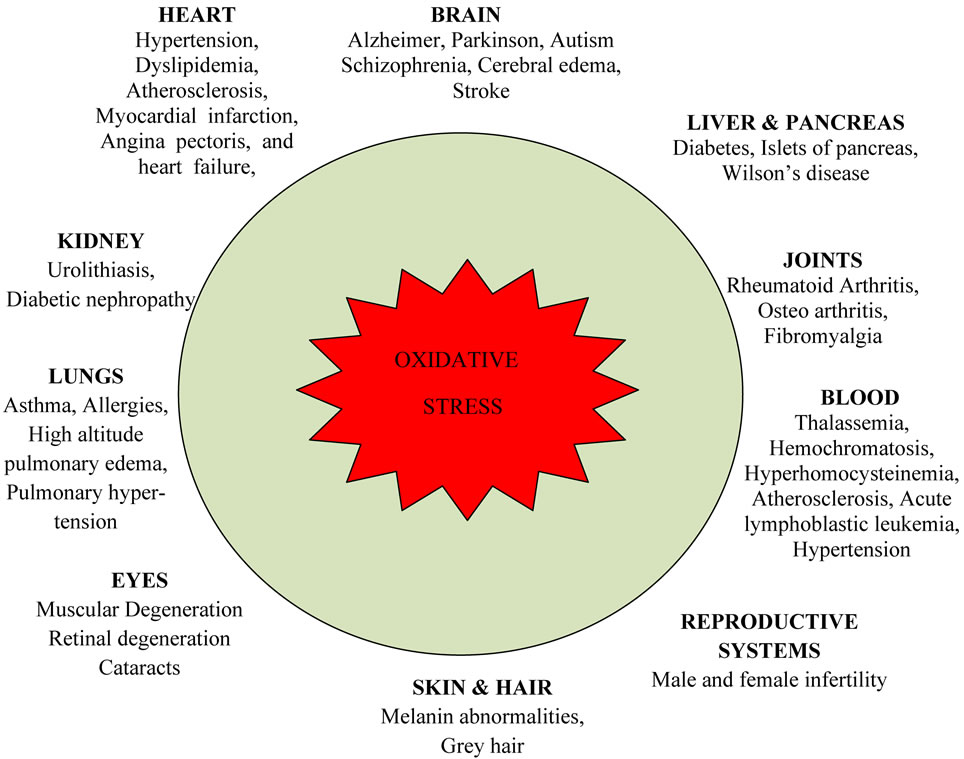
Figure 4. Deleterious effects of oxidative stress on human health.
in AD brains relative to age-matched controls [36]. DNA bases are susceptible to modification by oxidative stress involving hydroxylation [37] whereas increased levels of 8-hydroxyguanine and 8-hydroxy-2-deoxyguanosine are observed in PD brains; the selective attack on guanine bases implies OH radicals as the oxidative species [38]. In the AD brain, the activity of the antioxidant proteins catalase, superoxide dismutase (SOD), glutathione peroxidase and glutathione reductase are increased in the hippocampus and amygdale [39,40]. These are also used as markers of oxidative stress in different diseases.
3.1.2. Parkinson’s Disease (PD)
Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons of the substantia nigra and the deposition of intracellular inclusion bodies. The principal protein component of these deposits is α-synuclein [41], which is ubiquitously expressed in the brain; mutations of α-synuclein (A30P and A53T) contribute to familial forms of the disease [42]. A characteristic feature of the neurons within the substantia nigra is the age-dependent accumulation of neuromelanin [43]. In PD, these neuromelanin-containing cells are most likely to be lost [44]. Neuromelanin is a dark brown pigment that accumulates metal ions, particularly iron. Although the composition of neuromelanin has not been rigorously characterized, it is known that it consists primarily of the products of dopamine redox chemistry [45,46]. The oxidative stress associated with PD could be the result of a breakdown in the regulation of dopamine (neuromelanin)/iron biochemistry. A diverse array of evidence is emerging that α-synuclein has a role in modulating the activity of dopamine. The A53T mutation associated with familial PD impairs vesicular storage of dopamine [47,48], which leads to the accumulation of dopamine in the cytoplasm and subsequent generation of ROS through its interaction with iron, a process that increases with age. The mutations in α-synuclein have been shown to alter the expression of dihydropteridine reductase, which indirectly regulates the synthesis of dopamine [49]. Co-immuno-precipitation experiments have shown that α-synuclein forms stable complexes with the human dopamine transporter, thereby inhibiting uptake of dopamine by its transporter and that α-synuclein can regulate dopamine synthesis by inhibiting tyrosine hydroxylase [50,51] The link between α-synuclein and redox chemistry associated with iron-bound dopamine/neuromelanin has been given further credence by a study showing that initiation of Lewy body formation coincides with α-synuclein deposition exclusively within lipofuscin and neuro-melanin deposits [52].
3.1.3. Amyotrophic Lateral Sclerosis
ALS is distinguished by the loss of the lower motor neurons of the spinal cord and upper motor neurons in the cerebral cortex; and both sporadic and familial forms are possible. Like the other neurodegenerative diseases, ALS is characterized by the deposition of a misfolded protein in neural tissue, in this instance copper/zinc SOD [53]. There are more than 100 mutations of SOD associated with the familial forms of the disease. Through transgenic mouse studies it has been shown that these mutations lead to a toxic gain of function by SOD [54]. The nature of this gain of function is widely debated, and there are two main theories: one suggests that the toxicity is due to misfolded aggregated forms of SOD, whereas the other proposes that SOD becomes a pro-oxidant protein generating ROS. The merits or otherwise of these two hypotheses have recently been reviewed [55], and as the aggregation mechanism lies outside the scope of this review it will not be discussed here, except to note that the two theories might not be mutually exclusive.
Mutations of SOD can convert the protein from an anti-oxidant to a pro-oxidant capable of causing oxidative insults. Evidence that inappropriate metal-mediated redox chemistry is central to the progression of ALS includes the observation that copper chelators inhibit the course of the disease in both cell culture and mouse Models [56].
3.1.4. Mechanisms That May Link Oxidative Stress to Neuronal Dysfunction, Clinical Symptoms and Pathogenesis in Autism
Oxidative stress is known to be associated with premature aging of cells and can lead to tissue inflammation, damaged cell membranes, autoimmunity, and cell death [57]. Recent evidence has shown abnormalities in membrane lipid metabolism and an imbalance in immune and inflammatory responses in autism. A potential mechanism depicting association of oxidative stress in autism with membrane lipid abnormalities, immune dysregulation, inflammatory response, impaired energy metabolism, increased excitotoxicity, leading to clinical symptoms and pathology of the disease (Figure 5).
3.2. Oxidative Stress and Heart Disease
Several lines of evidence demonstrate that oxidative stress plays an important role in the pathogenesis and development of cardiovascular diseases, including hypertension, dyslipidemia, atherosclerosis, myocardial infraction, angina pectoris, and heart failure [58-61].
3.2.1. Coronary Heart Disease (CHD)
Traditional vascular risk factors, including hyperlipidemia (cholesterol, LDL, etc.), hypertension, cigarette smoking, diabetes, overweight, physical inactivity, age, male sex and familial predisposition, only partly explain the excess risk of developing cerebrovascular and Coro-
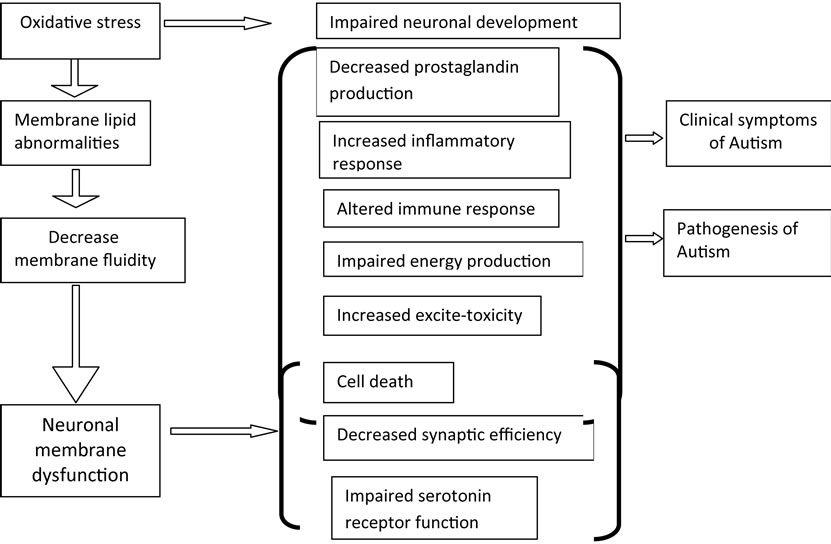
Figure 5. Schematic depletion of potential mechanisms that may mediate neuronal dysfunction and clinical symptoms in autism.
nary Heart Disease (CHD). Many studies also support the role of OS in disease pathogenesis. Paradoxically, although moderate exercise poses an acute oxidant stress, regular endurance exercise is associated with improved cardiovascular function and a reduction in traditional CHD risk factors. These findings are consistent with the hypothesis that adaptations induced by acute exposures to exercise-induced oxidative stress lead to long-term vascular protection. This occurs through activation of signalling pathways that lead to increased synthesis of intracellular antioxidants and antioxidant enzymes and decreased ROS production during exercise.
3.2.2. Atherosclerosis
Atherosclerosis is a complex process involving the deposition of plasma lipoproteins and the proliferation of cellular elements in the artery wall. This chronic condition advances through a series of stages leading to the atherosclerotic plaques formation that provide a barrier to arterial blood flow and may contribute to clinical events. Considerable evidence has been gathered in support of the hypothesis that free-radical-mediated oxidative processes and its specific products play a key role in atherogenesis [62]. At the centre of this hypothesis are low-density lipoproteins (LDL), which as part of their normal circulation, occasionally leaves the antioxidantreplete plasma, entering the sub-endothelial space of arteries. Here LDL lipids are oxidized. The oxidized form of LDL (oxLDL) is capable of initiating processes that contribute to the formation of atherosclerotic lesions. oxLDL is taken up by macrophages and induces the release of factors that recruit other cells and stimulate smooth muscle cell proliferation. oxLDL may also upregulate expression of cellular adhesion molecules that facilitate leukocyte binding. All of these events speed up the formation of plaque, which may results heart attack and stroke in many patients [63]. The oxidation theory is supported by the presence of oxLDL within atherosclerotic lesions and the correlation between the sensitivity of LDL to oxidation and risk of CVD. Moreover, LDL oxidization can be inhibited by nutritional antioxidants. Several epidemiological evidences and interventional studies correlate higher level of antioxidant-rich food uptake with lower incidence of CHD [64].
3.2.3. Stroke/Ischemia-Reperfusion
Stroke is one the main causes of disability and mortality in many countries. Especially the condition of ischemia and reperfusion occurring after stroke has been shown to be associated with free radical-mediated reactions potentially leading to cell death [65]. Although ischemic and hemorrhagic stroke have different risk factors and patho-physiological mechanisms, there is evidence of an increased generation of free radicals and other reactive species in both conditions, leading to oxidative stress. Ischemic stroke is the consequence of the interruption or severe reduction of blood flow in arteries followed by physiological and metabolic changes that appear within seconds of the cessation of blood flow. When anoxia is followed promptly by reperfusion, tissue can be saved but reperfusion might potentially have negative conesquences: upon reoxygenation, OS is rapidly built up and numerous non-enzymatic oxidation reactions take place both in the cytosol and/or in cellular organelles. Independent of the mechanisms responsible for ischemic stroke, ischemia causes a cascade of events that can increase free radical production via several different pathways, including inflammatory cells, xanthine oxidase, cyclooxygenase, and mitochondria. Also the large increases in glutamate and aspartate that accompany ischemia may contribute to free radical generation by excitotoxic mechanisms [66]. Upon reperfusion, the accumulation of blood borne inflammatory cells, such as neutrophils and monocytes/macrophages might further promote oxidative stress. Consequently, a large excess of O2-derived free radicals appears during the first minute of reperfusion and peaks some 4 to 7 minutes after the onset of reperfusion. Clinical trials try to assessing the efficacies of antioxidant treatment or of antioxidant-rich nutritional intervention in stroke are in progress. And these should always include the measurement of biomarkers of oxidative stress to delineate their ability for predicting long-term clinical outcome and therapeutic response.
