Journal of Biophysical Chemistry
Vol.3 No.2(2012), Article ID:19439,9 pages DOI:10.4236/jbpc.2012.32019
Role of endothelium on the abnormal Angiotensin-mediated vascular functions in epileptic rats
![]()
1Universidade de Ribeirão Preto—UNAERP. Curso de Medicina, Ribeirāo Preto, Brasil; *Corresponding Author: carolbaraldi@hotmail.com
2Faculdade de Medicina de Ribeirão Preto-USP, Ribeirão Preto, Brasil
3Faculdade de Ciências Farmacêuticas de Ribeirão Preto-USP, Ribeirão Preto, Brasil
Received 28 January 2012; revised 20 February 2012; accepted 14 March 2012
Keywords: Endothelium; Aorta Myogenic Tone; Angiotensin; Angiotensin II Receptor; Epilepsy
ABSTRACT
Epidemiological studies have found that the risk for cardiovascular disease is increased in patients with epilepsy. The Renin Angiontensin System (RAS), an important player in vascular tone control, is also involved in many neurological disorders, including seizures and epilepsy. Although it has been reported that Angiotensin II (Ang II) release and Angiotensin receptors expression are altered in many cerebral areas in patients/animal models with neurological disorders, there are no data on the vascular function. We evaluated Ang I and Ang II-mediated vascular responses and to correlate their contractile responses to the presence of endothelium and the protein levels of components of the RAS (AT1, AT2, Mas and ACE) in aorta isolated from genetically epileptic rats (WAR strain). The major finding was that the vascular contractile response induced by Ang I and Ang II is endothelium-dependent. Ang II induced contractions in aortas from Wistar rats either with intact endothelium (E+) (1.16 ± 0.04 g, n = 6) and endothelium-denuded (E–) (1.24 ± 0.04 g, n = 6). Maximum contractile response (ME) induced by Ang I was lower in Wistar E+ (0.45 ± 0.03 g, n = 6) compared with Wistar E– (1.13 ± 0.08 g, n = 6). Ang I and Ang II failed to induce contraction in WAR E+, whereas the ME induced by Ang I in WAR E– was lower (0.52 ± 0.04 g, n = 11) than in the Wistar. ME induced by Ang II in aortas from WAR was also lower (0.40 ± 0.03 g, n = 11) compared with Wistar. AT1 receptor expression in both E+ WAR and Wistar was lower than in both E– WAR and Wistar. AT2 and Mas receptor expression was higher in Wistar E– and E+ as compared to WAR E– and E+. ACE expression was higher in both E+ WAR and Wistar, but it was lower in both E– WAR and Wistar. Endothelium impairs the contractile response induced by Angiotensin in WAR, suggesting that endothelial relaxing factors play important role on the aorta contraction.
1. INTRODUCTION
Epidemiological studies have found that the risk for cardiovascular disease is increased in persons with epilepsy and it is related to premature death [1-6]. The Renin-Angiotensin System (RAS) is a player in many neurological disorders, including seizures and epilepsy. It has been pointed out that the RAS is a mediator in neurological problems such as the seizures observed in epilepsy and cardiovascular diseases [7].
Considering epilepsy as a neurodegenerative disorder, important studies of Tchekalarova and Georgiev [7] have demonstrated the brain involvement of the RAS in seizure genesis and/or control by activation of the AT1 receptor, thereby facilitating the discharge firing. Argañaraz et al. [8] have reported increased AT1 receptor expression in the cortex and in the hippocampus of patients with temporal lobe epilepsy, while AT2 receptor expression was elevated only in the hippocampus of these patients. According to these authors, changes in Angiotensin II (Ang II) release may occur via mechanisms that remain unknown. Ang II would have proor anti-epileptogenic function depending on its receptor activation. Their findings suggest that the up-regulation of the AT1 receptor is related to increased tissue excitability in the hippocampus and cortical areas.
Regarding to the peripheral actions of RAS it is important to consider that the RAS plays a relevant role in blood pressure and body fluid homeostasis regulation [9-12]. Angiotensin I (Ang I) is the main substrate for the Angiotensin Converting Enzyme (ACE), yielding the vasoconstrictor Ang II, the main RAS effector [13]. Ang I may also be hydrolyzed by prolyl-endopeptidase and carboxy-peptidases, to yield the vasodilator Angiotensin-1-7 (Ang 1-7) [14]. However, Ang II is the major substrate for (Ang 1-7) synthesis [15]. Besides the AT1 and AT2 receptors, the GPCR Mas has been identified as an endogenous receptor for the Angiotensin-(1-7) [16-18].
Ang II exerts its effect through activation of type 1 (AT1) and type 2 (AT2) receptors belonging to the family of G-protein coupled receptors (GPCRs). Both types of receptor trigger distinct and sometimes opposing signal transduction pathways [19,20]. The most prevalent physiological and pathological effects of Ang II, such as vascular smooth muscle cell contraction, are mediated by the AT1 receptor. On the other hand, AT2 receptors are generally assumed to counteract these actions. Activation of AT2 receptors leads to several biological processes, such as vascular relaxation [21-24].
It is well known that the RAS and the nitric oxide (NO) system play a crucial part in the regulation of cardiovascular physiology and pathology [25]. According to Yan et al. [25] there is a functional relationship between Ang Iotensin II and NO. Nitric Oxide is the main relaxing endothelial factor involved on the aorta relaxation [26,27]. Works using cultured neurons have demonstrated that NO is specifically involved in mediating Ang II actions [28,29]. Ang II is also considered a neurotransmitter/ neuromodulator, because it is responsible for the activetion of AT1 and AT2 receptors present in several brain regions [30-32]. Although the importance of animal models for the investigation of epilepsy and related disorders is recognized, few studies have been devoted to the relationship between epilepsy and peripheral vascular dysfunctions focusing on endothelium, whose co-morbidities are involved in this neurological disorder.
Growing evidence has indicated the participation of RAS in some neurodegenerative disorders such as Alzheimer’s [33,34], Parkinson’s [35] and Huntington’s [36] diseases. Genetic models of epilepsy have played a crucial role in the elucidation of the pharmacological, electrophysiological, and neuroethological aspects of generalized seizures [37,38]. Of our particular interest is the genetically audiogenic susceptible rodents, the so-called Wistar audiogenic rats (WAR), which present severe co-morbidities [39]. The WAR strain was first obtained by Garcia-Cairasco [40] and has been proven useful for characterization of behavioral sequences in acute audiogenic seizures [41-44], audiogenic kindling studies [45-48], screening of new anticonvulsant agents [49] and research on the neurochemistry of audiogenic seizures [50,51]. Multiple inductions of audiogenic seizures in the WAR strain are a model of temporal lobe epilepsy, due to the recruitment of limbic areas such as hippocampus and amygdale [52].
Although it has been reported that Ang II release and Angiotensin receptors function/expression are altered in many cerebral areas in patients/animal models with neurological disorders, there are no data on the vascular function to Angiotensin peptides of genetically audiogenic susceptible rodents such as the Wistar audiogenic rats (WAR), a model of temporal lobe epilepsy and therefore it has been considered to evaluate the vascular function (vascular reactivity to Angiotensin peptides in WARs.
Therefore, it was hypothesized that the contractile response to Ang I and Ang II is altered in the aorta isolated from rats of the WAR strain. Because of this, our work was designed to evaluate the importance of the endothelium on Angiotensin-mediated vascular responses and to investigate the mechanisms involved in the vascular function of Wistar audiogenic rats.
2. METHODS
2.1. Animals
Male WAR and Wistar rats (Control) aged 80 days were employed. WAR belonged to the inbred strain of the Physiology Department of Faculdade de Medicina de Ribeirão Preto, University of São Paulo, Brazil [23]. Water and food were available ad libitum and room temperature was controlled at 23˚C, in a 12 h light/dark circle. All the experiments were performed in accordance with the recommendations of the Brazilian Society for Neuroscience and Behavior for animal experimentation and were approved by the Ethics Commitee on Animal Research of the School of Medicine of Ribeirão Preto, University of São Paulo, Brazil (CETEA, protocol n. 124/2005). All efforts were made in order to avoid any unnecessary animal suffering and to reduce the number of animals employed in the experiments. In order to characterize the model, in its genetic component, it was employed only naïve animals, it was not our aim to obtain epileptic behavior after sound stimulation, i.e., and there were no audiogenic stimuli to induce seizures in the WAR.
2.2. Systolic Blood Pressure Measurement
Systolic Blood Pressure (SBP) was measured in conscious rats by an indirect tail-cuff method (MLT125R Pulse Transducer/Pressure Cuff coupled to the PowerLab 4/S analogue-to-digital converter, AD Instruments Pty Ltd).
2.3. Vascular Reactivity Studies
Rats were killed by decapitation. The thoracic aorta was quickly removed, dissected out, and cut into 4 mm long rings. In some experimental protocols, the effect of the drugs in endothelium-denuded arteries was studiedin order to avoid interference from endothelium factors. To this end, the endothelium was mechanically removed by gently rolling the lumen of the vessel on a thin wire. The aortic rings were placed between two stainless-steel stirrups and connected to an isometric force transducer (Letica Scientific Instruments; Barcelona-Spain), to measure the tension in the vessels. The rings were placed in a 10 mL organ chamber containing Krebs solution with the following composition (mmol/L): NaCl 130, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, NaHCO3 14.9, glucose 5.5, and CaCl2 1.6. The solution was maintained at pH 7.4 and was gassed with 95% O2 and 5% CO2 at 37˚C. The rings were stretched to a basal tension of 1.5 g before allowing them to equilibrate for 60 min in the bath fluid, which was changed every 15 min. Endothelial integrity was qualitatively assessed by the degree of relaxation caused by acetylcholine (1 μmol/L) in the presence of the contractile tone induced by norepinephrine (0.1 μmol/L). For the studies, which required endothelium-intact aortas (E+), the rings that presented more than 90% of relaxation when stimulated with acetylcholine were considered. The arteries were considered endothelium-denuded (E–) if there was not any degree of relaxation upon stimulation with acetylcholine. Then, cumulative effect-concentration curves were constructed for Ang I and Ang II (10 ρmol/L - 10 μmol/L).
2.4. AT1, AT2, Mas and ACE Expression
This experimental procedure was designed to quantify the protein expression of AT1 and AT2 receptors as well as ACE, as a function of the expression of β-actin in control and WAR. Briefly, the aortic rings were isolated from WAR and control. Protein extraction from the tissues with or without endothelium was performed by maceration using lysis buffer consisting of Tris-HCl 10 mmol/L, pH 7.5, NaCl 150 mmol/L, EDTA 1 mmol/L, EGTA 1 mmol/L, SDS 0.1%, Nonidet P-40 1% and the following protease inhibitors: PMSF 2 mmol/L, SBTI 100 mg/mL, leupeptin 10 mg/mL, aprotinin100 mg/mL, benzamidin 10 mmol/L, and sodium orthovanadate 2 mmol/L. After 30 min of homogenization at 4˚C, the lysate was centrifuged for 15 min at 4˚C and 13,000 rpm, and total protein was measured using a commercial kit (Anresco). Then, 5 - 20 mg of total protein was separated by SDS-PAGE (12% for the Ang II receptors and 10% for ACE) and transferred to a nitrocellulose membrane, and Western Blotting was performed for AT1, AT2, and Mas receptors and for ACE. The membrane blocked with 3% BSA (in TTBS). The primary antibodies were diluted in TTBS + 3% BSA, 1:1000. The secondary antibodies were diluted in 1:3000 (anti-rabbit, purchased from Santa Cruz). The bands were developed by using an enhanced chemiluminescence kit. The membranes were deblotted, and Western Blotting for β-actin was developed.
2.5. Drugs
Acetylcholine, norepinephrine, Angiotensin I, and angiotensin II were obtained from Sigma Chem. Co. (St Louis, MO-USA). Solutions of the drugs were prepared in deionized water. All the antibodies employed in the Western Blotting analyses were purchased from Santa Cruz Biotechnology, Inc.
2.6. Data Analysis
Data are expressed as mean ± SEM. In each set of experiments, n indicates the number of studied rats.
The Western Blotting data were expressed by the intensity of the bands, which were measured by densitometry. Densitometric values obtained with the aid of the Image J program (http://rsb.info.nhi.gov/ij/) were used to calculate the rate of AT1 and AT2 receptors and ACE as a function of the β-actin expression, and the corresponding results were plotted using the Graph-Pad software (GraphPad, San Diego, CA).
The data were statistically analyzed by Student’s t-test or by ANOVA (one-way), followed by the NewmanKeuls post-test. Tests were employed according to the data. A value of p < 0.05 was considered significantly different.
3. RESULTS
The systolic pressure measured by the tail-cuff method was higher in WAR (120 ± 2 mmHg, n = 15, p < 0.05) as compared to the Wistar (90 ± 2.0 mmHg, n = 16) (Figure not shown).