3.2.4. Obesity
Associations between obesity and markers of oxidative stress and the susceptibility of lipid to oxidative modifycation have been observed in humans [67]. Several hypotheses have been proposed for the observed associations between obesity and oxidative stress. For example, it has been suggested that oxidative stress in obesity may result, in part, from the accumulation of intracellular triglycerides [68]. Intracellular triglycerides are proposed to elevate superoxide radical production within the electron transport chain by inhibiting the mitochondrial adenosine nucleotide transporter. The inhibition of this transporter leads to a decrease in intra-mitochondrial adenosine diphosphate (ADP) that, in turn, reduces the flux of protons through the adenosine triphosphate-synthase reaction (i.e., the adenosine triphosphate-synthase reaction requires ADP as substrate). As a result, electrons build up within the electron transport chain, which can then reduce O2 to form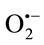 . Another hypothetical source of increased oxidative stress may be the presence of excessive adiposity as because adipocytes and preadipocytes have been identified as sources of inflammatory cytokines [69]. Cytokines are potent stimuli for the production of ROS/RNS by macrophages and monocytes. Specifically, cytokines up-regulate the activity of oxidant generating enzymes, including NAD(P)H-oxidase, inducible NOS, and myeloperoxidase. If the accumulation of intracellular triglycerides or tissue adiposity promotes increased oxidative stress, then reduction of total body fat through diet and/or exercise may be an effective means of reducing systemic inflammation and OS. Consistent with this prediction, reductions in plasma markers of oxidative stress and in ROS generation by isolated leukocytes have been observed after 4 weeks of energy restriction and weight loss [70]. However, the evidence is far from conclusive and the independent contribution of energy restriction and fat loss is remaining unclear.
. Another hypothetical source of increased oxidative stress may be the presence of excessive adiposity as because adipocytes and preadipocytes have been identified as sources of inflammatory cytokines [69]. Cytokines are potent stimuli for the production of ROS/RNS by macrophages and monocytes. Specifically, cytokines up-regulate the activity of oxidant generating enzymes, including NAD(P)H-oxidase, inducible NOS, and myeloperoxidase. If the accumulation of intracellular triglycerides or tissue adiposity promotes increased oxidative stress, then reduction of total body fat through diet and/or exercise may be an effective means of reducing systemic inflammation and OS. Consistent with this prediction, reductions in plasma markers of oxidative stress and in ROS generation by isolated leukocytes have been observed after 4 weeks of energy restriction and weight loss [70]. However, the evidence is far from conclusive and the independent contribution of energy restriction and fat loss is remaining unclear.
Since chronic hyperglycaemia is more prevalent in obese individuals, oxidative stress is also believed to play a major role in the development of obesity-related disorders including diabetes and hypertension, although the cause and effect relationships are not always clear.
3.3. Oxidative Stress and Kidney Disease
3.3.1. Urolithiasis
Urolithiasis is one of the most common disease of the urinary tract which has been afflicting human kind since antiquity. Urolith formation is a multifactorial process [71]. For most human diseases, increased formation of reactive oxygen species is secondary to primary disease process [72]. Similarly association of urolithiasis and free radicals has been reported [73]. Experiments performed on animals [74], cultures [75] and human sera [76] have revealed that there is presence of enhanced oxidative stress in stone forming conditions. Oxalate is known to induce lipid peroxidation by unknown mechanism which causes disruption of the structural integrity of the membranes [77]. Superoxide dismutase (SOD) is an inbuilt defense mechanism to fight back peroxidative stress. Along with SOD in response to the damaging peroxidative effect, α-tocopherol has proved to be an efficient protector to the membrane integrity [78]. The levels of serum malondialdehyde, nitrite, α-tocopherol, plasma ascorbate and erythrocyte superoxide dismutase are responsible for the pathogenesis of urolithiasis.
3.3.2. Diabetic Nephropathy
Recent years, diabetes and diabetic kidney disease continue to increase worldwide. In the USA, diabetes-associated kidney disease is a major cause of all new cases of end-stage kidney disease. All diabetic patients are considered to be at risk for nephropathy. Today we have not specific markers to expect development of end-stage renal disease. Clinically control of blood sugar level and blood pressure regulations are important two parameters to the prevention of diabetic nephropathy [79,80]. There are huge amount of in vitro and in vivo studies regarding explanation of mechanism of diabetes-mellitus-induced nephropathy. All of these mechanisms are a consequence of uncontrolled elevation of blood glucose level. Currently the proposed mechanism is the glomerular hyperfiltration/hypertension hypothesis. According to this hypothesis, diabetes leads to increased glomerular hyperfiltration and a resultant increased glomerular pressure. This increased glomerular pressure leads to damage to glomerular cells and to development of focal and segmental glomerulosclerosis [81,82]. Angiotensin II inhibitors reduce glomerular pressure and prevent albuminuria. Increased angiotensin II level induces OS through activation of NADPH oxidase, stimulating inflammatory cytokines, and so forth [83,84]. Increased blood glucose promotes glycosylation of circulator and cellular protein and may initiate a series of autooxidation reactions that culminate in the formation and accumulation of advanced glycosylation end-products (AGEs) in tissues. The AGEs have oxidizing potential and promote tissue damage by oxygen-free radicals [85]. In experimental studies, formation of OS increases because of high level of blood glucose. Sadi et al. showed that in diabetic rat kidney antioxidant enzyme, namely, catalase (CAT) and glutathion peroxidase (GSHPx), activities were found to be reduced; however, α-lipoic acid and vitamin C administration increased these antioxidant enzyme activities [86]. Increased OS is the common finding in tissues effecting from diabetes, including kidney. Reddi et al. showed that transforming growth factorβ1 (TGF-β1) is prooxidant and Se (selenium) deficiency increases OS via this growth factor. Chen et al. showed that nitrosative stress increases in diabetic rat model [87]. These results show the induction of oxidative and nitrosative stress in rat kidney. These may have a role in pathophysiology of diabetes-induced morphological and functional changes of kidney.
3.4. Oxidative Stress and Lung Disease
3.4.1. Asthma
Many observations suggest that oxidative stress plays an important role in the pathogenesis of asthma. In the airway, losing control of oxidants may bring about initiation of Th2-dominant immunity instead of inducing immune tolerance in the initial phase of development of airway allergic inflammation [88-90]. Furthermore, enhanced oxidative stress may contribute to the progression or perpetuation of existing airway inflammation through enhanced airway hyper responsiveness, stimulation of mucin secretion, and induction of various proinflammatory chemical mediators [91] all of which are believed to be related to severe asthma [92]. Higher incidences of bronchial asthma have been reported in areas with air pollution, which is a representative stimulus among exogenous oxidants [93,94]. Reduced intake of foods containing antioxidants is also related to the increased incidence of asthma [95]. Increased oxidative stress in asthmatic patients is also related to suppressed pulmonary function. You Sook Cho et al. showed that increased oxidative stress in the airway precedes the development of allergic inflammation, airway hyperresponsiveness, and other pivotal features of asthma such as enhanced mucus secretion. Therefore, it is strongly suggested that an increased level of ROS acts as a critical contributor to the induction of allergic airway inflammation. Controlling intracellular oxidative stress with appropriate timing, as opposed to simply focusing on the reduction of oxidative stress, is important for effectively managing bronchial asthma [96].
3.4.2. Pulmonary Fibrosis
Pulmonary fibrosis is the end result of a diverse group of lung disorders. Although there are multiple initiating agents for pulmonary fibrosis, including toxins, fibres/ particles, autoimmune reactions, drugs and radiation, the aetiology of the majority of cases of pulmonary fibrosis is unknown. Several studies have suggested that oxidantantioxidant imbalance in the airways plays a critical role in the pathogenesis of idiopathic pulmonary fibrosis (IPF) [97,98]. In addition, oxidants may contribute to the development of pulmonary fibrosis due to their effects on the production of cytokines and growth factors such as TGF-β, a key regulator of aberrant repair mechanisms that are characteristic of many fibrotic diseases including IPF. There are several potential interactions between TGF-β and oxidants/antioxidants in the lung. TGF-β not only induces ROS production by activation of NADPH oxidases and/or mitochondrial dysfunction, but also decreases natural cellular antioxidant production through decreased expression of both catalase and mitochondrial SOD [99,100]. Increased levels of oxidized proteins have been reported in human subjects with IPF [101,102]. Some studies have reported that various antioxidant enzyme systems protect against lung fibrosis [103]. But IPF subjects also have lower antioxidant capacity than healthy subjects [104]. Thus, oxidants and TGF-β seem to interact to enhance the fibrotic response in the lungs.
3.4.3. Lung Cancer
Reactive oxygen species have been suggested to stimulate oncogenes such as Jun and Fos. Overexpression of Jun is directly associated with lung cancer [105,106]. In lung cancers, p53, which is associated with the production of ROS, is often mutated and defective in inducing apoptosis. When mutated, p53 accumulates in the cytoplasm and functions as an oncogene [107]. Modification of proteins and lipids may increase the risk of mutagenesis, through formation of genotoxic lipid peroxidative by-products that react with DNA, oxidative modification of DNA polymerase or inhibition of DNA repair enzymes [108].
3.5. Oxidative Stress and Eye Disease
Oxidative stress has been implicated in the pathogenesis of several eye conditions such as cataract, macular degeneration, diabetic retinopathy and retinitis pigmentosa, corneal disease, [109-114].
3.5.1. Cataract
Contemporary hypothesis considers oxidative stress as an important factor in age-related processes in the body including senile cataract [115]. Production of ROS and reduction of endogenous antioxidants both contribute to cataract formation. UV-induced oxidation damage seems to play a major role in a number of specific pathological conditions of intraocular tissues such as cataract and retinal degeneration. The crystalline lens is constantly subjected to oxidative stress from radiation and others sources and this can damage the crystalline proteins, lipids, polysaccharides and nucleic acids [115]. However, it has several mechanisms to protect its components from oxidative stress and to maintain its redox state. These include enzymatic pathways and high concentration of ascorbate and reduced glutathione [116]. However, with ageing, accumulation of oxidized lens components and decreased efficiency of repair mechanisms can contribute to the development of cataract [116]. Chronic UV-induced ROS formation is believed to be responsible for various degenerative diseases in the eye including cataract formation [117]. According to Berthoud and Beyer oxidative stress-induced damage to lens gap junctions and consequent altered intercellular communication may contribute to cataract formation. Lipid peroxidation (LPO) has been proposed as a causative factor of cataract. Sawada et al. found a significant increase in superoxide dimutase activity and protein level in nuclear cataracts, suggesting the involvement of oxidative stress [118].
3.5.2. Age Related Macular Degeneration (AMD)
Age-related macular degeneration is a leading cause of blindness in the developed countries [119]. The retinal is highly susceptible to photochemical damage from the continuous exposure to UV, but the relationship between UV light exposure and AMD is unclear, although short wavelength radiation and blue light induce significant oxidative stress to the retinal pigment epithelium [119]. There are several reports that have associated oxidative stress with the cellular damage caused by ROS in the pathogenesis of AMD [120]. The retina is particularly susceptible to oxidative stress because of its high oxygen consumption, its high proportion of polyunsaturated fatty acids and its exposure to visible light [121]. Klein et al. found that cataract was associated with incidence of early AMD, soft indistinct drusen, increased retinal pigmentation and progression of AMD [122]. Several risk factors for AMD such as genetics, age, exposure to sunlight and smoking have been reported. In a review by Ambati et al. [176] it was found that photo-oxidative damage to the retina, mediated by ROS were implicated in the development of AMD. ROS including other free radicals have been implicated in the apoptotic cell death and the development of pathological changes in AMD [123-126]. Janick-Papis et al. stressed that oxidative stress is a major factor in the pathogenesis of AMD [127]. Retinal pigment epithelium cells are prone to ROS arising from intense oxygen metabolism. Also, the cells can be exposed to ROS as a consequence of accumulation of iron ions in the cells, sunlight exposure and tobacco smoke [127]. The macular pigment formed by two dihydroxycarotenoids, lutein and zeaxanthin is a natural barrier protecting the macula against oxidative stress [128]. However, retinal factors such as intense oxygen metabolism, exposure to ultraviolet radiation, high concentration of polyunsaturated fatty acids and presence of photosensitizers may increase the production of ROS [128]. There is experimental evidence in support of oxidative damage to the retinal pigment epithelium and the choriocapillaries that is both light and oxygen-dependent, owever, the precise linkage between oxygen-induced events and the progression of AMD remains unclear [129,130]. It is considered that ROS including free radicals are responsible for apoptotic cell death and the development of pathological changes in AMD [130]. Research findings have demonstrated that a diet poor in antioxidant micronutrients (vitamin C, E, carotenoids, zinc) and low plasma levels of antixidants may favor the development of the AMD. San Giovanni and Chew [131] have reported that mega-3 long-chain polyunsaturated fatty acids exhibit cytoprotective and cytotherapeutic actions providing anti-angiogenic and neuroprotective mechanisms within the retina and this may have a protective role against ischemic-, light-, oxygen-, inflammatory-, and age-related pathology of the ascular and neural retina. It has been reported that micronutrient supplementation enhances antioxidant defense and might prevent or retard AMD or modify the course of the disease.