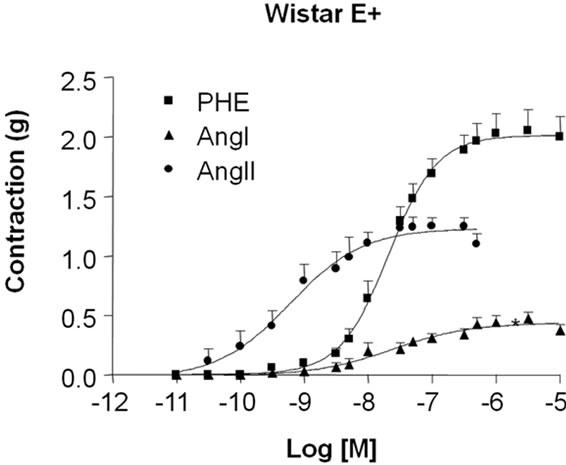
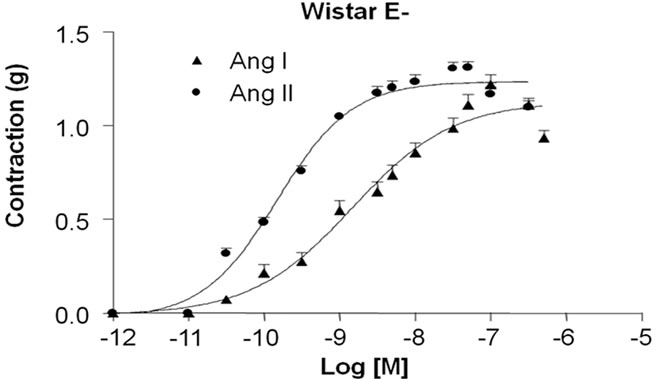
In order to study the vascular reactivity to the agonists of the renin-Angiotensin system (RAS), it was constructed concentration-effect curves for Ang I and Ang II. The influence of endothelium on the contractile responses induced by Ang I and Ang II in aortic rings isolated from WAR and Wistar was examined by using intact-endothelium (E+, Figures 1(a) and 1(b)) and denuded (E–, Figures 2(a) and (b)) arteries.
As shown in Figure 1(a), in Wistar E+ both Ang I and Ang II induced contractile response with the maximum effect (ME) of 0.45 ± 0.03 g (n = 6) and 1.16 ± 0.04 g (n = 6), respectively. This response was significantly higher for Ang II than Ang I (p < 0.01). On the other hand, Ang I (ME: 0.10 ± 0.008 g, n = 10) and Ang II-induced contractile responses (ME: 0.09 ± 0.008 g, n = 7) were almost abolished in aortic rings from WAR E+ (Figure 1(b)).
In order to investigate if this response was abolished only to RAS agonists, the contractile response to phenylephrine as an adrenergic agonist was also evaluated in intact endothelium aortic rings. Differently of the Ang I and Ang II-induced responses, PHE was able to trigger contractile response in both Wistar (ME: 2.0 ± 0.06 g, n = 6) (Figure 1(a)) and WAR (ME: 1.50 ± 0.02 g, n = 9) (Figure 1(b)).
As depicted in Figure 2, it was possible to obtain contractile response for both Ang I and Ang II in denuded aortic rings. However, the contractile responses induced by Ang I (ME: 0.52 ± 0.04 g, n = 11, p < 0.001) and Ang II (ME: 0.40 ± 0.03 g, n = 11, p < 0.05) were lower in WAR (Figure 2(b)) than the response observed in the Wistar (Figure 2(a)) for Ang I (ME: 1.13 ± 0.08 g, n = 11) and Ang II (ME: 1.24 ± 0.04 g, n = 11).
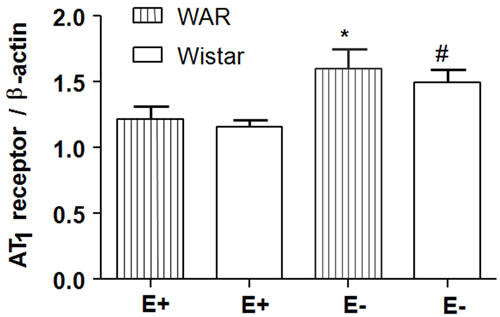
It was evaluated whether this differential vascular reactivity was due to the different expression of the RAS components in the aortic tissue with intact endothelium and in endothelium-denuded aortic rings isolated from WAR and Wistar rats. As illustrated in Figure 3, the AT1 receptor protein expression was increased upon endothelium removal in both WAR and Wistar. Thus, AT1 expression in WAR E+ (1.21 ± 0.09, n = 4) and Wistar E+ (1.16 ± 0.05, n = 4) were lower (p < 0.05) compared with WAR E– (1.60 ± 0.14, n = 4) and Wistar E– (1.50 ± 0.09, n = 4), respectively. There were no statistical differences
 (a)
(a)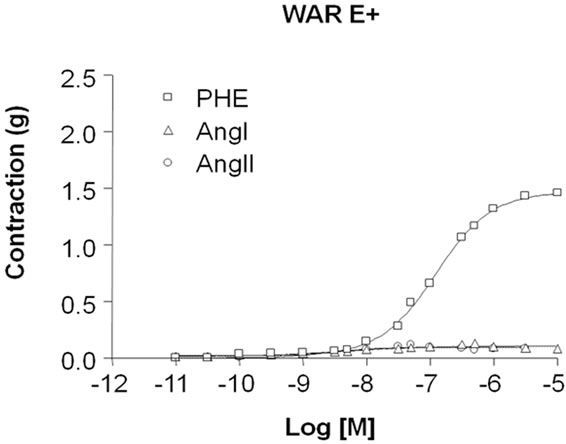 (b)
(b)
Figure 1. Contractile response induced by angiotensin I (Ang I), Angiotensin II (Ang II) and phenylephrine (PHE) in intact endothelium (E+) aortic rings isolated from Wistar (a) and WAR (b) rats. Cumulative concentrationeffect curves for Ang I, Ang II and PHE were constructed in E+ aortic rings isolated from Wistar (control, n = 6) and WAR (n = 10) rats. Results are presented as mean ± SEM and are expressed as grams (g) of tension.
 (a)
(a)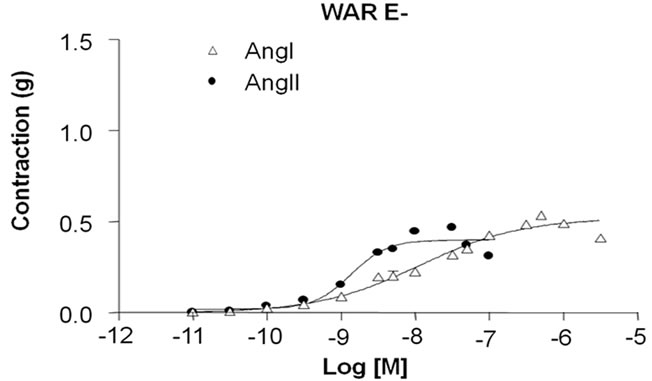 (b)
(b)
Figure 2. Contractile response induced by Angiotensin I (Ang I) and Angiotensin II (Ang II) in denuded endothelium (E–) aortic rings isolated from Wistar (A) and WAR (B) A: Cumulative concentration-effect curves for Ang I and Ang II were constructed in E– aortic rings isolated from Wistar rats (control, n = 11) and WAR rats (n = 11). Results are presented as mean ± SEM and are expressed as grams (g) of tension.