3.5.3. Diabetic Retinopathy (DR)
Diabetes has been implicated in the increased oxidative stress which is thought to play an important role in the pathogenesis of various diabetic complications [132-134]. However, the source of the hyperglycemia induced oxidative stress is not clear. Yue et al. investigated the correlation between redox status and oxidative stress in the eyes, aorta and kidneys of streptozotocin (STZ)-induced diabetic rats [135]. The primary endogenous antioxidants, glutathione (GSH) and malondialdehyde (MDA) (markers of oxidative stress) in those tissues were measured after strepto-zotocin (STZ) injection at different times and it was observed that oxidative stress occurred in the eyes and aorta, but not the kidneys of diabetic rats. It was found that ROS generation and lipid peroxidation may play a vital role in the development of diabetic complications in the eyes and aorta and that the administration of vitamin E may prevent ROS-induced lipid peroxidation and thereby limit the development of diabetic complications in the aorta and eyes [135]. Possible sources of oxidative stress and damage to proteins in diabetes include free radicals generated by auto-oxidation reactions of sugars and sugar adducts to proteins [136]. The oxidative stress may be amplified by a continuing cycle of metabolic stress, tissue damage and cell death, leading to increased free radical production and compromised free radical inhibitory and scavenger systems, which further exacerbate the oxidative stress [136].
3.5.4. Autoimmune Uveitis (AIU)
Autoimmune and inflammatory uveitis are a group of potentially blinding diseases that arise without a known infectious trigger and are often associated with immunological responses to unique retinal proteins [137]. Experimental studies have implicated free radicals in the pathogenesis of this eye condition [137,138] suggesting that free radicals and oxidative stress play a role in the pathogenesis of the disease. Photoreceptor mitochondrial oxidative stress has been considered to be the initial pathological event in experimental autoimmune uveitis. Determination of alterations in retinal mitochondrial levels in response to oxidative stress during the early phase of experimental autoimmune uveitis showed the presence of mitochondrial specific oxidative stress-related proteins in the retina along with down regulation of ATP synthase; providing evidence of stress related retinal damage [139].
3.5.5. Retinitis Pigmentosa (RP)
Retinitis pigmentosa, a heterogeneous group of inherited retinal disorders characterized by progressive photoreceptor apoptosis, is the leading cause of inherited retinal degeneration-associated blindness worldwide [140,141]. It is a disease in which one of a variety of mutations selectively causes rod photoreceptor cell death, followed by gradual death of cone cells resulting in blindness [140]. According to Komeima et al. although RP is commonly considered to be genetic in origin, oxidative stress plays a role in its pathogenesis [140]. The authors suggested that protection from oxidative damage may be a broadly applicable treatment strategy in RP. According to Usui et al. [141] following the death of the rod photoreceptors from a variety of mutations, the level of tissue oxygen in the outer retina becomes elevated and there was a progressive oxidative damage to cones that ultimately triggers apoptosis. The authors [141] further reported that NADPH oxidase (Nox) plays a critical role in generation of the oxidative stress that leads to cone cell death in RP and proposed that inhibition of Nox provides a new treatment strategy.
3.6. Oxidative Stress and Skin Disease
Since the skin is always in contact with oxygen in the presence of surface lipid, it is one of the best target organs of environmental oxidative damage. Among the various insults brought by UV irradiation, free radicals and lipid peroxides are one of the most reasonable candidates for explaining injuries. Besides direct absorption of UVB photons by DNA and subsequent structural changes, generation of ROS following irradiation with UVA and UVB requires the absorption of photons by endogenous molecules called photosensitizers. When a molecule absorbs UV radiation, it become electronically excited and becomes a short-lived free radical. This process is called photosensitization; several cellular constituents (e.g., porphyrins, flavins, quinones and others) and biologically active drugs (e.g., tetracyclines, thiazides) can act as photosensitizers within skin cells. Because most photosensitized reactions are oxygen dependent, UV irradiation absorption results in the generation of ROS. Free radicals are also produced by neutronphils (white blood cells having immune functions) that are increased in photodamaged skin and contribute to the overall prooxidant state. Thus, UV-induced generation of ROS in the skin develops oxidative stress and causes many skin disease.
3.7. Oxidative Stress and Reproductive System Disease
Oxidative stress (OS) has been identified as one factor that affects fertility status and thus, has been extensively studied in recent years [142]. Tobacco smoke consists of approximately 4000 compounds such as alkaloids, nitrosamines and inorganic molecules, and many of these substances are reactive oxygen or nitrogen species. Significant positive association has been reported between active smoking and sperm DNA fragmentation [143], as well as axonemal damage and decreased sperm count [144]. Sperm from smokers have been found to be significantly more sensitive to acidinduced DNA denaturation than those from non smokers because the smokers’ sperm have been shown to contain higher levels of DNA strand breaks [145]. Thus, smoking does, in fact, affect the quality and quantity of sperm present within a male.
Several studies have examined the role of oxidative stress and pregnancy complications. Two retrospective studies [146,147] reported that reduced antioxidant status increased risk of spontaneous abortion. Vural et al. [147] demonstrated that plasma levels of ascorbic acid (vitamin C) and α-tocopherol (vitamin E) were significantly lower in women with recurrent spontaneous abortion, although a prospective investigation is needed to rule out the possibility that lower levels of plasma antioxidants are not a result of the spontaneous abortion rather than the reverse. ROS have been implicated in the development of premature rupture of the fetal membranes [148,149] and evidence suggests that oxidative stress may be associated with preeclampsia [149,150].
3.8. Oxidative Stress and Blood Disorder
3.8.1. Beta-Thalassemia
Production of free radicals by iron overload, alteration in serum trace elements, and antioxidant enzymes status play an important role in the pathogenesis of beta thalassemia major [151]. Beta thalassemia major is an inherited disease resulting from reduction or total lack of beta globin chains. In patients with beta thalassemia major where frequent blood transfusions are required due to severe anemia, oxidative stress occurs as a result of increased levels of lipid peroxides and free-radical intermediates, as well as the decrease in total antioxidant capacity. Use of iron chelatory agents in combination with antioxidants can be helpful in the regulation of the antioxidant status in patients with beta thalassemia major. Oxidative stress and disturbance in antioxidant balance in beta thalassemia major has been studied extensively [152,153]. One study group found a significant increase in the levels of lipid peroxide and iron and significant decrease in levels of vit E and total antioxidant capacity. Serum zinc was significantly increased while copper levels decreased and there is a nonsignificant increase in erythrocyte superoxide dismutase. The results suggested that the oxidative stress and decreased antioxidant defence mechanism play an important role in the pathogenesis of beta thalassemia major.
3.8.2. Acute Lymphoblastic Leukemia (ALL)
Impairment of antioxidant role of CAT and SOD may support the accumulation of free radicals. Alternatively, it is possible that the antioxidant system is disturbed as a consequence of an abnormality in the antioxidative metabolism due to the cancer process. This effect could be enhanced by the characteristic increase in the production of H2O2 by the cancer cells [154]. El-Shabag et al. study revealed that the levels of glutathione (GSH) was statistically significant decreased in just diagnose ALL patients and in treatment group compared to controls. This finding may indicate a possible link between decreased GSH and increased levels of cells alterations due to oxidative damage, supporting the idea that there is a persistence of oxidative stress in acute lymphoblastic leukemia [155,156] With respect to Vitamin C levels in plasma, it was demonstrated that Vit. C was significantly decreased in both ALL diagnosed patients and patients after treatment compared to controls.
3.9. Oxidative Stress and Joint Disorder
Oxidative stress and impaired antioxidant systems have an important role in etiology of rheumatoid arthritis (RA), pathogenesis of joint tissue injury and chronic inflammation [157-159] and may lead to dyslipidimia, atherosclerosis, connective tissue degradation and joint and periarticular deformities [160-162]. It causes immunomodulation, which may lead to autoimmune diseases such as systemic lupus erythematosus (SLE) and anti-phospholipid syndrome [163,164].
3.9.1. Rheumatoid Arthritis
An increase in reactive oxygen species (ROS) plays an important role in the pathogenesis of rheumatoid arthritis (RA) [165-167] and antioxidants and antioxidative enzymes have been shown to reduce cartilage damage in animal models of RA [168,169]. Excessive production of ROS can damage protein, lipids, nucleic acids and matrix components [170]. Increased ROS levels in RA result in a pro-oxidation environment, which in turn could result in decreased antioxidant activity and increased malondialdehyde (MDA) levels (an oxidative stress parameter) [171]. In RA, ROS are important intracellular signaling molecules in the cells of the immune system that amplify the synovial inflammatory-proliferative response [170]. T-cells are exposed to increased oxidative stress and become refractory to growth and death stimuli, which further contributes to the perpetuation of the immune response [172]. The chronic inflammation of RA has been associated with oxidative stress and a decline in the levels of cellular antioxidant sulfhydryls [173].
Persistent inflammation results in destruction of cartilage and bone. This occurs through a number of mechanisms, including oxidative and proteolytic breakdown of collagen and proteoglycans [174-177]. Once sequestered within the joint space, neutrophils degranulate and release a variety of potentially harmful enzymes and peptides [178]. They may also undergo a respiratory burst and generate several reactive oxygen species, including superoxide, hydrogen peroxide, hypohalous acids, and possibly hydroxyl radical [179]. Although these destructtive oxidants have often been held partly responsible for joint destruction, compelling evidence that they are in fact produced within the synovium is lacking.
3.9.2. Temporomandibular (TMB) Joint Disorders
It has been suggested that mechanical stresses can lead to ROS-induced oxidative stress of the temporomandibular joint (TMJ) [180]. Excessive oxidative stress in the TMJ thus results in tissue damage, which further propagates to temporomandibular disorder(TMD). Using electron spin resonance (ESR) spin trapping technique, it has been previously demonstrated that hydroxyl radical (HO•) could be generated by catalytic reaction of H2O2 with free iron ions (Fenton reaction) in the IL-1a induced TMJ rat arthritis model or in synovial fluid (SF) of the TMD patients [181,182]. ROS can be generated in the TMJ by several pathways: they include 1) direct mechanical injury [183], 2) hypoxia-reperfusion [184] and 3) arachidonic acid catabolism to the articular tissues (Figure 6).
ROS affect various molecular species of the TMJ and deteriorate the TMJ function: they include 1) reduction of SF viscosity by depolymerization and/or molecular configuration of hyaluronic acid (HA) [185,186]; 2) reduction of lubrication of the articular surface by dete-
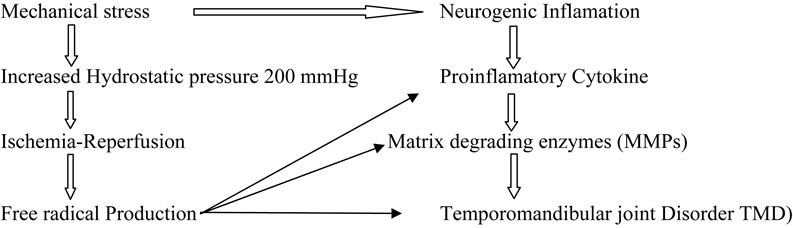
Figure 6. Proposed mechanism s of degenerative TMJ disease.
rioration of the surfaceactive phospholipid (SAPL) layer, which acts as an extremely efficient boundary lubricant and protector of articular surfaces [187]; 3) breakdown of collagen proteoglycans [188]; 4) activation of cartilage degrading enzymes such as matrix metalloproteinases. The ROS, especially HO• is responsible for lipid peroxidation and disruption of cellular homeostasis. Nitzan reported that lysis of SAPL layer by phospholipase A2 (sPLA2) together with the depolymerization of HA caused by free radicals may result in a deteriorated lubrication of the articular surface, thus further proceeding to the internal derangement (ID) of the TMJ [189]. The HO• also degrades collagen and proteoglycans (Pgs) into low molecular masses, which act as immunogens for synoviocytes or chondrocytes [190]. These denatured Pgs may induce proinflammatory cytokines from various cells in the TMJ compartment. Evidence suggests that HO• can show dose dependent activatation of pro MMP 9 by affecting its molecular configuration. These data suggest that free radicals may cause molecular deterioration, which further proceeds to degenerative changes in the TMJ. Cytokines, IL-1, and TNF-alpha have been shown to increase the release of 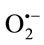 from cultured chondrocytes and synovial cells in a dose-dependent fashion thereby causing DNA damage [191,192].
from cultured chondrocytes and synovial cells in a dose-dependent fashion thereby causing DNA damage [191,192].