 (a)
(a)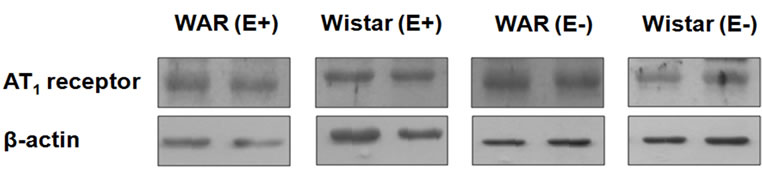 (b)
(b)
Figure 3. AT1 receptor protein expression in intact endothelium (E+) and denuded (E–) aorta from WAR and Wistar rats. Representative image of Western Blotting analysis (a) and densitometric group data (b) evaluating protein expression of the AT1 receptor in aortas from WAR and Wistar rats. Results are presented as mean ± SEM (n = 4). *Indicates difference between the group WAR E– compared with WAR E+ (p < 0.05). #Indicates difference between the group Wistar E– compared with Wistar E+ (p < 0.05). Data were analyzed by using one-way ANOVA (post test: Newman-Keuls multiple comparisons test).
between WAR E– and Wistar E– or WAR E+ and Wistar E+.
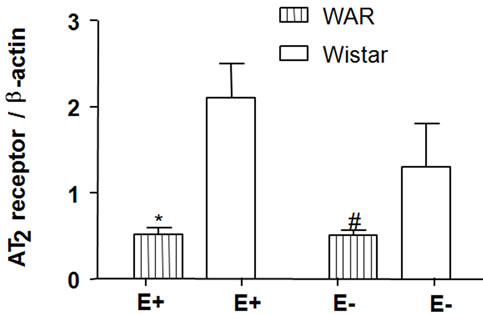
According to the Western Blotting analysis, the AT2 receptor expression was lower in both WAR E– (0.51 ± 0.06, n = 4, p < 0.05) and WAR E+ (0.52 ± 0.07, n = 4, p < 0.001) than in Wistar E– (1.30 ± 0.5, n = 4) and Wistar E+ (2.10 ± 0.4, n = 4), respectively (Figure 4). The presence or the absence of endothelium did not alter the expression of AT2 receptor either WAR or Wistar rats.
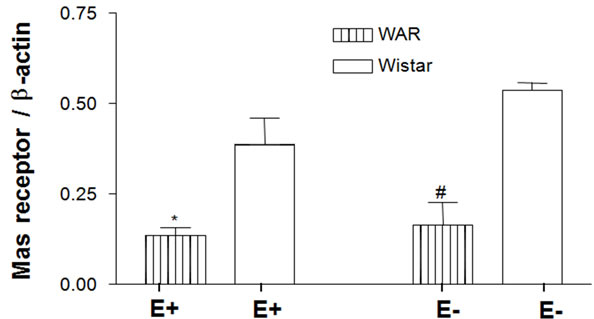
As represented in Figure 5 the expression of the Mas receptor in WAR E+ (0.13 ± 0.02 n = 4) and WAR E– (0.16 ± 0.06, n = 4) was lower than its expression in Wistar E+ (0.39 ± 0.07, n = 4, p < 0.05) and Wistar E– (0.54 ± 0.02, n = 4, p < 0.001), respectively. The presence or the absence of endothelium did not alter the expression of Mas receptor either WAR or Wistar rats.
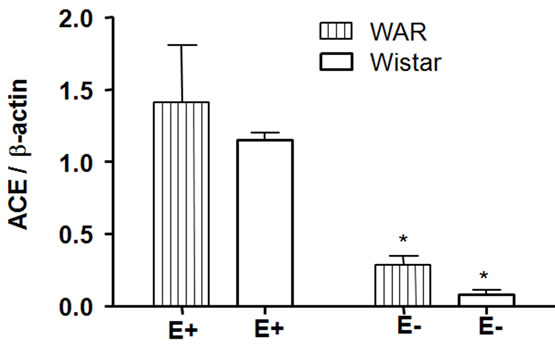
As shown in Figure 6, ACE expression was higher in both WAR E+ (1.41 ± 0.4, n = 4, p < 0.001) and Wistar E+ (1.15 ± 0.05, n = 4, p < 0.001) than in denuded aortas from WAR and Wistar, respectively. Endothelium removal decreased ACE expression in both WAR (0.28 ± 0.06, n = 4) and Wistar (0.11 ± 0.03, n = 4).
4. DISCUSSION
This is the first study concerning the involvement of the RAS on the peripheral vascular activity in an animal model to study epilepsy. It was evaluated the implications of the agonist of the RAS in the contractile dysfunction on the animal model of epilepsy, the WAR strain.
 (a)
(a)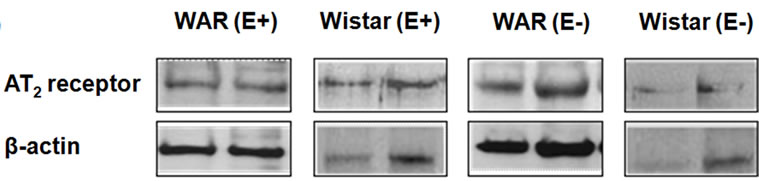 (b)
(b)
Figure 4. AT2 receptor protein expression in intact endothelium (E+) and denuded aorta (E–) from WAR and Wistar rats. Representative image of Western Blotting analysis (a) and densitometric group data (b) evaluating protein expression of the AT2 receptor in aortas from WAR and Wistar (control) rats. Results are presented as mean ± SEM (n = 4). *Indicates difference between E+ WAR and E+ Wistar (p < 0.001). #Indicates difference between E– WAR and E– Wistar (p < 0.05). Data were analyzed by using one-way ANOVA (post test: Newman-Keuls multiple comparisons test).
 (a)
(a)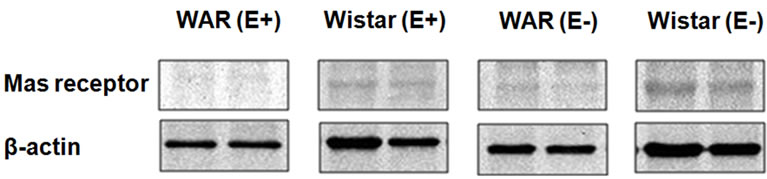 (b)
(b)
Figure 5. Mas receptor protein expression in intact endothelium (E+) and denuded (E–) aorta from WAR and Wistar rats. Representative image of Western Blotting analysis (a) and densitometric group data (b) evaluating protein expression of the Mas receptor in aortas from WAR and Wistar E+ and E–. Results are presented as mean ± SEM (n = 4). *Indicates difference between E+ WAR and E+ Wistar (p < 0.05). #Indicates difference between E– WAR and E– Wistar (p < 0.001). Data were analyzed by using one-way ANOVA (post test: Newman-Keuls multiple comparisons test).