3.9.3. Systemic Lupus Erythematosus (SLE)
Some studies have found that oxidative stress is increased in SLE [193-196] but others have reported conflicting results [197]. An increase in malondialdehyde (MDA), a product of lipid peroxidation, has been reported in serum/plasma [198,199] and erythrocytes [200,201] in patients with SLE. Anti-oxidant enzymes such as SOD, CAT and GPx have been studied intensively in the serum and erythrocytes of SLE patients but controversies still exist. Recently, the role of the thiol in the progression of inflammatory and autoimmune disease has been intensively studied [202].The cellular level of GSH affects T helper cell maturation and Th1/Th2 balance in the cell [203]. The low GSH has been found in SLE and changes in the production of cellular GSH play a major role in the modulation of Th1/Th2 balance in the cell [204].
Measurement of plasma F2-isoprostane with highly sensitive and specific mass spectroscopy/gas chromatography (MS/GC) methodology provides a window into the balance between the production of ROS and antioxidant defences in vivo. Isoprostanes are generated by a free radical mediated peroxidation of arachidonic acid, independent of cyclooxygenase activity [204]. Elevated levels of F2-isoprostane are associated with oxidant injury to tissue in a variety of disorders including ischemiareperfusion injury, atherosclerosis, diabetes and autoimmune disease [205,206]. In lupus, plasma F2-isoprostane levels have been linked to high disease activity, as well as to the presence of renal disease and anti-phospholipid syndrome [207-210]. Evidence of increased oxidative stress has also been observed in disorders in which fatigue is a prominent symptom, including chronic fatigue and fibromyalgia [211-215].
3.10. Oxidative Stress and Liver & Pancreatic Disease
The liver is an important organ which has a central role in metabolic homeostasis, because it is responsible for the metabolism, synthesis, storage and redistribution of nutrients, carbohydrates, fats and vitamins [216]. The liver is also an important place for free radicals. Liver enzymes, including diamine oxidase, aldehyde dehydrogenase, tryptophan dual oxidase, liver dehydrogenase and the cytochrome P450 enzyme system, induce oxidation and uncoupling [217]. Hepatocytes have more mitochondria than hepatic stellate cells (HSC), so they produce a large number of highly unstable ROS. Moreover endoplasmic reticulum and peroxisomes have a greater capacity to produce ROS than mitochondria, at least in the liver [218]. Nitric oxide synthase is present in liver parenchymal cells and Kupffer cells [219]. Inflammation of the liver occurs when nitrogen oxides exist in the form of free thiol with a reversible group through the role of glutathione-S-transferase (GSH-ST) [220]. Nitrogen oxides can form reactive intermediates, such as nitro-cool acid, which causes necrosis of liver cells, inhibition of mitochondrial function and consumption of cell pyridine nucleotides, leading to breakdown of DNA. Nitric oxide and peroxynitrite ion radicals can combine to form hydrogen peroxide compound, which causes inhibition of mitochondrial respiration, Na+/K+ pump function and the phosphorylation of kinases, leading to cell damage [221]. Overproduction of RNS may lead to nitrosylation reactions that can alter the structure of proteins and thus impair their normal functions [222].
Polyunsaturated fatty acids and hydrogen peroxide radicals act as intermediates of the formation of lipid peroxides on cell membrane [223]. Peroxides can also be formed by aldehyde compounds, causing inflammation and organ fibrosis, as in the case of alcoholic liver disease [224]. In addition, human non-alcoholic liver disease and hepatitis C show different quantities of thiobarbiturates which also indicates high level of lipid peroxides in these patients [225]. The activity of superoxide dismutases (SOD), glutathione peroxidase (GSH-PX) in chronic liver cirrhosis and hepatitis is significantly lower than control, which is negatively correlated with serum alanine aminotransferase (ALT) level [226]. By testing different types of patients with liver disease, previous studies showed serum SOD decreased significantly [227]. Oxidative stress is closely related to the pathological damage of hepatic fibrosis [228,229]. In chronic hepatitis C, oxidative stress is also increased and antioxidant defense mechanisms are inadequate. Serum levels of oxidized protein products, such as MDA, nitric oxide (NO), and the activities of myeloperoxidase (MPO), arylesterase (AE) and paraoxonase-1 (PON1), are higher in chronic hepatitis C patients when compared to healthy individuals, which indicates that these molecules might be useful tools as biomarkers [230]. Several studies suggested that markers of oxidative stress increase in patients with alcoholic hepatitis, such as CYP2E, which increased 15 - 20-fold, leading to excessive electron leakage and release of ROS, causing oxidative stress and immune responses [231,232]. Redox reaction followed by imbalance and oxidative damage often leads to subclinical hepatitis without jaundice, inflammatory necrotic hepatitis, liver cirrhosis and cancer [233].
3.10.1. Diabetes
Oxidative stress and oxidative damage to the tissue are common end points of some chronic diseases, like diabetes [234]. During diabetes, persistent hyperglycemia causes increased production of free radicals, especially reactive oxygen species (ROS), for all tissues from glucose auto-oxidation and protein glycosylation. The increase in the level of ROS in diabetes could be due to their increased production and/ or decreased destruction by nonenzymic and enzymic catalase (CAT), reduced glutathione (GSH), and superoxide dismutase (SOD) antioxidants. The level of these antioxidant enzymes critically influences the susceptibility of various tissues to oxidative stress and is associated with the development of complications in diabetes [235].
3.10.2. Wilson’s Disease
Many studies suggest that oxidative stress is responsible for the pathogenesis of liver injury [236]. Wilson’s disease (WD), a copper overload disease, is inherited as an autosomal recessive trait [237]. While it is a relatively rare disorder in European countries, it is a widespread cause of chronic liver disease in childhood in Turkey, where consanguineous marriages occur in 25% of the population [238]. Oxidative stress resulting from an increased production of free radicals via the copper accumulation and defects in antioxidant defenses may be central to the toxic processes in WD [239-241]. Previous studies have demonstrated that malondialdehyde (MDA) levels increase and antioxidant capacity decreases in acute and chronic hepatitis [242]. Sokol et al. and Mansouri et al. suggest that mitochondrial lipid peroxidation takes place at varying levels in Wilson disease [243].
4. CONCLUSION
Extensive data suggest that oxidative damage may play a major role in the causation of a number of human diseases. Data are emerging that implicate aberrant redox interactions with key proteins in the neurodegenerative diseases AD, PD and ALS, and the subsequent induction of oxidative stress leading to neurodegeneration. Not only limited to neurodegenerative disorders, oxidative stress manifests its toxic effects through a variety of different pathways and other diseases. In order to maintain defense against the destructive oxygen species, an appropriate balance of enzymatic and non-enzymatic anti-oxidative defenses is necessary.
REFERENCES
- Sies, H. (1985) Oxidative stress. Academic Press, San Diego, 1-8.
- Sies, H. (1986) Biochemistry of oxidative stress. Angewandte Chemie International Edition in English, 25, 1058-1071. doi:10.1002/anie.198610581
- Sies, H. and Cadenas, E. (1985) Oxidative stress: Damage to intact cells and organs. Philosophical Transactions of the Royal Society of Lond: Biological Sciences, 311, 617-631. doi:10.1098/rstb.1985.0168
- Nathan, C. (2003) Specificity of a third kind: Reactive oxygen and nitrogen intermediates in cell signalling. Journal of Clinical Investigation, 111, 769-778.
- Kröncke, K.D. (2003) Nitrosative stress and transcription. Biological Chemistry, 384, 1365-1377. doi:10.1515/BC.2003.153
- Garrido, N., Meseguer, M., Simon, C., Pellicer, A. and Remohi, J. (2004) Pro-oxidative and anti-oxidative imbalance in human semen and its relation with male fertileity. Asian Journal of Andrology, 6, 59-65.
- Fridovich, I. (1997) Superoxide anion radical, superoxide dismutase, and related matters. The Journal of Biological Chemistry. 272, 18515-18517. doi:10.1074/jbc.272.30.18515
- Halliwell, B. and Gutteridge, J.M.C. (1999) Free radicals in biology and medicine. 3rd Edition, Clarendon Press, Oxford.
- Beckman, K.B. and Ames, B.N. (1999) Endogenous oxidative damage of mtDNA. Mutation Research, 424, 51- 58. doi:10.1016/S0027-5107(99)00007-X
- Wei, Y.-H., Lu, C.-Y., Wei, C.-Y., Ma, Y.-S. and Lee, H.-C. (2001) Oxidative stress in human aging and mitochondrial disease-consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chinese Journal of Physiology, 44, 1-11.
- Evans, J.L., Goldfine, I.D., Maddux, B.A. and Grodsky, G.M. (2005) Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes, 52, 1-8. doi:10.2337/diabetes.52.1.1
- Finkel, T. and Holbrook, N.J. (2000) Oxidants, oxidative stress and the biology of ageing. Nature, 408, 239-247. doi:10.1038/35041687
- Vaziri, N.D., Dicus, M., Ho, N.D., Boroujerdirad, L. and Sindhu, R.K. (2003) Oxidative stress and dysregulation ofsuperoxide dismutase and NADPH oxidase in renal insufficiency. Kidney International, 63, 179-185. doi:10.1046/j.1523-1755.2003.00702.x
- Apel, K. and Hirt, H. (2004) Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annual Review of Plant Biology, 55, 373-399. doi:10.1146/annurev.arplant.55.031903.141701
- Meister, A. and Anderson, M.E. (1983) Glutathione. Annual Review of Biochemistry, 52, 711-760. doi:10.1146/annurev.bi.52.070183.003431
- Raghuvanshi, R., Kaul, A., Bhakuni, P., Mishra, A. and Misra, M.K. (2007) Xanthine oxidase as a marker of myocardial infarction. Indian Journal of Clinical Biochemistry, 22, 90-92. doi:10.1007/BF02913321
- Sevanian, A. and Hochstein, P. (1985) Mechanisms and consequences of lipid peroxidation in biological systems. Annual Review of Nutrition, 5, 365-390. doi:10.1146/annurev.nu.05.070185.002053
- Jamieson, D. (1986) The relation of Free radical production to Hyperoxia. Annual Review of Physiology, 48, 703-719. doi:10.1146/annurev.ph.48.030186.003415
- Sharma, R.K. and Agarwal, A. (2004) Role of reactive oxygen species in gynecologic diseases. Reproductive medicine and Biology, 4, 177-199.
- Kunwar, A., et al. (2011) Free radicals, oxidative stress and importance of antioxidants in human health. Journal of Medical and Allied Sciences, 1, 53-60.
- Chaitanya, K.V., Pathan, A.A.K., Mazumdar, S.S., Chakravarthi, G.P., Parine, N. and Bobbarala, V. (2010) Role of oxidative stress in human health: An overview. Journal of Pharmacy Research, 3, 1330-1333.
- Zhang, J., Perry, G. and Smith, M.A. (1999) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. American Journal of Pathology, 154, 1423-1429.
- Yudoh, K., Trieu, N.V., Nakamura, H., Kayo, H.-M., Tomohiro, K. and Kusuki, N. (2005) Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Research & Therapy, 7, 380-391. doi:10.1186/ar1499
- Haydent, M.R. and Tyagi, S.C. (2002) Neural redox stress and remodeling in metabolic syndrome, type 2 diabetes. Journal of the Pancreas, 3, 126-138.
- Vasavidevi, V.B., Kishor, H.D., Adinath, N.S., Rajesh, D.A. and Raghavendra, V.K. (2006) Depleted nitrite and enhanced oxidative stress in urolithiasis. Indian Journal of Clinical Biochemistry, 21, 177-180. doi:10.1007/BF02912938
- Yudoh, K., Trieu, N.V., Nakamura, H, Kayo, H.-M., Tomohiro, K. and Kusuki, N. (2005) Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Research & Therapy, 7, 380-391. doi:10.1186/ar1499
- Haydent, M.R. and Tyagi, S.C. (2002) Neural redox stress and remodeling in metabolic syndrome, type 2 diabetes. Journal of the Pancreas, 3, 126-138.
- Peña-Silva, R.A., Miller, J.D., Chu, Y. and Heistad, D.D. (2009) Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. American Journal of Physiology Heart and Circulatory Physiology, 297, 1354-1360. doi:10.1152/ajpheart.00570.2009
- Vasavidevi, V.B., Kishor, H.D., Adinath, N.S., Rajesh, D.A. and Raghavendra, V.K. (2006) Depleted nitrite and enhanced oxidative stress in urolithiasis. Indian Journal of Clinical Biochemistry, 21, 177-180. doi:10.1007/BF02912938
- Verzola, D., Maria, B.B., Barbara, V., Luciano, O., Franco, D., Francesca, S., Valeria, B., Maria, T.G., Giacomo, G. and Giacomo, D. (2004) Oxidative stress mediates apoptotic changes induced by hyperglycemia in human tubular kidney cells. Journal of the American Society of Nephrology, l15, S85-S87. doi:10.1097/01.ASN.0000093370.20008.BC
- Bailey, D.M. and Davies, B. (2001) Acute mountain sickness; prophylactic benefits of antioxidant vitamin supplementation at high altitude. High Altitude Medicine & Biology, 2, 21-29. doi:10.1089/152702901750067882
- Baumgartner, R.W., Eichenberger, U. and Bartsch, P. (2002) Postural ataxia at high altitude is not related to mild to moderate acute mountain sickness. European Journal of Applied Physiology, 86, 322-326. doi:10.1007/s00421-001-0534-8
- Chao, W.H., Askew, E., Roberts, D.E., Wood, S.M. and Perkins, J.B. (1999) Oxidative stress in humans during work at moderate altitude. Journal of Nutritiion, 129, 2009-2012.
- Selley, M.L., Close, D.R. and Stern, S.E. (2002) The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiology of Aging, 23, 383-388. doi:10.1016/S0197-4580(01)00327-X
- Butterfield, D.A., Castegna, A., Lauderback, C.M. and Drake, J. (2002) Evidence that amyloid β-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiology of Aging, 23, 655-664. doi:10.1016/S0197-4580(01)00340-2
- Arlt, S., Beisiegel, U. and Kontush, A. (2002) Lipid peroxidation in neurodegeneration: New insights into Alzheimer’s disease. Current Opinion in Lipidology, 13, 289- 294. doi:10.1097/00041433-200206000-00009
- Gabbita, S.P., Lovell, M.A. and Markesbery, W.R. (1998) Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. Journal of Neurochemistry, 71, 2034- 2040. doi:10.1046/j.1471-4159.1998.71052034.x
- Alam, Z.I., et al. (1997) Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8- hydroxyguanine levels in substantia nigra. Journal of Neurochemistry, 69, 1196-1203. doi:10.1046/j.1471-4159.1997.69031196.x
- Zemlan, F.P., Thienhaus, O.J. and Bosmann, H.B. (1989) Superoxide dismutase activity in Alzheimer’s disease: Possible mechanism for paired helical filament formation. Brain Research, 476, 160-162. doi:10.1016/0006-8993(89)91550-3
- Pappolla, M.A., Omar, R.A., Kim, K.S. and Robakis, N.K. (1992) Immunohistochemical evidence of oxidative stress in Alzheimer’s disease. American Journal of Pathology, 140, 621-628.
- Spillantini, M.G., et al. (1997) α-synuclein in Lewy bodies. Nature, 388, 839-840. doi:10.1038/42166
- Gasser, T. (2001) Genetics of Parkinson’s disease. Journal of Neurology, 248, 833-840. doi:10.1007/s004150170066
- Marsden, C.D. (1983) Neuromelanin and Parkinson’s disease. Journal of Neural Transmission (Supplementa), 19, 121-141.
- Hirsch, E., Graybiel, A.M. and Agid, Y.A. (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature, 334, 345- 348. doi:10.1038/334345a0
- Wakamatsu, K., Fujikawa, K., Zucca, F.A., Zecca, L. and Ito, S. (2003) The structure of neuromelanin as studied by chemical degradative methods. Journal of Neurochemistry, 86, 1015-1023. doi:10.1046/j.1471-4159.2003.01917.x
- Zecca, L., et al. (2003) The neuromelanin of human substantia nigra: Structure, synthesis and molecular behaveiour. Journal of Neural Transmission. Supplementum, 65, 145-155. doi:10.1007/978-3-7091-0643-3_8
- Lotharius, J., et al. (2002) Effect of mutant α-synuclein on dopamine homeostasis in a new human mesencephalic cell line. The Journal of Biological Chemistry, 277, 38884-38894. doi:10.1074/jbc.M205518200
- Lotharius, J. and Brundin, P. (2002) Impaired dopamine storage resulting from α-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Human Molecular Genetics, 11, 2395-2407. doi:10.1093/hmg/11.20.2395
- Baptista, M.J., et al. (2003) Co-ordinate transcriptional regulation of dopamine synthesis genes by α-synuclein in human neuroblastoma cell lines. Journal of Neurochemistry, 85, 957-968. doi:10.1046/j.1471-4159.2003.01742.x
- Wersinger, C., Prou, D., Vernier, P. and Sidhu, A. (2003) Modulation of dopamine transporter function by α-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. The FASEB Journal, 17, 2151-2153.
- Perez, R.G., et al. (2002) A role for α-synuclein in the regulation of dopamine biosynthesis. The Journal of Neuroscience, 22, 3090-3099.
- Braak, E., et al. (2001) α-synuclein immunopositive Parkinson’s disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathology, 101, 195-201.
- Bruijn, L.I., et al. (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science, 281, 1851-1854. doi:10.1126/science.281.5384.1851
- Gurney, M.E., et al. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science, 264, 1772-1775. doi:10.1126/science.8209258
- Valentine, J.S. and Hart, P.J. (2003) Misfolded CuZn SOD and amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of USA, 100, 3617-3622. doi:10.1073/pnas.0730423100
- Wiedau-Pazos, M., et al. (1996) Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science, 271, 515-518. doi:10.1126/science.271.5248.515
- Klein, J.A. and Ackerman, S.L. (2003) Oxidative stress, cell cycle, and neurodegeneration. Journal of Clinical Investigation, 111, 785-793.
- Linke, A., Adams, V., Schulze, P.C., Erbs, S. and Gielen, S., et al. (2005) Antioxidative effects of exercise training in patients with chronic heart failure: Increase in radical scavenger enzyme activity in skeletal muscle. Circulation, 111, 1763-1770. doi:10.1161/01.CIR.0000165503.08661.E5
- Cristina Polidori, M., Pratico, D., Savino, K., Rokach, J., Stahl, W. and Mecocci, P. (2004) Increased F2 isoprostane plasma levels in patients with congestive heart failure are correlated with antioxidant status and disease severity. Journal of Cardiac Failure, 10, 334-338. doi:10.1016/j.cardfail.2003.11.004
- [61] de Champlain, J., Wu, R., Girouard, H., et al. (2004) Oxidative stress in hypertension. Clinical and Experimental Hypertension, 26, 593-601.
- [62] Rizzo, M., Kotur-Stevuljevic, J., et al. (2009) Atherogenic dyslipidemia and oxidative stress. Translational Research, 153, 217-223. doi:10.1016/j.trsl.2009.01.008
- [63] Steinberg, D. (1997) Low density lipoprotein oxidation and its pathobiological significance. Journal of Biological Chemistry, 272, 20963-20966.
- [64] Devasagayam, T.P.A., Tilak, J.C., Boloor, K.K., Sane, K.S., Ghaskadbi, S.S. and Lele, R.D. (2004) Free radicals and antioxidants in human health: Current status and future prospects. Journal of the Association of Physicians of India, 52, 794-804.
- [65] Tribble, D.L. (1999) Antioxidant consumption and risk of coronary heart disease: Emphasis on vitamin C, vitamin E, and β-carotene. Circulation, 99, 591-595. doi:10.1161/01.CIR.99.4.591
- [66] Alexandrova, M., Bochev, P., Markova, V., Bechev, B., Popova, M., Danovska, M. and Simeonova, V. (2004) Dynamics of free radical processes in acute ischemic stroke: Influence on neurological status and outcome. Journal of Clinical Neuroscience, 11, 501-506. doi:10.1016/j.jocn.2003.10.015
- [67] Piantadosi, C.A. and Zhang, J. (1996) Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke, 27, 327-332. doi:10.1161/01.STR.27.2.327
- [68] Van Gaal, L.F., Vertommen, J. and De Leeuw, I.H. (1998) The in vitro oxidizability of lipoprotein particles in obese and non-obese subjects. Atherosclerosis, 137, S39-S44. doi:10.1016/S0021-9150(97)00316-X
- [69] Bakker, S.J., IJzerman, R.G., Teerlink, T., Westerhoff, H.V., Gans, R.O. and Heine, R.J. (2000) Cytosolic triglycerides and oxidative stress in central obesity: The missing link between excessive atherosclerosis, endothelial dysfunction, and beta cell failure? Atherosclerosis, 148, 17-21. doi:10.1016/S0021-9150(99)00329-9
- [70] Coppack, S.W. (2001) Pro-inflammatory cytokines and adipose tissue. Proceedings of the Nutrition Society, 60, 349-356. doi:10.1079/PNS2001110
- [71] Dandona, P., Mohanty, P., Ghanim, H., Aljada, A., Browne, R., Hamouda, W., Prabhala, A., Afzal, A. and Garg, R. (2001) The suppressive effect of dietary restricttion and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. The Journal of Clinical Endocrinology & Metabolism, 86, 355-362. doi:10.1210/jc.86.1.355
- [72] Menon, M. and Resnick, M.I. (2002) Urinary lithiasis: Etiology, diagnosis and medical management. In: Walsh, P.C., Ed., Campbell’s Urology, Saunder’s, 4, 3229-3305.
- [73] Halliwell, B. (1991) Reactive oxygen species in living systems: Source, biochemistry and role in human disease. American Journal of Medicine, 91, 14s-21s. doi:10.1016/0002-9343(91)90279-7
- [74] Selvam, R. and Kalaiselvi, P. (2001) Studies on calcium oxalate binding proteins: Effect of lipid peroxidation. Nephron, 88, 163-167. doi:10.1159/000045978
- [75] Muthukumar, A. and Selvam, R. (1997) Renal injury mediated calcium oxalate nephrolithiasis: Role of lipid peroxidation. Renal Failure, 19, 401-408. doi:10.3109/08860229709047726
- [76] Thamilselvan, S., Khan, S.R. and Menon, M. (2003) Oxalate and calcium oxalate mediated free radical toxicity in renal epithelial cells: Effect of antioxidants. Urological Research, 31, 3-9.
- [77] Singh, P.P. and Barjatia, M.K. (2002) Peroxidative stress and antioxidant status in relation to age in normal population and renal stone formers. Indian Journal of Nephrology, 12, 10-15.
- [78] Thamilselvan, S., Hackett, R.L. and Khan, S.R. (1997) Lipid peroxidation in ethylene glycol induced hyperoxaluria and calcium oxalate nephrolithiasis. Journal of Urology, 157, 1059-1063. doi:10.1016/S0022-5347(01)65141-3
- [79] Pillai, C.K. and Pillai, K.S. (2002) Antioxidants in health. Indian Journal of Physiology and Pharmacology, 46, 1-5.
- [80] Adler, A.I, Stevens, R.J, et al. (2003) Development and progression of nephropathy in type 2 diabetes: The Unitsed Kingdom Prospective Diabetes Study (UKPDS 64). Kidney International, 63, 225-232. doi:10.1046/j.1523-1755.2003.00712.x
- [81] Robert, C. and Stanton, T. (2011) Oxidative stress and diabetic kidney disease. Current Diabetes Reports, 11, 330-336. doi:10.1007/s11892-011-0196-9
- [82] Anderson, S. and Brenner, B.M. (1988) Pathogenesis of diabetic glomerulopathy: Hemodynamic considerations. Diabetes/Metabolism Reviews, 4, 163-177. doi:10.1002/dmr.5610040206
- [83] Zatz, R., Dunn, B.R, Meyer, T.W., et al. (1986) Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. Journal of Clinical Investigation, 77, 1925-1930. doi:10.1172/JCI112521
- [84] Garrido, A.M. and Griendling, K.K. (2009) NADPH oxidases and angiotensin II receptor signaling. Molecular and Cellular Endocrinology, 302, 148-158. doi:10.1016/j.mce.2008.11.003
- [85] Mehta, P.K. and Griendling, K.K. (2007) Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. American Journal of Physiology, 292, C82-C97. doi:10.1152/ajpcell.00287.2006
- [86] Mansouri, E., Panahi, M., Ghaffari, M.A. and Ghorbani, A. (2011) Effects of grape seed proanthocyanidin extract on oxidative stress induced by diabetes in rat kidney. Iranian Biomedical Journal, 15, 100-106.