 (a)
(a)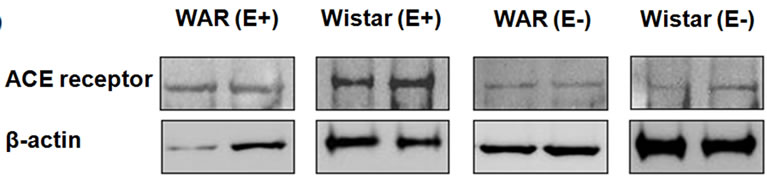 (b)
(b)
Figure 6. Angiotensin-Converting Enzyme (ACE) protein expression in intact endothelium (E+) and denuded (E–) aorta from WAR and Wistar rats. Representative image of Western Blotting analysis (a) and densitometric group data (b) evaluating protein expression of ACE in aortas from WAR and Wistar E+ and E–. Results are presented as mean ± SEM (n = 4). *Indicates difference between the WAR/Wistar E– group compared to the WAR/Wistar E+ group (p < 0.001), respectively. Data were analyzed by using one-way ANOVA (post test: Newman-Keuls multiple comparisons test).
It is well known that central and peripheral structures are affected by RAS activity in a way that one response influences others and vice-versa. Considering the epilepsy disorder, this relationship could contribute to the co-morbidities, including cardiovascular alterations. Although Ang II has been assumed to be the main peptide acting in the periphery, a large number of studies support the concept that it acts as a neurotransmitter or neuromodulator in specific neuronal pathways in the central nervous system [53-56]. In the present study, our experimental approach was focused on the peripheral vascular function of WAR a genetically developed strain [39], in animals who had not been stimulated for developing audiogenic seizures. Although it is not classified as a hypertensive status, in this study it was demonstrated that the level of systolic blood pressure of WAR was higher than that of the Wistar control group. This increased blood pressure level may be of concern because the impaired cardiovascular system is one of the possible mechanisms of cause or effect of the development of such seizures and the prevalence of sudden unexplained death in epileptic patients-SUDEP [57,58]. According to Scorza et al. [59], the abnormalities in cardiovascular system are involved in SUDEP. Since hypertension is an important risk factor for cardiovascular diseases, the increasing systolic blood pressure in epileptics could represent a point of the surveillance in the clinical practice. Adaptive cardiovascular reflexes could explain increased arterial pressure in WAR during seizures contributing to the cardiac manifestations that are integrated with the sympathetic and/or parasympathetic discharges triggered by seizures [60].
Our results show that the Ang II-induced vasoconstriction was potentiated by endothelium removal. These data are in accordance to Boulanger et al. [61]; Caputo et al. [62]; Gruetter et al. [63]; and Zhang et al. [64], who have demonstrated that this vasoconstriction is decreased by the release of endothelium-derived relaxing factors such as NO in non-epileptic vessels. In the present study, endothelium impaired the contractile response induced by both Ang I and Ang II. It has been reported that NO and Ang II have opposite effects on vascular responses as blood pressure regulators [65]. Since endothelium reduced the contractile response to Ang I and Ang II in aorta isolated from control (non-epileptic) rats, we suggest that an endothelium-relaxing factor could be involved in the lower response to these peptides. Otherwise, in Wistar rats relaxing endothelium-derived factors do not influence the contractile response induced by Ang II, since its response was similar in the presence and absence of endothelium. Other studies have also shown the dependence on endothelium of the contractile response induced by both Ang I and Ang II [66]. In addition, our results suggest that the RAS system rather than the adrenergic response is affected by endothelium in aorta from epileptic rats.
In the present study, it is interesting to note that the expression of AT1 receptors in E+/E– WAR aorta was similar to their expression in E+/ E– Wistar aorta, despite the differences in the contractile responses to Ang I/Ang II observed in E– WAR and in the E+ WAR aortas. Considering the AT1 receptor expression, one can suggest that it contributes to the higher contractile response in E– WAR as compared to the one detected in E+ WAR.
Due to vascular responses, AT1 receptor activation and AT2 receptor activation have opposite effects on blood pressure [65]. The activation of AT2 receptors by Ang II induces endothelium-dependent vasodilation in vascular beds [66]. In this sense, we have evaluated the AT2 receptor expression. As reported by Pueyo & Michel [67] and Harbel et al. [68], the Ang II receptors are mainly of the AT1 type in rat endothelial cells, but AT2 receptors have also been found. Herein, AT2 receptor expression in WAR aorta was not altered by the presence or absence of endothelium. AT2 receptor expression was higher in the Wistar animals compared with WAR, regardless of E+ or E– aortas. Otherwise, a tendency to lower AT2 expression was observed for Wistar E– aorta, which could explain the lower amplitude of the contractile response induced by Ang I and Ang II.
Although AT1 receptor activation and AT2 receptor activation have opposite effects, it has not yet been confirmed that the AT1/AT2 receptor heterodimer inhibits AT1 signals [67]. Therefore, Mas receptor investigation could be helpful in explaining our results about the absence of the contractile response to Ang I and Ang II stimulation in the presence of endothelium since Ang (1-7) acts as an endogenous antagonist of Ang I/Ang II and blocks Ang II-mediated vasoconstriction and cell proliferation [69,70]. In our hands, Mas expression was higher in control Wistar compared with WAR aorta, in an endothelium-independent way. These results support the statement that the Mas receptor is not responsible for the differences observed in the vascular reactivity responses in WAR.
In the present investigation, ACE expression was higher in E+ WAR and E+ Wistar aorta compared with E– WAR and E– Wistar. These results suggest that a higher level of Ang I could be converted to Ang II in WAR.
The removal of the endothelium suppresses relaxing regulator factors, mainly NO. To some extent, Ang II acting on smooth muscle cells triggers a contraction that is not counterbalanced by its vasodilatory effects endothelium-dependent. It seems that in WAR animal, endothelium exerts a vasodilatory control stronger than that existing in control Wistar rats. In fact, Ang II contractile response is modified in WAR rat and the regulation by endothelium is in part involved in that alteration of Ang II response.
5. CONCLUSION
The presence of endothelium impairs the contractile response induced by both peptides Ang I and Ang II in WAR aortas. It suggests that endothelial factors could have an important role in protecting the aorta vascular smooth muscle contraction stimulated by these peptides in this genetically epileptic rat strain, whose systolic blood pressure is lightly increased in naïve conditions (without developing seizures).