- [87] Sadi, G., Eryilmaz, N., Tütüncüoğlu, E., Cingir, S. and Güray, T. (2012) Changes in expression profiles of antioxidant enzymes in diabetic rat kidneys. Diabetes/Metabolism Research and Reviews, 28, 228-235. doi:10.1002/dmrr.1302
- [88] Chen, Y.J. and Quilley, J. (2008) Fenofibrate treatment of diabetic rats reduces nitrosative stress, renal cyclooxygenase-2 expression, and enhanced renal prostaglandin release. Journal of Pharmacology and Experimental Therapeutics, 324, 658-663. doi:10.1124/jpet.107.129197
- [89] Peterson, J.D., Herzenberg, L.A., Vasquez, K. and Waltenbaugh, C. (1998) Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proceedings of the National Academy of Sciences of USA, 95, 3071-3076. doi:10.1073/pnas.95.6.3071
- [90] Kim, H.J., Barajas, B., Chan, R.C. and Nel, A.E. (2007) Glutathione depletion inhibits dendritic cell maturation and delayed-type hypersensitivity: Implications for systemic disease and immunosenescence. Journal of Allergy and Clinical Immunologyl, 119, 1225-1233. doi:10.1016/j.jaci.2007.01.016
- [91] Murata, Y., Shimamura, T. and Hamuro, J. (2002) The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. International Immunology, 14, 201-212. doi:10.1093/intimm/14.2.201
- [92] Rahman, I. (2003) Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. Journal of Biochemistry and Molecular Biology, 36, 95-109. doi:10.5483/BMBRep.2003.36.1.095
- [93] Comhair, S.A., Ricci, K.S., Arroliga, M., et al. (2005) Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. American Journal of Respiratory and Critical Care Medicine, 172, 306-313. doi:10.1164/rccm.200502-180OC
- [94] Battaglia, S., den Hertog, H., Timmers, M.C., Lazeroms, S.P., Vignola, A.M., Rabe, K.F., Bellia, V., Hiemstra, P.S. and Sterk, P.J. (2005) Small airways function and molecular markers in exhaled air in mild asthma. Thorax, 60, 639-644. doi:10.1136/thx.2004.035279
- [95] Ercan, H., Birben, E., Dizdar, E.A., Keskin, O., Karaaslan, C., Soyer, O.U., Dut, R., Sackesen, C., Besler, T. and Kalayci, O. (2006) Oxidative stress and genetic andepidemiologic determinants of oxidant injury in childhood asthma. Journal of Allergy and Clinical Immunology, 118, 1097-1104. doi:10.1016/j.jaci.2006.08.012
- [96] Fogarty, A., Lewis, S., Weiss, S. and Britton, J. (2000) Dietary vitamin E, IgE concentrations, and atopy. Lancet, 356, 1573-1574. doi:10.1016/S0140-6736(00)03132-9
- [97] Cho, Y.S. and Moon, H.-B. (2010) The role of oxidative stress in the pathogenesis of asthma. Allergy, Asthma & Immunology Research, 2, 183-187. doi:10.4168/aair.2010.2.3.183
- [98] Cantin, A.M., North, S.L., Fells, G.A., Hubbard, R.C. and Crystal, R.G. (1987) Oxidant-mediated epithelial cell injury in idiopathic pulmonary fibrosis. Journal of Clinical Investigation, 79, 1665-1673. doi:10.1172/JCI113005
- [99] Montuschi, P., Ciabattoni, G., Paredi, P., Pantelidis, P., du Bois, R.M., et al. (1998) 8-Isoprostane as a biomarker of oxidative stress in interstitial lung diseases. American Journal of Respiratory and Critical Care Medicine, 158, 1524-1527.
- [100] Fattman, C.L. (2008) Apoptosis in pulmonary fibrosis: Too much or not enough? Antioxidants & Redox Signaling, 10, 379-385. doi:10.1089/ars.2007.1907
- [101] Sturrock, A., Cahill, B., Norman, K., Huecksteadt, T.P., Hill, K., et al. (2006) Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species dependent proliferation in human pulmonary artery smooth muscle cells. American Journal of Physiology Lung Cellular and Molecular, 290, L661-L673. doi:10.1152/ajplung.00269.2005
- [102] Rottoli, P., Magi, B., Cianti, R., Bargagli, E., Vagaggini, C., et al. (2005) Carbonylated proteins in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics, 5, 2612-2618. doi:10.1002/pmic.200401206
- [103] Lenz, A.G., Costabel, U. and Maier, K.L. (1996) Oxidized BAL fluid proteins in patients with interstitial lung diseases. European Respiratory Journal, 9, 307-312. doi:10.1183/09031936.96.09020307
- [104] Gao, F., Kinnula, V.L., Myllarniemi, M. and Oury, T.D. (2008) Extracellular superoxide dismutase in pulmonary fibrosis. Antioxidants & Redox Signaling, 10, 343-354. doi:10.1089/ars.2007.1908
- [105] Rahman, I., Skwarska, E., Henry, M., Davis, M., O’Connor, C.M., et al. (1999) Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radical Biology & Medicine, 27, 60-68. doi:10.1016/S0891-5849(99)00035-0
- [106] Szabo, E., Riffe, M.E., Steinberg, S.M., Birrer, M.J. and Linnoila, R.I. (1996) Altered cJUN expression: An early event in human lung carcinogenesis. Cancer Research, 56, 305-315.
- [107] Volm, M., van Kaick, G. and Mattern, J. (1994) Analysis of c-fos, c-erbB1, c-erbB2 and c-myc in primary lung carcinomas and their lymph node metastases. Clinical and Experimental Metastasis, 12, 329-334. doi:10.1007/BF01753840
- [108] Stewart, Z.A. and Pietenpol, J.A. (2001) p53 Signaling and cell cycle checkpoints. Chemical Research in Toxicology, 14, 243-263. doi:10.1021/tx000199t
- [109] Roberts, L.J. 2nd and Morrow, J.D. (1995) The isoprostanes: Novel markers of lipid peroxidation and potential mediators of oxidant injury. Advances in Prostaglandin, Thromboxane, and Leukotriene Research, 23, 219-224.
- [110] Babizhayev, M.A. (1996) Failure to withstand oxidative stress induced by phospholipid peroxides as a possible cause of lens opacities in systemic diseases and aging. Biochimica et Biophysica Acta, 1315, 87-99. doi:10.1016/0925-4439(95)00091-7
- [111] Lodovici, M., Raimondi, L., Guqlielmi, F., Gemignani, S. and Dolara, P. (2003) Protection against ultraviolet Binduced oxidative DNA damage in rabbit corneal-derived cells (SIRC) by 4-counmaric acid. Toxicology, 184, 141- 147. doi:10.1016/S0300-483X(02)00572-3
- [112] Ho, M.-C., Peng, Y.-J., Chen, S.-J. and Chiou, S.-H. (2010) Senile cataract and oxidative stress. Journal of Clinical Gerontology and Geriatrics, 1, 17-21. doi:10.1016/j.jcgg.2010.10.006
- [113] Lodovici, M., Caldini, S., Morbidelli, L., Ziche, M. and Dolara, P. (2009) Protective effect of 4-coumaric acid from UVB ray damage in the rabbit eye. Toxicology, 255, 1-5. doi:10.1016/j.tox.2008.09.011
- [114] Ohira, A., Ueda, T., Ohishi, K., Hiramitsu, T., Akeo, K. and Obara, Y. (2008) Oxidative stress in ocular disease. Nippon Ganka Gakkai Zasshi, 112, 22-29.
- [115] Vinson, J.A. (2006) Oxidative stress in cataract. Pathophysiol, 13, 151-162.
- [116] Zoric, L. (2003) Parameters of oxidative stress in the lens. Aqueous humor and blood in patients with diabetes and senile cataracts. Srpski Arhiv za Celokupno Lekarstvo, 131, 137-142. doi:10.2298/SARH0304137Z
- [117] Berthoud, V.M. and Beyer, E.C. (2009) Oxidative stress, lens gap junctions, and cataract. Antioxid Redox Signal, 11, 339-353. doi:10.1089/ars.2008.2119
- [118] Javitt, J.C. and Taylor, H.R. (1994) Cataract and latitude. Documenta Ophthalmologica, 88, 307-325. doi:10.1007/BF01203684
- [119] Sawada, H., Fukuchi, T. and Abe, H. (2009) Oxidative stress in aqueous humor of patients with senile macular degeneration. Current Eye Research, 34, 36-41. doi:10.1080/02713680802500960
- [120] Chalam, K.V., Khetpal, V., Rusovici, R. and Balaiy, S. (2011) A review: Role of ultraviolet radiation in age-related macular degeneration. Eye & Contact Lens, 37, 225- 232. doi:10.1097/ICL.0b013e31821fbd3e
- [121] Ohia, S.E., Opere, C.A., Leday, A.M. (2005) Pharmacological consequences of oxidative stress in ocular tissues. Mutatation Research, 579, 22-36. doi:10.1016/j.mrfmmm.2005.03.025
- [122] Beatty, S., Koh, H., Henson, D. and Boulton, M. (2000) The role of oxidative stress in the pathogenesis of agerelated macular degeneration. Survey of Ophthalmology, 45, 115-134. doi:10.1016/S0039-6257(00)00140-5
- [123] Klein, R., Klein, B.E., Wong, T.Y., Tomany, S.C. and Cruickshanks, K.J. (2002) The association of cataract and cataract surgery with long term incidence of age-related maculopathy. The beaver dam study. Archives of Ophthalmology, 120, 1551-1558.
- [124] Ambati, J., Ambati, B., Yoo, S.H., Lanchuley, S. and Adamis, A.P. (2003) Age-related macular degeneration: Etiology, pathogenesis and therapeutic strategies. Survey of Ophthalmology, 48, 257-293. doi:10.1016/S0039-6257(03)00030-4
- [125] Gottsch, J.D., Bynoe, L.A., Harlan, J.B., Rencs, E.V. and Green, W.R. (1993) Light-induced deposits in the Bruch’s membrane of proptoporphyric mice. Archives of Ophthalmology, 111, 126-129. doi:10.1001/archopht.1993.01090010130039
- [126] Nowak, M., Gnitecki, W. and Jurowski, P. (2005) The role of retinal oxygen in the origin of macular degeneration (AMD). Klinika Oczna, 107, 715-718.
- [127] Wu, J., Seregard, S. and Algvere, P.V. (2006) Photochemical damage of the Retina. Survey of Ophthalmology, 51, 461-481. doi:10.1016/j.survophthal.2006.06.009
- [128] Janik-Papis, K., Ulińska, M., Krzyzabowska, A., Stockzyńska, E., Borucka, A., Woźniak, K., Małgorzata, Z., Szaflik, J.P. and Blasiak, J. (2009) Role of oxidative mechanisms in the pathogenesis of age-related macular degeneration. Klinika Oczna, 111, 168-173.
- [129] Drobek-Slowik, M., Karczewicz, D. and Safranow, K. (2007) The potential role of oxidative stress in the pathogenesis of the age-related macular degeneration (AMD). Postępy Higieny i Medycyny Doświadczalnej, 61, 28-37.
- [130] Winkler, B.S., Boulton, M.E., Gottsch, J.D. and Sternberg, P. (1999) Oxidative damage and age-related macular degeneration. Molecular Vision, 5, 32-42.
- [131] Roehlecke, C., Schaller, A., Knels, L. and Funk, R.H. (2009) The influence of sublethal blue light exposure on human RPE cells. Molecular Vision, 15, 1929-1938.
- [132] SanGiovanni, J.P. and Chew, E.Y. (2005) The role of omega-3 long chain polyunsaturated fatty acids in health and disease of the retina. Progress in Retinal and Eye Research, 24, 87-138. doi:10.1016/j.preteyeres.2004.06.002
- [133] Feldmen, E.L. (2003) Oxidative stress and diabetic retinopathy: A new understanding of an old problem. Journal of Clinical Investigation, 111, 431-433.
- [134] Brownlee, M. (2005) The pathophysiology of diabetic implications: A unifying mechanism. Diabetes, 54, 1615- 1625. doi:10.2337/diabetes.54.6.1615
- [135] Kowluru, R.A. and Kanwar, M. (2009) Oxidative stress and the development of diabetic retinopathy: Contributory role of matrix metalloproteinase-2. Free Radical Biology & Medicine, 46, 1677-1685. doi:10.1016/j.freeradbiomed.2009.03.024
- [136] Yue, K.K.M., Chung, W.S., Leung, A.W.N. and Cheung, C.H.K. (2003) Redox changes precede the occurrence of oxidative stress in eyes and aorta, but not kidneys of diabetic rats. Life Sciences, 73, 2557-2570. doi:10.1016/S0024-3205(03)00662-3
- [137] Baynes, J.W. (1991) Role of oxidative stress in development of complications in diabetes. Diabetes, 40, 405-412. doi:10.2337/diabetes.40.4.405
- [138] Caspi, R.R. (2010) A look at autoimmunity and inflamemation of the eye. Journal of Clinical Investigation, 120, 3073-3083. doi:10.1172/JCI42440
- [139] Zamir, E., Zhang, Z., Samuni, A., Kogan, M. and Pe’er, J. (1999) Nitroxide stable radical suppresses autoimmune uveitis in rats. Free Radical Biology & Medicine, 27, 7- 15. doi:10.1016/S0891-5849(99)00026-X
- [140] Sarawathy, S. and Rao, N.A. (2009) Mitochondrial proteomics in experimental autoimmune uveitis oxidative stress. Investigative Ophthalmology & Visual Science, 50, 5559-5566.