REFERENCES
- Earnest, M.P., Thomas, G.E., Eden, R.A. and Hossack, K.F. (1992) The sudden unexplained death syndrome in epilepsy: Demographic, clinical, and postmortem features. Epilepsia, 33, 310-316. doi:10.1111/j.1528-1157.1992.tb02321.x
- Ficker, D.M., So, E.L., Shen, W.K., Annegers, JF, O’Brien, P.C., Cascino, G.D. and Belau, P.G. (1998) Populationbased study of the incidence of sudden unexplained death in epilepsy. Neurology, 51, 1270-1274.
- Bell, G.S. and Sander, J.W. (2006) Sudden unexpected death in epilepsy. Risk factors, possible mechanisms and prevention: A reappraisal. Acta Neurologica Taiwanica, 15, 72-83.
- Nei, M. and Hays, R. (2010) Sudden unexpected death in epilepsy. Current Neurology and Neuroscience Reports, 10, 319-326. doi:10.1007/s11910-010-0116-4
- Scorza, F.A., Arida, R.M., Terra, V.C. and Cavalheiro, E.A. (2010) What can be done to reduce the risk of SUDEP? Epilepsy Behavior, 18, 137-138. doi:10.1016/j.yebeh.2010.04.046
- Shorvon, S. and Tomson, T. (2011) Sudden unexpected death in epilepsy. Lancet, 378, 2028-2038. doi:10.1016/S0140-6736(11)60176-1
- Tchekalarova, J. and Georgiev, V. (2005) Angiotensin pep tides modulatory system: How is it implicated in the control of seizure susceptibility? Life Science, 76, 955-970. doi:10.1016/j.lfs.2004.10.012
- Argañaraz, G.A., Konno, A.C., Perosa, S.R., Santiago, J.F.C., Boim, M.A., Vidotti, D.B., Varella, P.P., Costa, L.G., Canzian, M., Porcionatto, M.A., Yacubian, E.M., Sakamoto, A.C., Carrete, H. Jr., Centeno, R.S., Amado, D., Cavalheiro, E.A., Silva J.A. Jr. and Mazzacoratti, M.G. (2008) The renin-Angiotensin system is upregulated in the cortex and hippocampus of patients with temporal lobe epilepsy related to mesial temporal sclerosis. Epilepsia, 49, 1348-1357. doi:10.1111/j.1528-1167.2008.01581.x
- Peach, M.J. (1977) Renin-Angiotensin system: Biochemistry and mechanisms of action. Physiological Reviews, 57, 313-370.
- Ferrario, C.M., Jessup, J., Chappell, M.C., Averill, D.B., Brosnihan, K.B., Tallant E.A., Diz, D.I., and Gallagher, P.E. (2005) Effect of Angiotensin-converting enzyme inhibition and Angiotensin II receptor blockers on cardiac Angiotensin-converting enzyme 2, Circulation, 111, 2605- 2610. doi:10.1161/CIRCULATIONAHA.104.510461
- Ferrario, C.M. and Strawn W.B. (2006) Role of the reninAngiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. The American Journal of Cardiology, 98, 121-128. doi:10.1016/j.amjcard.2006.01.059
- Mascareno, E., Galatioto, J., Rozenberg, I., Salciccioli, L., Kamran, H., Lazar, J.M., Liu, F., Pedrazzini, T. and Siddiqui, M.A. (2012) Cardiac lineage protein-1 (CLP-1) regulates cardiac remodeling via transcriptional modulation of diverse hypertrophic and fibrotic responses and Angiotensin II-transforming growth factor β (TGFβ1) signaling axis. Journal of Biological Chemistry, 287, 13084- 13093. doi:10.1074/jbc.M111.288944
- Matsusaka, T. and Ichikawa, I. (1997) Biological functions of Angiotensin and its receptors. Annual Review of Physiology, 59, 395-412. doi:10.1146/annurev.physiol.59.1.395
- Santos, R.A.S., Campagnole-Santos, M.J. and Andrade, S. (2000) Angiotensin-(1-7): Na update. Regulatory Peptides, 91, 45-62. doi:10.1016/S0167-0115(00)00138-5
- Vickers, C., Hales, P., Kaushik, V., Dick, L., Gavin, J., Tang, J., Godbout, K., Parsons, T., Baronas, E., Hsieh, F., Acton, S., Patane, M., Nichols, A. and Tummino, P. (2002) Hydrolysis of biological peptides by human Angiotensinconverting enzyme related carboxypeptidase. Journal of Biological Chemistry, 277, 14838-14843. doi:10.1074/jbc.M200581200
- Oudit, G.Y., Crackower, M.A., Backx, P.H. and Penninger, J.M. (2003) The role of ACE2 in cardiovascular physiology. Trends in Cardiovascular Medicine, 13, 93-101. doi:10.1016/S1050-1738(02)00233-5
- Ferrario, C.M., Chappell, M.C., Tallant, E.A., Brosnihan, K.B. and Diz, D.I. (1997) Counterregulatory actions of Angiotensin-(1-7). Hypertension, 30, 535-541.
- Santos, R.A., Simoes e Silva, AC.,. Maric, C., Silva, D.M., Machado, R.P., de Buhr, I., Heringer-Walther, S., Pinheiro, S.V., Lopes, M.T., Bader, M., Mendes, E.P., Lemos, V.S., Campagnole-Santos, M.J., Schultheiss H.P., Speth, R. and Walther, T. (2003) Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proceedings of the National Academy of Sciences, 100, 8258-8263. doi:10.1073/pnas.1432869100
- De gasparo, M., Catt, C.J., Inagami, Wright, T.J.W. and Unger, T.H. (2000) International union of pharmacology. XXIII. The Angiotensin II receptors. Pharmacological Reviews, 52, 415-472.
- Ishii, K., Takekoshi, K., Shibuya, S., Kawakami, Y., Isobe, K. and Nakai, T. (2001) Angiotensin subtype-2 receptor (AT2) negatively regulates subtype-1 receptor (AT1) in signal transduction pathways in cultured porcine adrenal medullary chromaffin cells. Journal of Hypertension, 19, 1991-1999. doi:10.1097/00004872-200111000-00009
- Carey, R.M. (2005) Cardiovascular and renal regulation by the Angiotensin type 2 receptor: The AT2 receptor comes of age. Hypertension, 45, 840-844. doi:10.1161/01.HYP.0000159192.93968.8f
- Gendron, L., Payet, M.D. and Gallo-Payet, N. (2003). The Angiotensin type 2 receptor of Angiotensin II and neuronal differentiation: From observations to mechanisms. Journal of Molecular Endocrinology, 31, 359-372. doi:10.1677/jme.0.0310359
- Horiuchi, M., Akishita, M. and Dzau, V.J. (1999) Recent progress in Angiotensin II type 2 receptor research in the cardiovascular system. Hypertension, 33, 613-621.