- [141] Komeima, K., Rogers, B.S. and Campochiaro, P.A. (2007) Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. Journal of Cellular Physiology, 213, 809-815. doi:10.1002/jcp.21152
- [142] Usui, S., Ovesson, B.C., Lee, S.Y., Jo, Y.J., Yoshida, T., Miki, A., Miki, K., Iwase, T., Lu, L. and Compochiaro, P.A. (2009) NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. Journal of Neurochemistry, 110, 1028-1037. doi:10.1111/j.1471-4159.2009.06195.x
- [143] Makker, K., Agarwal, A. and Sharma, R. (2009) Oxidative stress and male infertility. Indian Journal of Medical Research, 129, 357-367.
- [144] Sun, J.G., Jurisicova, A. and Casper, R.F. (1997) Detection of deoxyribonucleic acid fragmentation in human sperm: Correlation with fertilization in vitro. Biology of Reproduction, 56, 602-607. doi:10.1095/biolreprod56.3.602
- [145] Vine, M.F., Tse, C.K., Hu, P. and Truong, K.Y. (1996) Cigarette smoking and semen quality. Fertility and Sterility, 65, 835-842.
- [146] Potts, R.J., Newbury, C.J., Smith, G., Notarianni, L.J. and Jefferies, T.M. (1999) Sperm chromatin damage associated with male smoking. Mutation Research, 423, 103- 111. doi:10.1016/S0027-5107(98)00242-5
- [147] Barrington, J.W., Lindsay, P., James, D., et al. (1996) Selenium deficiency and miscarriage: A possible link? British Journal of Obstetrics and Gynaecology, 103, 130- 132. doi:10.1111/j.1471-0528.1996.tb09663.x
- [148] Vural, P., Akgul, C., Yildirim, A. and Canbaz, M. (2000) Antioxidant defence in recurrent abortion. Clinica Chimica Acta, 295, 169-177. doi:10.1016/S0009-8981(99)00255-7
- [149] Plessinger, M.A., Woods Jr., J.R. and Miller, R.K. (2000) Pretreatment of human amnion-chorion with vitamins C and E prevents hypochlorous acid-induced damage. American Journal of Obstetrics & Gynecology, 183, 979-985. doi:10.1067/mob.2000.106676
- [150] Bilodeau, J.F. and Hubel, C.A. (2003) Current concepts in the use of antioxidants for the treatment of preeclampsia. Journal of Obstetrics and Gynaecology Canada, 25, 742- 750.
- [151] Chappell, L.C., Seed, P.T., Briley, A., et al. (2002) A longitudinal study of biochemical variables in women at risk of preeclampsia. American Journal of Obstetrics & Gynecology, 187, 127-136. doi:10.1067/mob.2002.122969
- [152] Shazia, Q., Mohammad, Z.H., Rahman, T. and Shekhar, H.U. (2012) Correlation of oxidative stress with serum trace element levels and antioxidant enzyme status in beta thalassemia major patients: A review of the literature. Anemia, 2012, 7 p. doi:10.1155/2012/270923
- [153] Pavlova, L.E., Savov, V.M., Petkov, H.G. and Charova, I.P. (2007) Oxidative stress in patients with beta-thalassemia major. Prilozi, 28, 145-154.
- [154] Suryakar, A.N., Katkam, R.V. and Joshi, N.G. (2008) Oxidative stress and disturbance in antioxidant balance in beta thalassemia major. Indian Journal of Clinical Biochemistry, 23, 337-340. doi:10.1007/s12291-008-0074-7
- [155] Battisti, V., Maders, L.D., Bagatini, M.D., Santos, K.F., Spanevello, M., Maldonado, P.A., Brulé, A.O., Araújo Mdo, C., Schetinger, M.R. and Morsch, V.M. (2008) Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clinical Biochemistry, 41, 511-518. doi:10.1016/j.clinbiochem.2008.01.027
- [156] Papiez, M.A., Dybala, M., Sowa-Kucma, M., Krzysciak, W., Taha, H.M. and Alicja, G.N. (2009) Evaluation of oxidative status and depres-sion-like responses in Brown Norway rats with acute mye-loid leukemia. Progress Neuro-Psychopharmacology and Biological Psychiatry, 33, 596-604.
- [157] El-Sabagh, M.E., Ramadan, K.S., El-slam, I.M.A. and Ibrahim, A.M. (2011) Antioxidants status in acute lymphoblastic leukemic patients. American Journal of Medicine and Medical Sciences, 1, 1-6.
- [158] Altindag, O., Karakoc, M., Kocyigit, A., Celik, H. and Soran, N. (2007) Increased DNA damage and oxidative stress in patients with rheumatoid arthritis. Clinical Biochemistry, 40, 167-171. doi:10.1016/j.clinbiochem.2006.10.006
- [159] Pedersen-Lane, J.H., Zurier, R.B. and Lawrence, D.A. (2007) Analysis of the thiol status of peripheral blood leukocytes in rheumatoid arthritis patients. Journal of Leukocyte Biology, 81, 934-941. doi:10.1189/jlb.0806533
- [160] Kalpakcioglu, B. and Senel, K. (2008) The interrelation of glutathione reductase, catalase, glutathione peroxidase, superoxide dismutase, and glucose-6-phosphate in the pathogenesis of rheumatoid arthritis. Clinical Rheumatology, 27, 141-145. doi:10.1007/s10067-007-0746-3
- [161] Vasanthi, P., Nalini, G. and Rajasekhar, G. (2009) Status of oxidative stress in rheumatoid arthritis. International Journal of Rheumatic Diseases, 12, 29-33. doi:10.1111/j.1756-185X.2009.01375.x
- [162] Ku, I.A., Imboden, J.B., Hsue, P.Y. and Ganz, P. (2009) Rheumatoid arthritis: Model of systemic inflammation driving atherosclerosis. Circulation Journal, 73, 977-985. doi:10.1253/circj.CJ-09-0274
- [163] Rho, Y.H., Chung, C.P., Oeser, A., et al. (2010) Interaction between oxidative stress and high-density lipoprotein cholesterol is associated with severity of coronary artery calcification in rheumatoid arthritis. Arthritis Care & Research, 62, 1473-1480. doi:10.1002/acr.20237
- [164] Khan, F. and Siddiqui, A.A. (2006) Prevalence of anti-3-nitrotyrosine antibodies in the joint synovial fluid of patients with rheumatoid arthritis, osteoarthritis and systemic lupus erythematosus. Clinica Chimica Acta, 370, 100-107. doi:10.1016/j.cca.2006.01.020
- [165] Leitinger, N. (2008) The role of phospholipid oxidation products in inflammatory and autoimmune diseases: Evidence from animal models and in humans. Subcellular Biochemistry, 49, 325-350. doi:10.1007/978-1-4020-8830-8_12
- [166] Lemarechal, H., Allanore, Y., Chenevier-Gobeaux, C., Ekindjian, O.G., Kahan, A. and Borderie, D. (2006) High redox thioredoxin but low thioredoxin reductase activities in the serum of patients with rheumatoid arthritis. Clinica Chimica Acta, 367, 156-161. doi:10.1016/j.cca.2005.12.006
- [167] Hitchon, C.A. and El-Gabalawy, H.S. (2004) Oxidation in rheumatoid arthritis. Arthritis Research & Therapy, 6, 265-278. doi:10.1186/ar1447
- [168] Fay, J., Varoga, D., Wruck, C.J., et al. (2006) Reactive oxygen species induce expression of vascular endothelial growth factor in chondrocytes and human articular cartilage explants. Arthritis Research & Therapy, 8, R189. doi:10.1186/ar2102
- [169] De Bandt, M., Grossin, M., Driss, F., et al. (2002) Vitamin E uncouples joint destruction and clinical infl ammation in a transgenic mouse model of rheumatoid arthritis. Arthritis & Rheumatism, 46, 522-532. doi:10.1002/art.10085
- [170] Zwerina, J., Tzima, S., Hayer, S., et al. (2005) Heme oxygenase 1 (HO-1) regulates osteoclastogenesis and bone resorption. FASEB Journal, 19, 2011-2013
- [171] Hitchon, C.A. and El-Gabalawy, H.S. (2004) Oxidation in rheumatoid arthritis. Arthritis Research & Therapy, 6, 265-278. doi:10.1186/ar1447
- [172] Baskol, G., Demir, H. and Baskol, M. (2005) Assessment of paraoxonase 1 activity and malondialdehyde levels in patients with rheumatoid arthritis. Clinical Biochemistry, 38, 951-955. doi:10.1016/j.clinbiochem.2005.06.010
- [173] Griffiths, H.R. (2005) ROS as signalling molecules in T cells-evidence for abnormal redox signalling in the autoimmune disease, rheumatoid arthritis. Redox Report, 10, 273-280. doi:10.1179/135100005X83680
- [174] Pedersen-Lane, J.H., Zurier, R.B. and Lawrence, D.A. (2007) Analysis of the thiol status of peripheral blood leukocytes in rheumatoid arthritis patients. Journal of Leukocyte Biology, 81, 934-941. doi:10.1189/jlb.0806533
- [175] Wright, H.L., Moots, R.J., Bucknall, R.C. and Edwards, S.W. (2010) Neutrophil function in inflammation and inflammatory diseases. Rheumatology, 49, 1618-1631. doi:10.1093/rheumatology/keq045
- [176] Frears, E.R., Zhang, Z., Blake, D.R., O’Connell, J.P. and Winyard, P.G. (1996) Inactivation of tissue inhibitor of metalloproteinase-1 by peroxynitrite. FEBS Letters, 381, 21-24. doi:10.1016/0014-5793(96)00065-8
- [177] Nissim, A., Winyard, P.G. and Corrigall, V. (2005) Generation of neoantigenic epitopes after posttranslational modification of type II collagen by factors present within the inflamed joint. Arthritis & Rheumatism, 52, 3829- 3838. doi:10.1002/art.21479
- [178] Eggleton, P., Haigh, R. and Winyard, P.G. (2008) Consequence of neo-antigenicity of the “altered self”. Rheumatology, 47, 567-571. doi:10.1093/rheumatology/ken014
- [179] Edwards, S.W. and Hallett, M.B. (1997) Seeing the wood for the trees: The forgotten role of neutrophils in rheumatoid arthritis. Immunology Today, 18, 320-324. doi:10.1016/S0167-5699(97)01087-6
- [180] Klebanoff, S.J. (1999) Oxygen metabolites from phagocytes: In Inflammation: Basic principles and clinical correlates. Lippincott Williams and Wilkins, Philadelphia, 721-768.
- [181] Milam, S.B., Zardeneta, G. and Schmitz, J.P. (1998) Oxidative stress and degenerative temporomandibular joint disease: A proposed hypothesis. Journal of Oral and Maxillofacial Surgery, 56, 214-223. doi:10.1016/S0278-2391(98)90872-2
- [182] Kawai, Y., Kubota, E. and Okabe, E. (2000) Reactive oxygen species participation in experimentally induced arthritis of the temporomandibular joint in rats. Journal of Dental Research, 79, 1489-1495. doi:10.1177/00220345000790071001
- [183] Lee, M.C., Kawai, Y., Shoji, H., Yoshino, F., Miyazaki, H. and Kato, H. (2004) Evidence of reactive oxygen species generation in synovial fluid from patients with temporomandibular disease by electron spin resonance spectroscopy. Redox Report, 9, 331-336. doi:10.1179/135100004225006830
- [184] Chandra, H. and Symons, M.C. (1987) Sulphur radicals formed by cutting alpha-keratin. Nature, 328, 833-834. doi:10.1038/328833a0
- [185] Nitzan, D.W. (1994) Intraarticular pressure in the functioning human temporomandibular joint and its alteration by uniform elevation of the occlusal plane. Journal of Oral and Maxillofacial Surgery, 52, 671-679. doi:10.1016/0278-2391(94)90476-6
- [186] Matsuo, W., Kimura, H., Komatsu, K., Kobayashi, W., Sakuraba, Y. and Suzuki, M. (1993) The scavenging effects of TMJ synovial fluids onactive oxygen generation. Journal of Japanese Society for TMJ, 5, 347-355.
- [187] Guarda, N.L., Oliviero, F., Ramonda, R. and Ferronato, G. (2004) Influence of intra-articular injections of sodium hyaluronate on clinical features and synovial fluid nitric oxide levels of temporomandibular osteoarthritis. Reumatismo, 56, 272-277.
- [188] Nitzan, D.W. (2001) The process of lubrication impairment and its involvement in temporomandibular joint disc displacement: A theoretical concept. Journal of Oral and Maxillofacial Surgery, 59, 36-45. doi:10.1053/joms.2001.19278
- [189] Roberts, C.R., Roughley, P.J. and Mort, J.S. (1989) Degradation of human proteoglycan aggregate induced by hydrogen peroxide. Protein fragmentation, amino acid modification and hyaluronic acid cleavage. Biochemical Journal, 259, 805-811.