- Stoll, M. and Unger, T. (2001) Angiotensin and its AT2 receptor: New insights into an old system. Regulatory Peptides, 99, 175-182. doi:10.1016/S0167-0115(01)00246-4
- Yan, C., Kim, D., Aizawa, T. and Berk, B.C. (2003) Functional interplay between Angiotensin II and nitric oxide: Cyclic GMP as a key mediator. Arteriosclerosis, Thrombosis, and Vascular Biology, 23, 26-36. doi:10.1161/01.ATV.0000046231.17365.9D
- Konishi, M. and Su, C. (1983) Role of endothelium in dilator responses of spontaneously hypertensive rat arteries. Hypertension, 5, 881-886.
- Wu, C.C., Chen, S.J. and Yen, M.H. (1993) Different responses to acetylcholine in the presence of nitric oxide inhibitor in rat aortae and mesenteric arteries. Clinical and Experimental Pharmacology and Physiology, 20, 405-412. doi:10.1111/j.1440-1681.1993.tb01717.x
- Osaka, T., Yamashita, H. and Koizumi, K. (1992) Inhibitory inputs to the subfornical organ from the AV3V: Involvement of GABA. Brain Research Bulletin, 29, 581- 587. doi:10.1016/0361-9230(92)90126-I
- Dampney, R.A.L., Hirooka, Y., Potts, P.D. and Head G.A. (1996) Functions of Angiotensin peptides in the rostral ventrolateral medulla. Clinical and Experimental Pharmacology and Physiology, 3, S105-S111. doi:10.1111/j.1440-1681.1996.tb02822.x
- Head, G.A. (1996) Role of AT1 receptors in the central control of sympathetic vasomotor function. Clinical and Experimental Pharmacology and Physiology, 3, S93-S98. doi:10.1111/j.1440-1681.1996.tb02820.x
- Muratami, H. (1996) Brain Angiotensin and circulatory control. Clinical and Experimental Pharmacology and Physiology, 23, 458-464. doi:10.1111/j.1440-1681.1996.tb02761.x
- Unger, T., Becker, H., Petty, M., Demmert, G., Schneider, B., Ganten, D. and Langer, R.E. (1985) Differential effects of central Angiotensin II and substance P on sympathetic nerve activity in conscious rats. Implications for cardiovascular adaptation to behavioral responses. Circulation Research, 56, 563-575.
- Barnes, N.M., Cheng, C.H., Costall, B., Naylor, R.J., Williams, T.J. and Wischik, C.M. (1991) Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer’s disease. European Journal of Pharmacology, 200, 289-292.
- Tian, J., Shi, J., Bailey, K., Harris, J.M., Pritchard, A., Lambert, J.C., Chartier-Harlin, M.C., Pickering-Brown, S.M., Lendon, C.L. and Mann, D.M. (2004) A polymerphism in the Angiotensin 1-converting enzyme gene is associated with damage to cerebral cortical white matter in Alzheimer’s disease. Neuroscience Letters, 354, 103-106.
- Grammatopoulos, T.N., Ahmadi, F., Jones, S.M., Fariss, M.W., Weyhenmeyer, J.A. and Zawada, W.M. (2005) Angiotensin II protects cultured midbrain dopaminergic neurons against rotenone-induced cell death. Brain Research, 1045, 64-71. doi:10.1016/j.brainres.2005.03.038
- Bird, E.D. (1980) Chemical pathology of Huntington’s disease. Annual Review of Pharmacology and Toxicology, 20, 533-551. doi:10.1146/annurev.pa.20.040180.002533
- Fisher, R.S. (1989) Animal models of epilepsy. Brain Research, 14, 254-278. doi:10.1016/0165-0173(89)90003-9
- Mishra, P.K., Kahle, E.H., Bettendorf, A.F., Dailey, J.W. and Jobe, P.C. (1993) Anticonvulsant effects of intracerebroventricularly administered norepinephrine are potentiated in the presence of monoamine oxidase inhibition in severe seizure genetically epilepsy-prone rats (GEPR- 9s). Life Sciences, 52, 1435-1441. doi:10.1016/0024-3205(93)90067-D
- Doretto, M.C., Fonseca, C.G., Lôbo, R.B., Terra, V.C., Oliveira, J.A.C. and Garcia-Cairasco, N. (2003) Quantitative study of the response to genetic selection of the Wistar Audiogenic Rat Strain (WAR). Behavior Genetics, 33, 33-41. doi:10.1023/A:1021099432759
- Garcia-Cairasco N. (1989) Neural and ethological relations in the evaluation of motor control changes. II. Experimental models. Arquivos de Neuro-Psiquiatria, 47, 172-181. doi:10.1590/S0004-282X1989000200008
- Terra, V.C. and Garcia-Cairasco, N. (1992) Neuroetho-logical evaluation of audiogenic seizures and audiogeniclike seizures induced by microinjection of bicuculline into the inferior colliculus II. Effects of nigral clobazam microinjections. Behavioural Brain Research, 52, 19-28. doi:10.1016/S0166-4328(05)80321-3
- Terra, V.C. and Garcia-Cairasco, N. (1994) NMDA-dependent audiogenic seizures are differentially regulated by inferior colliculus subnuclei. Behavioural Brain Research, 62, 29-39. doi:10.1016/0166-4328(94)90035-3
- Tsutsui, J., Terra, V.C., Oliveira, J.A. and Garcia-Cairasco, N. (1992) Neuroethological evaluation of audiogenic seizures and audiogenic-like seizures induced by microinjection of bicuculline into the inferior colliculus I. Effects of midcollicular knife cuts. Behavioural Brain Research, 52, 7-17. doi:10.1016/S0166-4328(05)80320-1
- Garcia-Cairasco, N., Terra, V.C. and Doretto, M.C. (1993) Midbrain substrates of audiogenic seizures in rats. Behavioural Brain Research, 58, 57-67. doi:10.1016/0166-4328(93)90090-D
- Garcia-Cairasco, N., Wakamatsu, H., Oliveira, J.A., Gomes, E.L., Del Bel, E.A. and Mello LE. (1996) Neuroethological and morphological (Neo-Timm staining) correlates of limbic recruitment during the development of audiogenic kindling in seizure susceptible Wistar rats. Epilepsy Research, 26, 177-192. doi:10.1016/S0920-1211(96)00050-2
- Moraes, M.F., Del Vecchio, F., Terra, V.C. and GarciaCairasco, N. (2000) Time evolution of acoustic information processing in the mesencephalon of Wistar rats. Neuroscience Letters, 284, 13-16. doi:10.1016/S0304-3940(00)00978-2
- Romcy-Pereira, R.N. and Garcia-Cairasco, N. (2003) Hippocampal cell proliferation and epileptogenesis after audiogenic kindling are not accompanied by mossy fiber sprouting or Fluoro-Jade staining. Neuroscience, 119, 533- 546. doi:10.1016/S0306-4522(03)00191-X
- Galvis-Alonso, O.Y., Cortes De Oliveira, J.A. and GarciaCairasco, N. (2004) Limbic epileptogenicity, cell loss and axonal reorganization induced by audiogenic and amygdala kindling in wistar audiogenic rats (WAR strain). Neuroscience, 125, 787-802. doi:10.1016/j.neuroscience.2004.01.042
- Doretto, M.C., Garcia-Cairasco, N., Pimenta, N.J., Souza, D.A. and Tatsuo M.A. (1998) Dipyrone, a novel anticonvulsant agent? Insights from three experimental epilepsy models. NeuroReport, 9, 2415-2421. doi:10.1097/00001756-199807130-00048
- Ribeiro, A.M., dos Santos, W.F. and Garcia-Cairasco, N. (2000) Neuroethological analysis of the effects of spider venom from Scaptocosa raptoria (Lycosidae: Araneae) microinjected in the lateral ventricle of Wistar rats. Brain Research Bulletin, 52, 581-588. doi:10.1016/S0361-9230(00)00300-2
- Rossetti, F., Rodrigues, M.C., De Oliveira, J.A. and Garcia-Cairasco, N. (2006) EEG wavelet analyses of the striatum-substantia nigra pars reticulata-superior colliculus circuitry: audiogenic seizures and anticonvulsant drug administration in Wistar audiogenic rats (War strain). Epilepsy Research, 72, 192-208. doi:10.1016/j.eplepsyres.2006.08.001
- Pereira, M.G., Gitaí, D.L., Paçó-Larson, M.L., Pesquero, J.B., Garcia-Cairasco, N. and Costa-Neto CM. (2008) Modulation of B1 and B2 kinin receptors expression levels in the hippocampus of rats after audiogenic kindling and with limbic recruitment, a model of temporal lobe epilepsy. International Immunopharmacology, 8, 200-205. doi:10.1016/j.intimp.2007.07.028
- Allen, A.M., Zhuo, J. and Mendelsohn, F.A. (2001) AT1-receptors in the central nervous system. Journal of the ReninAngiotensin-Aldosterone System, 2, S95-S101. doi:10.1177/14703203010020011701
- Wright, J.W. and Harding, J. (2004) The brain Angiotensin system and extracellular matrix molecules in neural plasticity, learning and memory. Progress in Neurobiology, 72, 263-293. doi:10.1016/j.pneurobio.2004.03.003
- Wright, J.W., Reichert, J.R., Davis, C.J. and Harding, J.W. (2002) Neuronal plasticity and the brain renin-Angiotensin system. Neuroscience & Biobehavioral Reviews, 26, 529- 552. doi:10.1016/S0149-7634(02)00019-2
- Pan, H.L. (2004) Brain Angiotensin II and synaptic transmission. The Neuroscientist, 10, 422-431. doi:10.1177/1073858404264678
- Sundt, T.M., Sharborough, F.W., Piegras, D.G., Kearns, T.P., Messick, J.M. and O’Fallon, W.M. (1981) Correlation of cerebral flow and electroencephalographic changes during carotid endarterectomy. Mayo Clinic Proceedings, 56, 533-543.
- Lhatoo, S.D., Langan, Y. and Sander, J.W. (1999) Sudden unexpected death in epilepsy. Postgraduate Medical Journal, 75, 706-709.
- Scorza, F.A., Arida, R.M., Cavalheiro, E.A. and Silva, M.R. (2012) Because scientists are unable to explain the unexplained, screening for cardiovascular abnormalities is a good method to protect against sudden unexpected death in patients with epilepsy. Clinics (Sao Paulo), 67, 1-2. doi:10.6061/clinics/2012(01)01
- [61] Sevcencu, C. and Struijk, J.J. (2010) Autonomic alterations and cardiac changes in epilepsy. Epilepsia, 51, 725- 737.
- [62] Boulanger, C.M., Caputo, L. and Levy B.I. (1995) Endothelial AT,-mediated release of nitric oxide decreases angiotensin contraction in rat carotid artery. Hypertension, 26, 752-757.
- [63] Caputo, L., Benessiano, J., Boulanger, C.M. and Levy, B. (1995) Angiotensin II increases cGMP content via endothelial angiotensin II AT1 subtype receptors in the rat carotid artery. Arteriosclerosis, Thrombosis, and Vascular Biology, 15, 1646-1651. doi:10.1161/01.ATV.15.10.1646
- [64] Gruetter, C.A., Ryan, C.A., Lemke, S.M., Bailly, D.A., Fox, M.K. and Schoepp, D.D. (1988) Endothelium-dependent modulation of angiotensina II-induced contraction in blood vessels. European Journal of Pharmacology, 146, 85-95. doi:10.1016/0014-2999(88)90489-X
- [65] Zhang, J., Van Meel, C.A., Pfaffendorf, M., Zhang, J. and Van Zwiten, P.A. (1994) Endothelium-dependent, nitric oxide-mediated inhibition of Angiotensina II-induced contraction in rabbit aorta. European Journal of Pharmacology, 262, 247-253. doi:10.1016/0014-2999(94)90738-2
- [66] Harada, S., Nakata, T., Oguni, A., Kido, H., Hatta, T., Fukuyama, R., Fushiki, S., Sasaki, S. and Takeda, K. (2002) Contrasting Effects of Angiotensin Type 1 and 2 Receptors on Nitric Oxide Release under Pressure. Hypertension Research, 25, 779-786. doi:10.1291/hypres.25.779
- [67] Lemos, V.S., Côrtes, S.F., Silva, D.M.R., CampagnoleSantos, M.J. and Santos, R.A.S. (2002) Angiotensin-(1-7) is involved in the endothelium-dependent modulation of phenylephrine-induced contraction in the aorta of mRen-2 transgenic rats. British Journal of Pharmacology, 135, 1743-1748. doi:10.1038/sj.bjp.0704630
- [68] Pueyo, M.E. and Michel, J.B. (1997) Angiotensin II receptors in endothelial cells. General Pharmacology, 29, 691-696. doi:10.1016/S0306-3623(97)00021-9
- [69] Haberl, R., Anneser, F., Villringer, A. and Einhiupl, K.M. (1990) Angiotensin induces endothelium-dependent vasodilatation of rat cerebral arterioles. American Journal of Physiology, 258, 1840-1846.
- [70] Abdalla, S., Lother, H., Abdel-Tawab, A.M. and Quitterer, U. (2001) The Angiotensina II AT2 receptor is an AT1 receptor antagonist. The Journal of Biological Chemistry, 276, 39721-29726. doi:10.1074/jbc.M105253200
- [71] Tallant, E.A., Diz, D.I. and Ferrario, C.M. (1999) Stateof-the-Art lecture. Antiproliferative actions of Ang Iotensin-(1-7) in vascular smooth muscle. Hypertension, 34, 950-957.