- [190] Yoshiaki, K., Lee, M.C. and Kubota, E. (2008) Oxidative stress and temporomandibular joint disorders. Japanese Dental Science Review, 44, 145-150. doi:10.1016/j.jdsr.2008.08.001
- [191] Xie, D. and Homandberg, G.A. (1993) Fibronectin fragments bind to and penetrate cartilage tissue resulting in proteinase expression and cartilage damage. Biochimica et Biophysica Acta, 1182, 189-196. doi:10.1016/0925-4439(93)90140-V
- [192] Finkelstein, E., Rosen, G.M. and Rauckman, E.J. (1982) Production of hydroxyl radical by decomposition of superoxide spin-trapped adducts. Molecular Pharmacology, 21, 262-265.
- [193] Buettner, G.R. (1987) Spin trapping: ESR parameters of spin adducts. Free Radical Biology & Medicine, 3, 259- 303. doi:10.1016/S0891-5849(87)80033-3
- [194] Ames, P.R., Alves, J., Murat, I., Isenberg, D.A. and Nourooz-Zadeh, J. (1999) Oxidative stress in systemic lupus erythematosus and allied conditions with vascular involvement. Rheumatology, 38, 529-534. doi:10.1093/rheumatology/38.6.529
- [195] Nuttall, S.L., Heaton, S., Piper, M.K., Martin, U. and Gordon, C. (2003) Cardiovascular risk in systemic lupus erythematosus-evidence of increased oxidative stress and dyslipidaemia. Rheumatology, 42, 758-762. doi:10.1093/rheumatology/keg212
- [196] Taysi, S., Gul, M., Sari, R.A., Akcay, F. and Bakan, N. (2002) Serum oxidant/antioxidant status of patients with systemic lupus erythematosus. Clinical Chemistry and Laboratory Medicine, 40, 684-688. doi:10.1515/CCLM.2002.117
- [197] Iuliano, L., Pratico, D., Ferro, D., et al. (1997) Enhanced lipid peroxidation in patients positive for antiphospholipid antibodies. Blood, 90, 3931-3935.
- [198] Soep, J.B., Mietus-Snyder, M., Malloy, M.J., Witztum, J.L. and von Scheven, E. (2004) Assessment of atherosclerotic risk factors and endothelial function in children and young adults with pediatric-onset systemic lupus erythematosus. Arthritis & Rheumatism, 51, 451-457. doi:10.1002/art.20392
- [199] Mansour, R.B., Lassoued, S., Gargouri, B., El Gaid, A., Attia, H. and Fakhfakh, F. (2008) Increased levels of autoantibodies against catalase and superoxide dismutase associated with oxidative stress in patients with rheumatoid arthritis and systemic lupus erythematosus. Scandinavian Journal of Rheumatology, 37, 103-108. doi:10.1080/03009740701772465
- [200] Kurien, B.T. and Scofield, R.H. (2006) Lipid peroxidetion in systemic lupus erythematosus. Indian Journal of Experimental Biology, 44, 349-356.
- [201] Turgay, M., Durak, I., Erten, S., Ertugrul, E., Devrim, E. and Avci, A. (2007) Oxidative stress and antioxidant parameters in a Turkish group of patients with active and inactive systemic lupus erythematosus. APLAR Journal of Rheumatology, 10, 101-106. doi:10.1111/j.1479-8077.2007.00268.x
- [202] Turi, S., Nemeth, I., Torkos, A., Saghy, L., Varga, I. and Matkovics, B. (1997) Oxidative stress and antioxidant defense mechanism in glomerular diseases. Free Radical Biology & Medicine, 22, 161-168. doi:10.1016/S0891-5849(96)00284-5
- [203] Ballatori, N., Krance, S.M., Notenboom, S., Shi, S., Tieu, K. and Hammond, C.L. (2009) Glutathione dysregulation and the etiology and progression of human diseases. Biological Chemistry, 390, 191-214. doi:10.1515/BC.2009.033
- [204] Peterson, J.D., Herzenberg, L.A., Vasquez, K. and Waltenbaugh, C. (1998) Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proceedings of the National Academy of Sciences of USA, 95, 3071-3076. doi:10.1073/pnas.95.6.3071
- [205] Perl, A., Gergely Jr., P. and Banki, K. (2004) Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. International Reviews of Immunology, 23, 293-313. doi:10.1080/08830180490452576
- [206] Chang, D.M., Su, W.L. and Chu, S.J. (2002) The expression and significance of intracellular T helper cytokines in systemic lupus erythematosus. Immunological Investigations, 31, 1-12. doi:10.1081/IMM-120003217
- [207] Viallard, J.F., Pellegrin, J.L., Ranchin, V., Schaeverbeke, T., Dehais, J. and Longy-Boursier, M. (1999) Th1 (IL-2, interferon-gamma (IFN-gamma)) and Th2 (IL-10, IL-4) cytokine production by peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE). Clinical & Experimental Immunology, 115, 189-195. doi:10.1046/j.1365-2249.1999.00766.x
- [208] Liu, T.F. and Jones, B.M. (1998) Impaired production of IL-12 in systemic lupus erythematosus. I. Excessive production of IL-10 suppresses production of IL-12 by monocytes. Cytokine, 10, 140-147. doi:10.1006/cyto.1997.0268
- [209] Hochberg, M.C. (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis & Rheumatism, 40, 1725. doi:10.1002/art.1780400928
- [210] Bombardier, C., Gladman, D.D., Urowitz, M.B., Caron, D. and Chang, C.H. (1992) Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis & Rheumatism, 35, 630-640. doi:10.1002/art.1780350606
- [211] Lohr, G.W. and Waller, H.D. (1974) Glucose-6-phosphate dehydrogenase. Methods in Enzymology Analysis, 2, 636- 643.
- [212] Buege, J.A. and Aust, S.D. (1978) Microsomal lipid peroxidation. Methods in Enzymology, 52, 302-310. doi:10.1016/S0076-6879(78)52032-6
- [213] Kono, Y. (1978) Generation of superoxide radical during auto-oxidation of hydroxylamine and an assay for superoxide dismutase. Archives of Biochemistry and Biophysics, 186, 189-195. doi:10.1016/0003-9861(78)90479-4
- [214] Luck, H. (1971) Catalase. In: Hu, B., Ed., Methods of Enzymatic Analysis, 3, 279.
- [215] Paglia, D.E. and Valentine, W.N. (1967) Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. Journal of Laboratory and Clinical Medicine, 70, 158-169.
- [216] Beutler, E., Duron, O. and Kelly, B.M. (1963) Improved method for the determination of blood glutathione. Journal of Laboratory and Clinical Medicine, 61, 882-888.
- [217] Taub, R. (2004) Liver regeneration: From myth to mechanism. Nature Reviews Molecular Cell Biology, 5, 836-847. doi:10.1038/nrm1489
- [218] Irshad, M. (2002) Oxidative stress in liver diseases. Tropical Gastroenterology, 23, 6-8.
- [219] Brown, G.C. and Borutaite, V. (2012) There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion, 12, 1-4. doi:10.1016/j.mito.2011.02.001
- [220] Curran, R.D., Billiar, T.R., Stuehr, D.J., Hofmann, K. and Simmons, R.L. (1989) Hepatocytes produce nitrogen oxides from L-arginine in response to inflammatory products of Kupffer cells. The Journal of Experimental Medicine, 170, 1769-1774. doi:10.1084/jem.170.5.1769
- [221] Diesen, D.L. and Kuo, P.C. (2010) Nitric oxide and redox regulation in the liver: Part I. General considerations and redox biology in hepatitis. Journal of Surgical Research, 162, 95-109. doi:10.1016/j.jss.2009.09.019
- [222] Diesen, D.L. and Kuo, P.C. (2011) Nitric oxide and redox regulation in the liver: Part II. Redox biology in pathologic hepatocytes and implications for intervention. Journal of Surgical Research, 167, 96-112. doi:10.1016/j.jss.2009.10.006
- [223] Klatt, P. and Lamas, S. (2000) Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. European Journal of Biochemistry, 267, 4928-4944. doi:10.1046/j.1432-1327.2000.01601.x
- [224] Niki, E. (2009) Lipid peroxidation: Physiological levels and dual biological effects. Experimental and Molecular Pathology, 47, 469-484. doi:10.1016/j.freeradbiomed.2009.05.032
- [225] Stehbens, W.E. (2004) Oxidative stress in viral hepatitis and AIDS. Experimental and Molecular Pathology, 77, 121-132. doi:10.1016/j.yexmp.2004.04.007
- [226] Stehbens, W.E. (2003) Oxidative stress, toxic hepatitis, and antioxidants with particular emphasis on zinc. Experimental and Molecular Pathology, 75, 265-276. doi:10.1016/S0014-4800(03)00097-2
- [227] Osman, H.G., Gabr, O.M., Lotfy, S. and Gabr, S. (2007) Serum levels of bcl-2 and cellular oxidative stress in patients with viral hepatitis. Indian Journal of Medical Microbiology, 25, 323-329. doi:10.4103/0255-0857.37333
- [228] Halliwell, B. (1997) Antioxidants and human disease: A general introduction. Nutrition Reviews, 55, S44-S49, Discussion S9-S52.
- [229] Poli, G. (2000) Pathogenesis of liver fibrosis: Role of oxidative stress. Molecular Aspects of Medicine, 21, 49- 98. doi:10.1016/S0098-2997(00)00004-2
- [230] Rogowski, O., Shnizer, S., Wolff, R., Lewis, B.S. and Amir, O. (2011) Increased serum levels of oxidative stress are associated with hospital readmissions due to acute heart failure. Cardiology, 118, 33-37. doi:10.1159/000324192
- [231] Ali, E.M.M., Shehata, H.H., Ali-Labib, R. and Zahra, E.L.M. (2009) Oxidant and antioxidant of arylesterase and paraoxonase as biomarkers in patients with hepatitis C virus. Clinical Biochemistry, 42, 1394-1400. doi:10.1016/j.clinbiochem.2009.06.007
- [232] Lu, Y., Zhuge, J., Wang, X., Bai, J. and Cederbaum, A.I. (2008) Cytochrome P450 2E1 contributes to induced fatty liver in mice. Hepatology, 47, 1483-1494. doi:10.1002/hep.22222
- [233] Nanji, A.A. (2002) Role of Kupffer cells in alcoholic hepatitis. Alcohol, 27, 13-15. doi:10.1016/S0741-8329(02)00207-0
- [234] Zhu, R., Wang, Y., Zhang, L. and Guo, Q. (2012) Oxidative stress and liver disease. Hepatology Research, 42, 741-749. doi:10.1111/j.1872-034X.2012.00996.x
- [235] John, W.B. and Suzanne, R.T. (1999) Role of oxidative stress in diabetic complications. Diabetes, 48, 1-9. doi:10.2337/diabetes.48.1.1
- [236] Lipinski, B. (2001) Pathophysiology of oxidative stress in diabetes mellitus. Journal of Diabetes and Its Complications, 15, 203-210. doi:10.1016/S1056-8727(01)00143-X
- [237] Poli, G. (1993) Lever damage due to free radicals. British Medical Bulletin, 49, 604-620.
- [238] Loudianos, G. and Gitlin, J.D. (2000) Wilson’s disease. Seminars in Liver Disease, 20, 353-364. doi:10.1055/s-2000-9389
- [239] Ozsoylu, S. and Kocak, N. (1985) Wilson disease in Turkish children. European Journal of Pediatrics, 147, 334. doi:10.1007/BF00442714
- [240] Hochstein, P., Kuma, S. and Forman, S.J. (1980) Lipid peroxidation and cytotoxicity of copper. Annals of the New York Academy of Sciences, 355, 240-248. doi:10.1111/j.1749-6632.1980.tb21342.x
- [241] Sokol, R.J., Devereaux, M. and Mierau, G.W. (1990) Oxidant injury to hepatic mitochondrial lipids in rats with hereditary copper overload. Modification by vitamin E deficiency. Gastroenterology, 99, 1061-1071.
- [242] Halliwell, B., Gutteridge, J.M.C. and Cross, E.S. (1992) Free radicals and human disease: Where are we now? Journal of Laboratory and Clinical Medicine, 119, 598- 620.
- [243] Bianchi, G., Marchesini, G. and Fahbri, A. (1997) Lipoperoxide plasma levels in patients with liver cirrhosis. Hepato Gastroenterol, 44, 784-788.
- [244] Sokol, R.J., Twedt, D. and McKim, J.M. (1994) Oxidant injury to hepatic mitochodria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology, 107, 1788-1798.
NOTES
*Corresponding author.